

Abstract
Background
Salt sensitivity (SS) of blood pressure (BP) affects 25% of adults, shares comorbidity with hypertension, and has no convenient diagnostic test. We tested the hypothesis that urine-derived exfoliated renal proximal tubule cells (RPTCs) could diagnose the degree of an individual's SS of BP.
Methods
Subjects were selected who had their SS of BP determined 5 y prior to this study (salt-sensitive: ≥7 mm Hg increase in mean arterial pressure (MAP) following transition from a random weekly diet of low (10 mmol/day) to high (300 mmol/day) sodium (Na+) intake, N = 4; inverse salt-sensitive (ISS): ≥7 mm Hg increase in MAP transitioning from a high to low Na+ diet, N = 3, and salt-resistant (SR): <7 mm Hg change in MAP transitioned on either diet, N = 5). RPTC responses to 2 independent Na+ transport pathways were measured.
Results
There was a negative correlation between the degree of SS and dopamine-1 receptor (D1R) plasma membrane recruitment (y = −0.0107x + 0.68 relative fluorescent units (RFU), R2 = 0.88, N = 12, P < 0.0001) and angiotensin II-stimulated intracellular Ca++ (y = −0.0016x + 0.0336, R2 = 0.7112, P < 0.001, N = 10) concentration over baseline.
Conclusions
Isolating RPTCs from urine provides a personalized cell-based diagnostic test of SS index that offers advantages over a 2-week controlled diet with respect to cost and patient compliance. Furthermore, the linear relationship between the change in MAP and response to 2 Na+ regulatory pathways suggests that an individual's RPTC response to intracellular Na+ is personalized and predictive.
Keywords: Salt sensitivity, Cell-based assay, Renal cells, Urinalysis, Sodium diet, Personalized medicine
1. Introduction
The long-term effect of high dietary sodium (Na+) on blood pressure (BP) and overall health is still under considerable debate [1]. While it is well documented that a high Na+ diet may lead to increased BP and cardiovascular risk in some individuals, there is also a segment of the population that demonstrates an increase in BP and cardiovascular risk on a low Na+ diet [1,2]. Indeed, the relationship between mortality, end-stage renal disease, or hypertension and the amount of Na+ that is consumed in the diet is “J” shaped, indicating that there is cardiovascular risk associated with high or low Na+ intake in some individuals [3–5]. Thus, there is a significant need for definitive diagnostic tools in order to stratify test subjects according to their ability to excrete a salt load so that health outcomes can be properly predicted [6].
Cell-based assays are allowing the diagnosis, prediction of clinical outcomes, and response to therapy in an increasing number of diseases including cancer, organ rejection, and diabetes [7]. Exfoliated organ cells have the potential to supplement current biomarker-based analyses since cell-based assays can measure aberrations in cell activity that may be present in complex chronic diseases. The kidneys play a major role in regulating BP as they regulate water and electrolyte homeostasis [6,8–10]. Excessive renal Na+ reabsorption, especially in the renal proximal tubule and thick ascending limb of Henle, leads to essential hypertension [11,12]. Salt sensitivity (SS) of BP is related to but distinct from hypertension in that there is an abnormal increase in BP following high Na+ intake even in subjects with normal or optimal BP. SS, even if the increase in blood pressure does not fulfill the definition of hypertension, leads to the same negative health outcomes as subjects with hypertension [13], and is a major public health challenge that affects 25% of middle-aged Americans, with a high prevalence in African-Americans. Despite its high prevalence and significant morbidity and mortality, the diagnosis of SS can only be determined using an extensive dietary salt loading and salt depletion study performed in an inpatient setting (i.e., clinical research center). Furthermore, the results obtained with a simpler and more rapid protocol [13] correlate poorly with the more generally accepted 2-week controlled Na+ diet protocol [14–17].
Target organ damage (e.g., kidney failure, stroke, and heart disease) resulting from hypertension and SS could be a result of the higher BP required to excrete the same amount of Na+ [3–5]. An important mechanism in the increase in Na+ excretion following a salt load is the inhibition of renal Na+ reabsorption via dopamine and dopamine 1-like receptors (D1R/D5R) which regulate >50% of total renal Na+ transport [18]. Defects in dopamine-mediated natriuresis have been directly implicated in the etiology of hypertension [19] that impairs D1R/D5R function, most evident in the renal proximal tubule (RPT) and RPT cells (RPTCs) [20].
Dopamine receptor activation or an increase in intracellular Na+ has been shown to recruit the predominantly cytoplasmic D1R to the plasma membrane [21]. The recruitment of D1R to the plasma membrane is important in the D1R/D5R-mediated decrease in Na+ transport in RPTCs [21]. Defects in D1R action in RPTCs, including D1R recruitment to the plasma membrane in the RPTCs, have been shown in human essential hypertension [19,20,22]. An abnormal interaction between renal D1R and angiotensin type 1 receptor (AT1R) in RPTCs [23–25] in the renal renin–angiotensin system may also lead to Na+ retention in hypertension, including salt-sensitive hypertension [26,27]. There is an aberrant interaction between D1R and AT1R in RPTCs from humans with essential hypertension [28].
Since SS involves aberrant regulation of Na+ transport in the RPT, we hypothesized that the use of living RPTCs isolated from urine (i.e., a virtual renal biopsy) could serve as a convenient method to assess RPT physiology almost instantaneously. Studies using living RPTCs isolated from urine have provided important clues about the etiology of disease [7,29–32]. We hypothesized that the physiological consequences of stimulation of D1R and AT1R could be recapitulated in freshly isolated RPTCs. Specifically, we tested the hypothesis that the intracellular Na+-induced recruitment of the D1R from cytosol to the plasma membrane and an increase in intracellular Ca++ with angiotensin II may be associated with the salt-sensitive phenotype. We further hypothesized that the degree of SS (i.e., SS index) is proportional to the response to D1R and AT1R stimulation.
2. Methods
2.1. Subjects
Our study was performed on 12 subjects selected from a pool of subjects that had been characterized 1 to 5 y previously for their SS index using a 2-week randomized controlled diet protocol [33]. Appropriate power calculations were performed for within group and between group comparisons at a 2-tail P ≤ 0.05 level of significance. During their initial characterization for SS index, subjects were placed on an isocaloric constant diet containing 1 gram protein/kg body weight/day and either 300 mmol/day Na+ (high Na+) or 10 mmol/day Na+ (low Na+), and 60 mmol/day potassium (K+) for 7 days (on each diet) in a randomized alternating protocol. At the time of study, the isocaloric and protein controlled diet was used to eliminate effects of dietary protein variations on renal dopamine formation. Body weight was recorded, and heart rate and arterial blood pressure were measured in the sitting position (at least 5 min rest) with an automated BP device (DINAMAP ProCare, Critikon) which has been validated according to the International Protocol in an adult population. Mean arterial pressure (MAP) was calculated as MAP = diastolic pressure (DP) + [0.33 + (heart rate (HR) × 0.0012)] × [systolic pressure (SP)]. Twenty-four hour urine collections were analyzed daily for Na+, K+ and creatinine to determine the state of Na+ balance. Exclusion criteria included a history of accelerated or malignant hypertension, contraindications to discontinuing antihypertensive therapy, or renal dysfunction as evidenced by a serum creatinine >1.5 mg/dl or proteinuria >300 mg/day [34]. Subjects which had previously experienced a myocardial infarction, stroke or transient ischemic episode, congestive heart failure, severe small vessel disease, or concurrent pregnancy were also excluded as previously described [33].
