
Figure 1. Progression of Ischemia/reperfusion Injury.
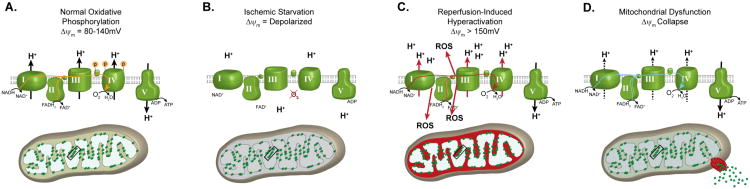
(A) Under normal, non-stressed conditions, OxPhos components are phosphorylated (as illustrated for Cytc and CcO), promoting controlled electron transfer and maintaining the ΔΨm in the 80-140mV range. This respiratory state is conducive to maximal ATP production and minimal ROS generation. (B) Ischemia induces a starvation state were the ETC cannot function due to lack of O2. Dephosphorylation of OxPhos during ischemia renders these enzymes ‘primed’ for hyperactivity, however they cannot operate due to lack of the terminal substrate for respiration, O2. (C) Reintroduction of O2 with reperfusion initiates electron transfer, proton pumping and ATP synthesis. However, in this hyperactive (dephosphorylated) state, OxPhos hyperpolarizes the ΔΨm, causing an exponential increase in ROS generation at complexes I and/or III. (D) This burst in ROS can act as a signal for triggering apoptosis. In addition, damage caused by ROS induces a mitochondrial dysfunction state, with reduced electron transfer kinetics and reduced ΔΨm levels, which results in energetic failure.