One to five years after their initial characterization, subjects were randomly selected from each pool of salt-sensitive, salt-resistant (SR), and inverse salt-sensitive (ISS) and then reenrolled in this follow-up study. Morning to mid-day voided urines were collected by normotensive subjects and brought to the laboratory for viable cell isolation and testing. There was no significant correlation between each subject's SS index and urine Na+, K+ or osmolarity indicating that the measured RPTC responses were not related to the amount of electrolytes in the urine.
2.2. Renal cell isolation
There is slow normal turnover of cells in normal healthy individuals which results in shedding of renal tubule cells [35]. The isolation and culture of RPTCs have been reported in the literature from rats and humans [7,29–32,36]. RPTCs were isolated to approximately 98% purity with a yield of 150–200 cells/void (approximately 120 ml). In order to validate that cells that were isolated were RPTCS, we used 2 independent isolation methods, as well as antibodies and lectins to identify markers on their outer membrane (vide infra).
Briefly, our method of isolating RPTCs from urine involved removing casts, cuboidal epithelial cells, and other contaminants using the following multi-step purification procedures. This procedure allowed >95% recovery of exfoliated RPTCs as determined from an internal control consisting of cultured RPTCs bioengineered to express tandem tomato fluorescent protein. The entire voided urine sample from a well hydrated individual (100 ml minimum) was centrifuged at 800 ×g for 5 min and allowed to decelerate without a brake in order not to disturb the pellet. The pellet was carefully aspirated with a 10 ml volume of urine and placed into a 15 ml tube. The pellet was washed 3 times in 10 ml PBS++ (PBS with Ca++ and Mg++, pH 7.4) and centrifuged to produce a new pellet following each wash. The final pellet was collected, filtered through a 40 μm filter, and placed into a 1.5 ml microfuge tube. RPTCs were isolated using 2 different methods with similar results. In the first method, 2 μg biotinylated BSA in 200 μl of serum-free media were added to the urine pellet for 30 min at 4 °C, followed by 25 μl Cellection Biotin Binder Kit, according to manufacturer's instructions (Invitrogen). This method utilizes the high affinity binding of cubulin/megalin complex for albumin and expressed in RPT but not in other nephron segments [37]. In the second method, we added 2 μg biotinylated CD13 monoclonal antibody in 200 μl of PBS++ (with 0.1% BSA and 2 mmol/l EDTA) to the urine pellet for 30 min at 4 °C. This was followed by the addition of 25 μl of Anti-Biotin Microbeads (Miltenyi Biotec), following manufacturer's instructions. Mixing with magnetic particles was performed for 30 min at 4 °C with end-over-end rotation, at 12 rotations/min. The supernatant containing the suspended cells was collected and centrifuged in a separate 1.5 ml microfuge tube at 200 ×g for 5 min. The isolated cells were RPTCs, as determined by gamma glutamyl transpeptidase (GGT) [38], aminopeptidase N (CD13) [39,40], and Lotus tetragonolobus agglutinin (LTA) [41] staining. In some experiments, for controls, we used cultured human RPTCs isolated from kidney tissue as previously published [42]. Na+ hydrogen exchanger 3 (NHE3) is expressed in the renal proximal tubule and thick ascending limb; Na+ potassium-2 chloride cotransporter (NKCC2) is expressed only in the thick ascending limb; Na+ chloride cotransporter (NCC) is only expressed in the distal convoluted tubule while epithelial Na+ channel (ENaC) is expressed only in late connecting tubule and collecting duct [43]. Therefore, the presence of NHE3 but not NKCC2, NCC, or ENaC indicates the presence of RPTCs not contaminated by cells from the other nephron segments. As indicated above, cubulin/megalin is only expressed in RPT and Tamm Horsfall protein (THP) in thick ascending limb of Henle [40].
2.3. Identification of collected RPTCs
Isolated RPTCs were imaged both by light microcopy and by an ImageStream (EMD Millipore) flow cytometer that is capable of taking photos of individually identifiable cells (Fig. 1). Flow images were obtained under bright-field illumination, dark-field illumination, or fluorescent illumination following CD13-Alexa-488 staining. RPTCs were stained using a variety of methods which have demonstrated selectivity for RPTCs (vide supra) [40,42].
Fig. 1.
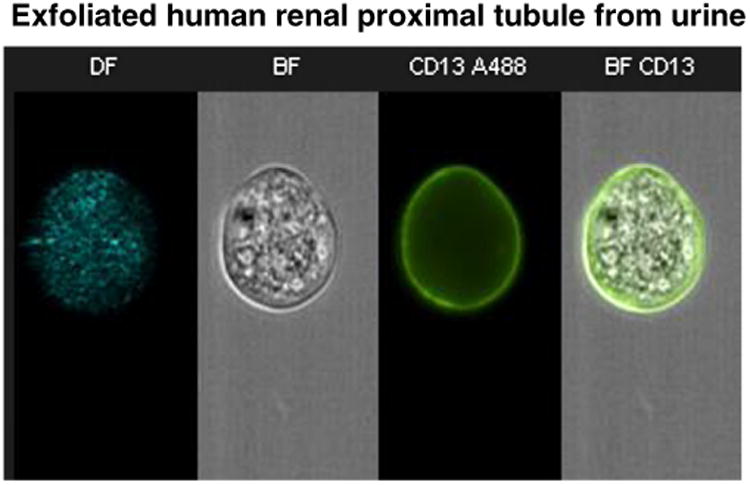
Individual exfoliated renal proximal tubule cells (RPTCs) were imaged in an ImageStream flow cytometer (one cell is shown with various imaging methods in each column). The first column shows dark field illumination (DF). The second column shows bright field illumination (BF). The third column shows staining with CD13 antibody labeled with Alexa-488. The final photo was produced by merging BF and CD13 images demonstrating a high degree of intracellular granularity in cells expressing CD13 which is characteristic of RPTCs.
2.4. Renal cell viability studies
RPTCs were isolated according to the protocol described (Section 2.2.), placed in RPTC culture media and incubated at 37 °C for 24 h. Cell viability was determined using Invitrogen's Live/Dead assay and trypan blue dye at 12 h, 24 h, and over several weeks. Previous studies by others have demonstrated that exfoliated urine cells could be maintained in a healthy state for several weeks and even reverted to immortalized progenitor stem cells [36]. In order to control for within subject variability, we measured D1R membrane recruitment and angiotensin II-stimulated increase in intracellular Ca++ for each subject multiple times over several months. We also collected random urines 18 times from one subject over 40 days and measured D1R receptor recruitment.
2.5. Intracellular Na+-mediated D1R recruitment
Intracellular Na+ was increased through the use of the Na+ ionophore monensin (10 μmol/l) as reported [21]. D1R recruitment was determined as the difference between cell surface D1R measured before and after monensin treatment, using anti-D1R antibodies labeled with 2 different fluorescent dyes, the specificities of which have been reported [20]. Urine-derived RPTCs, isolated using biotinylated BSA, were centrifuged (200 ×g, 5 min) onto a single well of a poly-D-lysine coated glass-bottomed 96-well plate. An extracellular epitope-specific antibody to the D1R was labeled with Alexa-488 and Alexa-647 using an antibody labeling kit (Life Technologies, Alexa Fluor ®, Grand Island, NY). The Alexa-488-labeled anti-D1R antibody was incubated with the isolated RPTCs at 37 °C for 30 min to measure the basal amount of plasma membrane D1R. The well was then washed 3 times with serum-free media. Monensin at 10 μmol/l (found to increase intracellular Na+ to 20 mmol/l in 30 min), along with the Alexa-647 labeled anti-D1R antibody, was added into the well and incubated at 37 °C for 30 min and then washed 3 times with PBS++. Binding of the second labeled antibody was used as a measure of the monensin-induced plasma membrane recruitment of D1R. Values were expressed as the relative fluorescent unit (RFU) ratio: Alexa-647/Alexa-488. Important controls included cells incubated with monensin at 4 °C instead of 37 °C, which had <10% of basal signal of the cells incubated at 37 °C since plasma membrane recruitment of G protein-coupled receptors is temperature-dependent. RPTCs possess an inherent small autofluorescent signal which was measured initially and subtracted from the final readings. Omitting either antibody produced no detectable signal after correcting for autofluorescence.
2.6. Angiotensin II-mediated intracellular Ca++ stimulation
The measurement of intracellular Ca++ using fluorescent dyes is well accepted [44]. We chose an angiotensin II concentration of 10 nmol/l for intracellular Ca++ stimulation to mimic previously reported endogenous levels of angiotensin II in the tubular lumen of rats [45,46]. Cells isolated using biotinylated CD13 monoclonal antibody were attached to a poly-D-lysine coated glass bottomed 96-well plate. The set-up was similar to the assay for D1R recruitment, except that the cells were allowed to recover for 1 h at 37 °C in serum-free media. Cells were loaded with Fura-2 AM (Ca++ sensitive dye) according to manufacturer's instructions (Invitrogen) in PBS plus calcium and magnesium (PBS++) at room temperature for 1 h. Cells were washed with PBS++ and confocal ratiometric time-lapse microscopy was performed for 30 min at 2-minute intervals with 10 nmol/l angiotensin II added at the 4 minute time point. One control was pre-incubation with 10 μmol/l losartan (AT1R antagonist), which completely blocked the rise in intracellular calcium. Another important control was the addition of vehicle control instead of angiotensin II, which similarly showed no significant change from baseline.
3. Results
3.1. RPTCs in urine
Isolation and purification of RPTCs were necessary since urine contains squamous, transitional, immune, and other cells derived from the kidney and urinary tract. The contaminating cells may offer future opportunities for additional cell based assays. Urinary RPTCs were positively identified by the presence of CD13 antibody binding, LTA-lectin binding, NHE3 and GGT immunostaining, and negative staining for other nephron segment cell markers (THP, NKCC2, NCC, and ENaC) [40,43]. We also imaged each cell in an ImageStream Flow Cytometer calibrated using cultured RPTCs (Fig. 1). ImageStreamphotographed cells demonstrated highly granular cytoplasm typical of RPTCs and demonstrated a positive stain with fluorescently labeled CD13. Cell viability was determined to be 95% (Invitrogen Live/Dead assay and trypan blue dye).
Inter-subject yield varied from 10 to 300 cells/dl. On average, 5% of urine specimens did not yield sufficient RPTCs for analysis. Our isolation procedure demonstrated a 92.3 ± 5.1% (N = 5) capture efficiency, and a 99.4 ± 0.4% (N = 5) capture selectivity using known quantities of cultured fluorescently-tagged cultured RPTCs spiked into urine specimens. Red fluorescently tagged human cultured RPTCs were generated by stably infecting tandem tomato fluorescent protein in the lentiviral vector pLVXpuro (Clontech Laboratories, Inc., Mountain View, CA). Infected cells were selected using puromycin (Sigma-Aldrich, St. Louis, MO). RPTCs expressing the highest amount of red fluorescent protein were sorted using an automated cell sorter in the Flow Cytometry Core laboratory at The University of Virginia.
Isolated exfoliated urine cells could be cryopreserved for at least one month in standard cell culture cryopreservation media in liquid nitrogen with a 57.16 ± 9.15% (N = 12) viability after thawing. However, all analytical results described here are from studies using freshly isolated RPTCs maintained in culture for several hours. We have demonstrated that RPTCs can be maintained for several weeks in culture, as previously demonstrated by others [36].
3.2. D1R membrane recruitment
We hypothesized that D1R-induced membrane recruitment would differ between SR and salt-sensitive individuals because of a defect in D1R function, which is important in the increased-RPT Na+ reabsorption in hypertension and/or SS [19]. We initially tested our hypothesis by measuring D1R recruitment to the plasma membrane in immortalized cultured human RPTCs that demonstrated a normal or an abnormal coupling of D1R to adenylyl cyclase (AC) [47]. A monensin-induced increase in intracellular Na+ produced an increase in plasma membrane D1R recruitment in D1R/AC-coupled cells (termed nRPTCs for normally-coupled RPTCs) but not in D1R/AC-uncoupled RPTCs (termed uRPTCs for uncoupled RPTCs [40]) (Fig. 2). We then tested our hypothesis in RPTCs isolated from the urine of normotensive human subjects. Urine-derived RPTCs demonstrated D1R plasma membrane recruitment as a continuous response that was negatively correlated with the subject's SS index (y = −0.0107x + 0.6784, R2 = 0.883, P < 0.0001, N = 12; Fig. 3). In order to test for intra-subject variability, we collected 18 random daily urine specimens from one of our test subjects with collection days spanning over a 40 day period. Intra-subject D1R membrane recruitment was inherently stable over time with a 3.8 ± 0.9% (coefficient of variation, N = 18) variability when measured over 40 days.
Fig. 2.

We tested our method of measuring D1R membrane recruitment in cultured renal proximal tubule cells (RPTCs) that were obtained from surgical specimens. We used cells that had been previously shown to demonstrate either a normal or abnormal D1R coupling to adenylyl cyclase, the latter due to a D1R defect. The normally coupled cells (nRPTC) demonstrated a robust recruitment of D1R to the plasma membrane following a monensin (MON, 10 μmol/l)-induced increase in intracellular Na+ (*P < 0.01 vs vehicle (VEH), N = 9, ANOVA, Holm-Sidak test). The uncoupled cells (uRPTC) did not demonstrate significant membrane D1R recruitment under the same experimental conditions. Data are mean ± standard error of the mean (SEM).
Fig. 3.
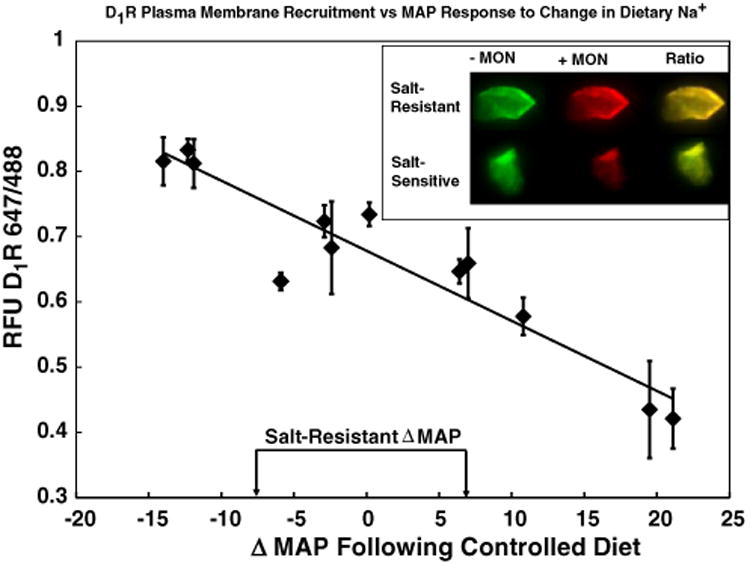
Correlation between the degrees of SS, expressed as a change in MAP in response to increased dietary Na+ and monensin (MON)-stimulated D1R membrane recruitment in urine-derived renal proximal tubule cells (RPTCs). The SS index of each individual subject is depicted on the X axis expressed as the change (Δ) in mean arterial pressure (MAP) vs. D1R 647/488 (representing D1R recruitment from the cytosol to the plasma membrane). Salt resistance was defined as a change in blood pressure of less than or equal to ±7 mm Hg [33]. In 12 subjects, previously tested for SS over a 2 week period, we observed a negative correlation (y = −0.0107x + 0.678, R2 = 0.88, N = 12, P < 0.0001) between a MON-induced increase in D1R recruitment from the cytosol to the plasma membrane in urine-derived RPTCs and the degree of SS. The inset picture demonstrates the change in fluorescence (D1R labeled with green dye before MON and red dye after the addition of MON) when cells from SR and salt-sensitive individuals were compared. Note that yellow indicates a higher ratio than green. Data are mean ± standard error of the mean (SEM) from 5 to 40 replicate measurements from each specimen.
3.3. Cryopreservation of exfoliated RPTCs
In preparation for significantly expanded studies in the future, we determined if cryopreservation of the exfoliated RPTC cells would preserve monensin-mediated D1R recruitment. We cryopreserved the cells using a standard cell culture cryopreservation method and upon thawing at 37 °C we found that these cells were viable (viability = 57.16% ± 9.15%, n = 12) and produced a consistent D1R recruitment response variability in 4 samples from one ISS subject (0.41 ± 0.01 RFU, standard error of the mean (SEM), N = 4). Thus, cryopreserved RPTCs compared favorably with cell viability from RPTCs, obtained from fresh urine, and were still capable of eliciting intracellular Na+-mediated D1R recruitment. We have routinely stored cryopreserved cultured RPTCs for over 10 years in a laboratory liquid nitrogen freezer.
3.4. Angiotensin II-stimulated increase in intracellular Ca++
Similar to the D1R plasma membrane recruitment, there was a negative correlation between the increase in intracellular Ca++ caused by angiotensin II and the change in MAP in response to a change in Na+ diet (y = −0.0016x + 0.0336, R2 = 0.7112, P < 0.001, N = 10; Fig. 4). These specimens were collected and analyzed for angiotensin II-mediated Ca++ stimulation over one year after the monensin-stimulated D1R recruitment experiments were performed, indicating that these were inherent physiological responses and not likely to be environmentally influenced. The angiotensin II-mediated stimulation of intracellular Ca++ was via the AT1R, because losartan, an AT1R antagonist, blocked the angiotensin II effect (Fig. 5).
Fig. 4.

Cells were stimulated with angiotensin II (10 nmol/l) to increase intracellular Ca++ that was then measured using Fura-2 fluorescence. The SS index of each individual subject is depicted on the X axis expressed as the change (Δ) in mean arterial pressure (MAP) vs. Fura-2 ratio (representing intracellular Ca++), similar to Fig. 3. There was a negative correlation between the SS index and intracellular Ca++ stimulation by angiotensin II (y = −0.0016x + 0.0336, R2 = 0.7112, P < 0.001, N = 10). The data are shown as mean ± SE of the mean from 5 to 40 replicate measurements from each specimen.
Fig. 5.

Intracellular Ca++ concentration in response to angiotensin II (10 nmol/l) stimulation was measured in cells obtained from a salt-resistant individual to validate the response in our experimental protocol. Intracellular Ca++ increased 1.78-fold (N = 3, *P < 0.05) over vehicle (VEH) in RPTCs. Losartan (AT1R antagonist, LOS, 10 μmol/l) alone showed no effect, but abrogated the angiotensin II effect (N = 3, **P < 0.01 vs angiotensin II alone) (ANOVA, Holm-Sidak test) demonstrating that a viable angiotensin type 1 receptor (AT1R) was present in these cells. The data are shown as mean ± SE of the mean.
4. Discussion
The kidney, with increased renal tubular Na+ transport, especially in the RPT and thick ascending limb of Henle, plays a critical role in the pathogenesis of essential or polygenic hypertension [11,12]. Hypertension is associated with increased cardiovascular risk and mortality [5,13]. However, independent of blood pressure, even in normotensive individuals, there is a positive relationship between SS and mortality [13] and salt intake increases cardiovascular risk [48,49]. Thus, consumption of a low salt diet has been advocated, especially with the relatively high prevalence of SS (vide infra) [13]. However, a low salt diet may not be beneficial to everyone as a low salt intake has also been associated with increased cardiovascular risk and mortality [1,3,4]. Therefore, such dietary guidelines should be based on whether or not a change in NaCl intake is deleterious or beneficial. Hence there is a need to determine a cost-effective method to screen for each person's SS index.
There is a lack of consensus on the accuracy of protocols used to diagnose SS [13–17,50]. One accepted definition of SS is a negative MAP change ≥10 mm Hg from baseline after infusion of 2 l of normal saline (baseline) followed by the administration of furosemide during a low Na+ diet. Under this protocol, 26% of 378 normotensive subjects and 51% of 198 essential hypertensive subjects were identified to have SS [50]. Another study, using a 14-day dietary protocol (7 days 20 mmol Na+ and 7 days 300 mmol Na+) showed that 18.4% of 163 normotensive subjects had SS [51]. An even more rigorous and extensive protocol was used by Kawasaki et al. [17]. In this protocol, 19 subjects with essential hypertension were fed initially 100 mmol NaCl for 7 days, followed by 9 mmol Na+ for 7 days, and then ending with 240 mmol NaCl for 7 days. The incidence of SS, defined as MAP >10% on day 6 of the high Na+ protocol relative to day 6 of the low Na+ protocol, was 48%. The most convenient method to determine SS is by the intravenous Na+ loading and furosemide method [13,50], but its accuracy of has been questioned [14–16] and thus the advocating of a 14 day test (7 days of low Na+ and 7 days of high Na+ intake) [14–16,51]. To overcome these dietary protocol limitations, we developed a robust and convenient SS diagnostic test, using urine specimens from subjects whose salt-sensitive, SR, and ISS phenotypes were based on the generally accepted 14-day oral salt loading protocol [14–16,51,52].
RPTCs exfoliated into the urine provide an instantaneous snapshot of renal cell physiology or pathology. Therefore, we hypothesized that these cells might retain their in vivo salt-sensitive phenotype as they do for other renal diseases [29,30,53–55]. Defects in the RPTC recruitment of D1R from the cytoplasm to the plasma membrane and D1R dysfunction were previously shown to be associated with defective renal Na+ handling and essential hypertension [22,56,57]. We now report that there is a negative correlation between D1R recruitment and SS, presumably accounting for a decreased ability to excrete Na+ in subjects with SS. We also report that the AT1R-induced stimulation of Ca++ is also negatively correlated with SS. These results suggest that SS is a continuous variable and provide a basis for personalized recommendations of Na+ intake based on an individual's unique cellular physiology. One's personal SS index does not appear to be related to diet since the index was relatively stable over the several months that the subjects were tested.
At the opposite extreme of the SS curve were some individuals who had a paradoxical increase in BP in response to a low Na+ diet. Overlack et al. have reported that 15% of 163 normotensive subjects had an increase in MAP with a low Na+ (20 mmol) diet [51]. These counter regulators or “inverse salt-sensitive” individuals had the highest increase in intracelllular Ca++ in response to AT1R stimulation and the highest D1R recruitment. Low concentrations of angiotensin II (1 nmol/l) increase renal tubular Na+ transport but near endogenous levels of angiotensin II (10 nmol/l) decrease renal tubular Na+ transport [58]. Placing ISS subjects on a low Na+ diet may be inadvisable because low Na+ intake has been associated with higher cardiovascular disease mortality [1–5]. Our data may explain the apparent paradoxical association between low Na+ intake and cardiovascular disease, including atherosclerosis [1–4] because the relative proportion of salt-sensitive, SR and ISS individuals in a given study population could result in vastly different conclusions.
The regulation of blood pressure is a result of many interacting systems. A primary role of neuroendocrine and cardiac mechanisms in the pathogenesis of hypertension is thought to be a result of abberrations in the regulation by the neuroendocrine and cardiac systems, while a secondary role is thought to result from vasoconstrictor and vasodilatory mechanisms originating in the endothelial cells [59,60]. Stress-induced increase in Na+ reabsorption occurs in the proximal part of the nephron that is modified by a family history of hypertension, indicating a role of genetics [61]. The primary regulator of blood pressure is the kidney [8,9,62], by its ability to regulate salt balance. However, extracellular fluid volume and exchangeable Na+ are not decreased in subjects with borderline hypertension and young hypertensive patients [63,64]. This apparent paradox can be explained by the temporal development of hypertension. Initially, hypertension develops slowly, followed by a relatively brief stage in which extracellular fluid volume is elevated. This stage is followed by an elevation of total peripheral resistance but the pressure-natriuresis normalizes the increased extracellular fluid volume. This could also be explained by recent demonstration that the interstitium, underneath the skin, can serve as a reservoir for the retained Na+ that is not accompanied by water retention, and blood pressure may not increase until after “saturation” of interstitial sequestration of Na+ [65].
Essential hypertension in humans and rodent models of essential hypertension are associated with increased Na+ transport in the renal proximal tubule and medullary thick ascending limb, although increased distal tubular transport has also been reported [11,12,66–68]. In contrast, monogenic hypertension is caused by increased Na+ transport mainly in the distal nephron [66,69]. The sympathetic nervous [70] and reninangiotensin systems [71] are important in the pathogenesis of essential hypertension and may regulate blood pressure by extra-renal mechanisms. However, the high blood pressure that develops in the F1 generation of normotensive and spontaneously hypertensive recipients of kidneys from parental spontaneously hypertensive rats is apparently not caused by abnormalities of the sympathetic nervous or reninangiotensin system, or mesenteric vascular smooth muscle reactivity to vasoconstrictors (e.g., vasopressin and norepinephrine) or vasodilators (e.g., acetylcholine and nitroprusside) [72]. Therefore, other vasoactive agents (e.g., dopamine), especially those that exert their effects independent of the vascular endothelium, must play a role in the development of hypertension. Inhibition of renal D1R/D5R receptors decreases renal Na+ excretion by about 50–60% in conscious rats and dogs on normal salt intake [18,73,74]. Systemic inhibition of D1R/D5R receptors in anesthetized rats with volume expansion equivalent to 2% of body weight decreased Na+ excretion by 40–67% [75]. Indeed, deletion of the aromatic amino acid decarboxylase which converts tyrosine to L-DOPA, the immediate precursor to dopamine, in renal proximal tubules in mice causes salt-sensitive hypertension [76]. Therefore, development of a convenient diagnostic test for an individual's SS index, based on actions of the renin-angiotensin system and the dopaminergic system could help delineate the contributions of these various blood pressure regulatory pathways to human disease.
This study has some limitations. The finding that both assays (monensin-stimulated D1R recruitment and angiotensin II-stimulated intracellular Ca++) demonstrated a significant correlation with SS of blood pressure reduces the chance of type 1 statistical error in these studies. The sample size is small and will have to be expanded in the future. Future studies will focus on the effect of short- and long-term dietary Na+ on the cell biology, biochemistry, and physiology of RPTCs recovered from the urine. Our test subjects were on an ad libitum diet and one cannot rule out habitually consistent Na+ or potassium consumption. However, it is unlikely that diets would remain consistently high or low over the year that was required to complete this study. Urines voided in the morning to mid-day were collected in order to reduce the probability of dietary consistencies.
In summary, urine-derived RPTCs provide an instantaneous “snapshot” of personalized SS index. Two markers associated with the renin-angiotensin and dopaminergic systems known to be responsible for a large percent of renal Na+ transport show a high degree of correlation with the SS index expressed by the test subjects. Since urine-derived RPTC responses correlate with each subject's degree of SS, we suggest that the renal cells maintain their genetically programmed physiological responses and thus can be viewed as a “virtual renal biopsy” for renal physiology. We have also demonstrated that RPTCs exfoliated in the urine survive cryopreservation with a similar recovery rate as cultured research cells, opening the door for storage and transportation of specimens to a testing facility.
Acknowledgments
Funding for these studies was provided by grants from National Institute of Heart, Lung, and Blood Institute P01HL074940 (Felder RA, Carey RM, and Jose PA) and National Institute of Diabetes, Digestive and Kidney Diseases R01DK039308 (Jose PA and Felder RA).
References
- 1.Alderman MH, Cohen HW. Dietary sodium intake and cardiovascular mortality: controversy resolved? Curr Hypertens Rep. 2012;14:193–201. doi: 10.1007/s11906-012-0275-6. [DOI] [PubMed] [Google Scholar]
- 2.Suematsu N, Ojaimi C, Recchia FA, et al. Potential mechanisms of low-sodium diet-induced cardiac disease: superoxide-NO in the heart. Circ Res. 2010;106:593–600. doi: 10.1161/CIRCRESAHA.109.208397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stolarz-Skrzypek K, Kuznetsova T, Thijs L, et al. Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. JAMA. 2011;305:1777–85. doi: 10.1001/jama.2011.574. [DOI] [PubMed] [Google Scholar]
- 4.O'Donnell MJ, Yusuf S, Mente A, et al. Urinary sodium and potassium excretion and risk of cardiovascular events. JAMA. 2011;306:2229–38. doi: 10.1001/jama.2011.1729. [DOI] [PubMed] [Google Scholar]
- 5.Dorresteijn JA, van der Graaf Y, Spiering W, et al. Relation between blood pressure and vascular events and mortality in patients with manifest vascular disease: J-curve revisited. Hypertension. 2012;59:14–21. doi: 10.1161/HYPERTENSIONAHA.111.179143. [DOI] [PubMed] [Google Scholar]
- 6.Felder RA, White MJ, Williams SM, Jose PA. Diagnostic tools for hypertension and salt sensitivity testing. Curr Opin Nephrol Hypertens. 2013;22:65–76. doi: 10.1097/MNH.0b013e32835b3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–34. doi: 10.2337/diabetes.54.12.3427. [DOI] [PubMed] [Google Scholar]
- 8.Crowley SD, Gurley SB, Herrera MJ, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–90. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asico L, Zhang X, Jiang J, et al. Lack of renal dopamine D5 receptors promotes hypertension. J Am Soc Nephrol. 2011;22:82–9. doi: 10.1681/ASN.2010050533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Iturbe B, Franco M, Johnson RJ. Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens. 2013;22:37–44. doi: 10.1097/MNH.0b013e32835b3d54. [DOI] [PubMed] [Google Scholar]
- 11.Aviv A, Hollenberg NK, Weder A. Urinary potassium excretion and sodium sensitivity in blacks. Hypertension. 2004;43:707–13. doi: 10.1161/01.HYP.0000120155.48024.6f. [DOI] [PubMed] [Google Scholar]
- 12.Doris PA. Renal proximal tubule sodium transport and genetic mechanisms of essential hypertension. J Hypertens. 2000;18:509–19. doi: 10.1097/00004872-200018050-00002. [DOI] [PubMed] [Google Scholar]
- 13.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–32. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 14.Kato N, Kanda T, Sagara M, et al. Proposition of a feasible protocol to evaluate salt sensitivity in a population-based setting. Hypertens Res. 2002;25:801–9. doi: 10.1291/hypres.25.801. [DOI] [PubMed] [Google Scholar]
- 15.Nichols J, Elijovich F, Laffer CL. Lack of validation of a same-day outpatient protocol for determination of salt sensitivity of blood pressure. Hypertension. 2012;59:390–4. doi: 10.1161/HYPERTENSIONAHA.111.185835. [DOI] [PubMed] [Google Scholar]
- 16.de la Sierra A, Giner V, Bragulat E, Coca A. Lack of correlation between two methods for the assessment of salt sensitivity in essential hypertension. J Hum Hypertens. 2002;16:255–60. doi: 10.1038/sj.jhh.1001375. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki T, Delea CS, Bartter FC, Smith H. The effect of high-sodium and low-sodium intakes on blood pressure and other related variables in human subjects with idiopathic hypertension. Am J Med. 1978;64:193–8. doi: 10.1016/0002-9343(78)90045-1. [DOI] [PubMed] [Google Scholar]
- 18.Siragy HM, Felder RA, Howell NE, Chevalier RL, Peach MJ, Carey RM. Intrarenal dopamine acts at the dopamine-1 receptor to control renal function. J Hypertens. 1988;6:S479–81. doi: 10.1097/00004872-198812040-00151. Suppl. [DOI] [PubMed] [Google Scholar]
- 19.Jose PA, Soares-da-Silva P, Eisner GM, Felder RA. Dopamine and G protein-coupled receptor kinase 4 in the kidney: role in blood pressure regulation. Biochim Biophys Acta. 1802;2010:1259–67. doi: 10.1016/j.bbadis.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felder RA, Sanada H, Xu J, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A. 2002;99:3872–7. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Efendiev R, Budu CE, Cinelli AR, Bertorello AM, Pedemonte CH. Intracellular Na+ regulates dopamine and angiotensin II receptors availability at the plasma membrane and their cellular responses in renal epithelia. J Biol Chem. 2003;278:28719–26. doi: 10.1074/jbc.M303741200. [DOI] [PubMed] [Google Scholar]
- 22.Sanada H, Jose PA, Hazen-Martin D, et al. Dopamine-1 receptor coupling defect in renal proximal tubule cells in hypertension. Hypertension. 1999;33:1036–42. doi: 10.1161/01.hyp.33.4.1036. [DOI] [PubMed] [Google Scholar]
- 23.Khan F, Spicarová Z, Zelenin S, Holtbäck U, Scott L, Aperia A. Negative reciprocity between angiotensin II type 1 and dopamine D1 receptors in rat renal proximal tubule cells. Am J Physiol Renal Physiol. 2008;295:F1110–6. doi: 10.1152/ajprenal.90336.2008. [DOI] [PubMed] [Google Scholar]
- 24.Zeng C, Luo Y, Asico LD, et al. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. Hypertension. 2003;42:787–92. doi: 10.1161/01.HYP.0000085334.34963.4E. [DOI] [PubMed] [Google Scholar]
- 25.Chugh G, Lokhandwala MF, Asghar M. Oxidative stress alters renal D1 and AT1 receptor functions and increases blood pressure in old rats. Am J Physiol Renal Physiol. 2011;300:F133–8. doi: 10.1152/ajprenal.00465.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams SM, Addy JH, Phillips JA, III, et al. Combinations of variations in multiple genes are associated with hypertension. Hypertension. 2000;36:2–6. doi: 10.1161/01.hyp.36.1.2. [DOI] [PubMed] [Google Scholar]
- 27.Sanada H, Yatabe J, Midorikawa S, et al. Single-nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin Chem. 2006;52:352–60. doi: 10.1373/clinchem.2005.059139. [DOI] [PubMed] [Google Scholar]
- 28.Gildea J, Yatabe J, Sasaki M, Wang X, Jose P, Felder R. Up-regulation of the angiotensin type II receptor by dopamine-1 receptor stimulation in normotensive but not hypertensive human renal proximal tubule cells block angiotensin II dependent down-regulation of caveolin 1. Hypertension; Abstract for 59th Annual Fall Conference and Scientific Sessions of Council for High Blood Pressure Research; Washington, DC. September 2005.2005. [Google Scholar]
- 29.Racusen LC, Fivush BA, Andersson H, Gahl WA. Culture of renal tubular cells from the urine of patients with nephropathic cystinosis. J Am Soc Nephrol. 1991;1:1028–33. doi: 10.1681/ASN.V181028. [DOI] [PubMed] [Google Scholar]
- 30.Racusen LC, Wilson PD, Hartz PA, Fivush BA, Burrow CR. Renal proximal tubular epithelium from patients with nephropathic cystinosis: immortalized cell lines as in vitro model systems. Kidney Int. 1995;48:536–43. doi: 10.1038/ki.1995.324. [DOI] [PubMed] [Google Scholar]
- 31.Inoue CN, Kondo Y, Ohnuma S, Morimoto T, Nishio T, Iinuma K. Use of cultured tubular cells isolated from human urine for investigation of renal transporter. Clin Nephrol. 2000;53:90–8. Erratum appears in Clin Nephrol 2000 Jun;53(6):492. [PubMed] [Google Scholar]
- 32.Laube GF, Haq MR, van't Hoff WG. Exfoliated human proximal tubular cells: a model of cystinosis and Fanconi syndrome. Pediatr Nephrol. 2005;20:136–40. doi: 10.1007/s00467-004-1703-x. [DOI] [PubMed] [Google Scholar]
- 33.Carey RM, Schoeffel CD, Gildea JJ, et al. Salt sensitivity of blood pressure is associated with polymorphisms in the sodium-bicarbonate cotransporter. Hypertension. 2012;60:1359–66. doi: 10.1161/HYPERTENSIONAHA.112.196071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levey AS, Eckardt KU, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2005;67:2089–100. doi: 10.1111/j.1523-1755.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 35.Prescott LF. The normal urinary excretion rates of renal tubular cells, leucocytes and red blood cells. Clin Sci. 1966;31:425–35. [PubMed] [Google Scholar]
- 36.Zhou T, Benda C, Dunzinger S, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7:2080–9. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- 37.Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21:1859–67. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helbert MJ, Dauwe SE, Van der Biest I, Nouwen EJ, De Broe ME. Immunodissection of the human proximal nephron: flow sorting of S1S2S3, S1S2 and S3 proximal tubular cells. Kidney Int. 1997;52:414–28. doi: 10.1038/ki.1997.348. [DOI] [PubMed] [Google Scholar]
- 39.Baer PC, Nockher WA, Haase W, Scherberich JE. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note Kidney Int. 1997;52:1321–31. doi: 10.1038/ki.1997.457. [DOI] [PubMed] [Google Scholar]
- 40.Gildea JJ, Shah I, Weiss R, et al. HK-2 human renal proximal tubule cells as a model for G protein-coupled receptor kinase type 4-mediated dopamine 1 receptor uncoupling. Hypertension. 2010;56:505–11. doi: 10.1161/HYPERTENSIONAHA.110.152256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grupp C, Hemprich U, John H, et al. Lectin staining for urine cytologic monitoring after kidney transplantation. Nephrol Dial Transplant. 2002;17:1491–6. doi: 10.1093/ndt/17.8.1491. [DOI] [PubMed] [Google Scholar]
- 42.Gildea JJ, Israel JA, Johnson AK, Zhang J, Jose PA, Felder RA. Caveolin-1 and dopamine-mediated internalization of NaKATPase in human renal proximal tubule cells. Hypertension. 2009;54:1070–6. doi: 10.1161/HYPERTENSIONAHA.109.134338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gildea JJ, McGrath HE, Van Sciver RE, Wang DB, Felder RA. Isolation, growth, and characterization of human renal epithelial cells using traditional and 3D methods. Methods Mol Biol. 2013;945:329–45. doi: 10.1007/978-1-62703-125-7_20. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79:1089–125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- 45.Seikaly MG, Arant BS, Seney FD. Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest. 1990;86:1352–7. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braam B, Mitchell KD, Fox J, Navar LG. Proximal tubular secretion of angiotensin II in rats. Am J Physiol. 1993;264:F891–8. doi: 10.1152/ajprenal.1993.264.5.F891. [DOI] [PubMed] [Google Scholar]
- 47.Gildea JJ, Wang X, Shah N, et al. Dopamine and Angiotensin type 2 receptors cooperatively inhibit sodium transport in human renal proximal tubule cells. Hypertension. 2012;60:396–403. doi: 10.1161/HYPERTENSIONAHA.112.194175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strazzullo P, D'Elia L, Kandala NB, Cappuccio FP. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ. 2009;339:b4567. doi: 10.1136/bmj.b4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He FJ, MacGregor GA. Reducing population salt intake worldwide: from evidence to implementation. Prog Cardiovasc Dis. 2010;52:363–82. doi: 10.1016/j.pcad.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–90. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 51.Overlack A, Ruppert M, Kolloch R, et al. Divergent hemodynamic and hormonal responses to varying salt intake in normotensive subjects. Hypertension. 1993;22:331–8. doi: 10.1161/01.hyp.22.3.331. [DOI] [PubMed] [Google Scholar]
- 52.Yatabe MS, Yatabe J, Yoneda M, et al. Salt sensitivity is associated with insulin resistance, sympathetic overactivity, and decreased suppression of circulating renin activity in lean patients with essential hypertension. Am J Clin Nutr. 2010;92:77–82. doi: 10.3945/ajcn.2009.29028. [DOI] [PubMed] [Google Scholar]
- 53.Racusen LC, Fivush BA, Li YL, Slatnik I, Solez K. Dissociation of tubular cell detachment and tubular cell death in clinical and experimental “acute tubular necrosis”. Lab Investig. 1991;64:546–56. [PubMed] [Google Scholar]
- 54.Racusen LC, Solez K. Ideas in pathology. Exfoliation of renal tubular cells. Mod Pathol. 1991;4:368–70. [PubMed] [Google Scholar]
- 55.Racusen LC, Monteil C, Sgrignoli A, et al. Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med. 1997;129:318–29. doi: 10.1016/s0022-2143(97)90180-3. [DOI] [PubMed] [Google Scholar]
- 56.Banday AA, Lokhandwala MF. Dopamine receptors and hypertension. Curr Hypertens Rep. 2008;10:268–75. doi: 10.1007/s11906-008-0051-9. [DOI] [PubMed] [Google Scholar]
- 57.O'Connell DP, Ragsdale NV, Boyd DG, Felder RA, Carey RM. Differential human renal tubular responses to dopamine type 1 receptor stimulation are determined by blood pressure status. Hypertension. 1997;29:115–22. doi: 10.1161/01.hyp.29.1.115. [DOI] [PubMed] [Google Scholar]
- 58.Hiranyachattada S, Harris PJ. Regulation of renal proximal fluid uptake by luminal and peritubular angiotensin II. J Renin Angiotensin Aldosterone Syst. 2004;5:89–92. doi: 10.3317/jraas.2004.016. [DOI] [PubMed] [Google Scholar]
- 59.DiBona GF. Sympathetic nervous system and the kidney in hypertension. Curr Opin Nephrol Hypertens. 2002;11:197–200. doi: 10.1097/00041552-200203000-00011. [DOI] [PubMed] [Google Scholar]
- 60.Iwamoto T, Kita S, Zhang J, et al. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med. 2004;10:1193–9. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 61.Ducher M, Bertram D, Pozet N, Laville M, Fauvel JP. Stress-induced renal alterations in normotensives offspring of hypertensives and in hypertensives. Am J Hypertens. 2002;15:346–50. doi: 10.1016/s0895-7061(01)02333-0. [DOI] [PubMed] [Google Scholar]
- 62.Davy KP, Hall JE. Obesity and hypertension: two epidemics or one? Am J Physiol Regul Integr Comp Physiol. 2004;286:R803–13. doi: 10.1152/ajpregu.00707.2003. [DOI] [PubMed] [Google Scholar]
- 63.Lebel M, Grose JH, Blais R. Abnormal relation of extracellular fluid volume and exchangeable sodium with systemic arterial pressure in early borderline essential hypertension. Am J Cardiol. 1984;54:1267–71. doi: 10.1016/s0002-9149(84)80078-8. [DOI] [PubMed] [Google Scholar]
- 64.Beretta-Piccoli C, Weidmann P. Circulatory volume in essential hypertension. Relationships with age, blood pressure, exchangeable sodium, renin, aldosterone and catecholamines. Miner Electrolyte Metab. 1984;10:292–300. [PubMed] [Google Scholar]
- 65.Titze J, Machnik A. Sodium sensing in the interstitium and relationship to hypertension. Curr Opin Nephrol Hypertens. 2010;19:385–92. doi: 10.1097/MNH.0b013e32833aeb3b. [DOI] [PubMed] [Google Scholar]
- 66.Ortiz PA, Garvin JL. Intrarenal transport and vasoactive substances in hypertension. Hypertension. 2001;38:621–4. doi: 10.1161/hy09t1.093361. [DOI] [PubMed] [Google Scholar]
- 67.Strazzullo P, Galletti F, Barba G. Altered renal handling of sodium in human hypertension: short review of the evidence. Hypertension. 2003;41:1000–5. doi: 10.1161/01.HYP.0000066844.63035.3A. [DOI] [PubMed] [Google Scholar]
- 68.Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Proximal sodium reabsorption: an independent determinant of blood pressure response to salt. Hypertension. 2000;36:631–7. doi: 10.1161/01.hyp.36.4.631. [DOI] [PubMed] [Google Scholar]
- 69.Lifton RP, Wilson FH, Choate KA, Geller DS. Salt and blood pressure: new insight from human genetic studies. Cold Spring Harb Symp Quant Biol. 2002;67:445–50. doi: 10.1101/sqb.2002.67.445. [DOI] [PubMed] [Google Scholar]
- 70.DiBona GF, Esler M. Translational medicine: the antihypertensive effect of renal denervation. Am J Physiol Regul Integr Comp Physiol. 2010;298:R245–53. doi: 10.1152/ajpregu.00647.2009. [DOI] [PubMed] [Google Scholar]
- 71.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–6. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grisk O, Heukaufer M, Steinbach A, Gruska S, Rettig R. Analysis of arterial pressure regulating systems in renal post-transplantation hypertension. J Hypertens. 2004;22:199–207. doi: 10.1097/00004872-200401000-00030. [DOI] [PubMed] [Google Scholar]
- 73.Wang ZQ, Felder RA, Carey RM. Selective inhibition of the renal dopamine subtype D1A receptor induces antinatriuresis in conscious rats. Hypertension. 1999;33:504–10. doi: 10.1161/01.hyp.33.1.504. [DOI] [PubMed] [Google Scholar]
- 74.Siragy HM, Felder RA, Howell NL, Chevalier RL, Peach MJ, Carey RM. Evidence that intrarenal dopamine acts as a paracrine substance at the renal tubule. Am J Physiol. 1989;257:F469–77. doi: 10.1152/ajprenal.1989.257.3.F469. [DOI] [PubMed] [Google Scholar]
- 75.Hansell P, Fasching A. The effect of dopamine receptor blockade on natriuresis is dependent on the degree of hypervolemia. Kidney Int. 1991;39:253–8. doi: 10.1038/ki.1991.30. [DOI] [PubMed] [Google Scholar]
- 76.Zhang MZ, Yao B, Wang S, et al. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest. 2011;121:2845–54. doi: 10.1172/JCI57324. [DOI] [PMC free article] [PubMed] [Google Scholar]