

Abstract
Biofilm-related infections are a major contributor to human disease, and the capacity for surface attachment and biofilm formation are key attributes for the pathogenesis of microbes. Serratia marcescens type I fimbriae-dependent biofilms are coordinated by the adenylate cyclase, CyaA, and the cyclic 3′,5′-adenosine monophosphate (cAMP)-cAMP receptor protein (CRP) complex. This study uses S. marcescens as a model system to test the role of cAMP-phosphodiesterase activity in controlling biofilm formation. Herein we describe the characterization of a putative S. marcescens cAMP-phosphodiesterase gene (SMA3506), designated as cpdS, and demonstrated to be a functional cAMP-phosphodiesterase both in vitro and in vivo. Deletion of cpdS resulted in defective biofilm formation and reduced type I fimbriae production, whereas multicopy expression of cpdS conferred a type I fimbriae-dependent hyper-biofilm. Together, these results support a model in which bacterial cAMP-phosphodiesterase activity modulates biofilm formation.
Introduction
The cyclic AMP (cAMP)-cAMP receptor protein (CRP) regulatory network exerts global control over processes of cellular physiology and virulence, such as cell division, catabolite repression, motility, and toxin production [1]–[3]. Bacterial catabolite repression modulates intracellular cAMP levels through adenylate cyclase (AC) and phosphoenolpyruvate: carbohydrate phosphotransferase (PTS) system activity [4], [5]. Degradation of cAMP to 5′-AMP is accomplished through enzymatic action of a cyclic 3′,5′-AMP phosphodiesterase (cAMP-PDE) [6]. The dynamic coordination of cAMP synthesis and degradation must be achieved for cAMP homeostasis and its effect on the cAMP-CRP network in response to fluctuating environments.
Many studies have highlighted an extraordinary role for ACs and cAMP-CRP in bacterial virulence-associated phenotypes. For instance, ACs and CRP-family proteins are required for full virulence in mouse infection models with the pathogens Mycobacterium tuberculosis, Pseudomonas aeruginosa, Salmonella typhimurium, and Yersinia enterocolitica [7]–[10]; in Vibrio cholerae, CRP is essential for in vivo colonization [11]. In Serratia marcescens, cAMP-CRP-dependent pathways control motility, hemolysis, production of flagella and the metalloprotease serralysin [12]–[14]. The function of cAMP-CRP is paramount to pathogenesis, yet the impact of altered cAMP equilibrium on surface attachment and biofilm formation is not well understood.
Biofilms, surface-adherent bacterial communities, are thought to afford microbes with the ability to persist in diverse environments, survive antimicrobials, and adapt to physiological stresses and host defenses [15]–[17]. Bacterial biofilms are of critical clinical significance and pose immense challenges to the eradication of multidrug-tolerant organisms, which likely support chronic and recurrent disease states that are recalcitrant to antimicrobial therapy [15], [18], [19]. An extensive spectrum of biofilm-related infections are implicated in human disease and foreign body-associated infections are intimately linked to biofilm formation [16]. Fimbriae, or type I pili, are crucial virulence factor(s) for biofilm development and bacterial attachment to host tissue surfaces [20]–[22]. Regulation of type I pili and/or biofilm formation by cAMP-CRP has been documented in Escherichia coli [23], [24] and V. cholerae [25], [26]. Moreover, V. cholerae PTS components are known to regulate biofilm formation [27].
S. marcescens engages several regulatory mechanisms for virulence, surface adhesion and biofilm formation [28]–[30]. Foremost among these are type I fimbrial adhesins [31], [32], which are critical mediators of surface attachment to biotic and abiotic surfaces [33], [34]. S. marcescens type I fimbriae-dependent biofilm formation is controlled by the cAMP-CRP complex; mutations inactivating positive regulators of cAMP, PTS enzyme IIAGlc (crr) and adenylate cyclase (cyaA), or the master cAMP effector, CRP (crp), resulted in a hyper-biofilm phenotype [35]. Notably, the effect of negative cAMP regulators, specifically cAMP-PDEs, on biofilm formation has not been determined for any bacterial species. Using S. marcescens as a model system, we investigated the role for bacterial cAMP-PDE activity in biofilm formation.
Materials and Methods
Cultures and media
Bacteria were cultured at 30°C in Lysogeny broth (LB, per liter: 5 g yeast extract, 10 g tryptone, 5 g NaCl) in test tubes on a tissue culture rotor (New Brunswick model TC-7, speed setting 8, 62 rpm). Kanamycin was added to both agar and broth when appropriate at a concentration of 100 µg/ml, gentamicin at 10 µg/ml, and tetracycline at 10 µg/ml. Saccharomyces cerevisiae was grown in YPD or SC-URA [36]. Strains, plasmids and oligonucleotide primers are chronicled in Table 1, 2, and 3 respectively. The S. marcescens strain (CMS376) used in this study was obtained from the Presque Isle Culture Collection (PIC strain number 3611). This is a pigmented strain that has been used for previous biofilm studies [34], [35].
Table 1. Strains used in this study.
| Strain | Description | Reference or source |
| InvSc1 | Saccharomyces cerevisiae strain uracil auxotroph | Invitrogen |
| S17-1 λpir | Escherichia coli strain used for conjugation and cloning | [69] |
| EC100D | E. coli strain used for cloning and protein purification | Epicentre |
| ER2566 | E. coli strain used for protein purification | New England Biolabs |
| JM3000-1 | E. coli K-20 rtlC cpdA::kan | E. coli Genetic Stock Center |
| CMS376 | Serratia marcescens wild type, PIC strain number 3611 | Presque Isle Cultures |
| CMS524 | cyaA-2 – CMS376 with transposon mutation | [35] |
| CMS629 | fimC-2 – CMS376 with transposon mutation | [35] |
| CMS2910 | CMS376 with cpdS deletion, ΔcpdS | This study |
| CMS3602 | CMS2910 with cyaA transposon mutation (prc-3) | This study |
Table 2. Plasmids used in this study.
| Plasmid | Description | Reference or source |
| pMQ131 | shuttle vector, oripBBR1, aphA-3 | [38] |
| pMQ132 | shuttle vector, oripBBR1, aacC1 | [38] |
| pMQ158 | pMQ132+ cyaA | this study |
| pMQ171 | pMQ131+ cpdS | this study |
| pMQ176 | pMQ131+ cpdS-N94A | this study |
| pMQ198 | pMQ131+ cpdS-His8 | this study |
| pMQ201 | pMQ131+ cpdS, N94A-His8 | this study |
| pMQ236 | Allelic replacement vector, oriR6K, nptII, I-SceI, rpsL | [38] |
| pMQ240 | I-SceI meganuclease expression vector for allelic replacement | [38] |
| pMQ247 | pMQ131+ PA4969-His8 | this study |
| pMQ360 | pMQ236+ SMA3501-9 with cpdS-Δ deletion allele | this study |
| pEJK3 | pMQ132+ cpdA from E. coli | [44] |
| pEJK5 | pMQ132+ PA4969 from P. aeruginosa | this study |
| pEJK7 | pMQ131+ cpdA-His8 from E. coli | this study |
Table 3. Oligonucleotide primers used in this study.
| Primer number | Primer sequencea |
| 1140 | caaattctgttttatcagaccgcttctgcgttctgatGCATCAGTATCCATCCGAGTCC |
| 1141 | tgtgagcggataacaatttcacacaggaaacagcttttATGGAAAGCCTGTTTAAACTGC |
| 1359 | GACCATCGCCGGTTGAAAATCGTGggcGCCCGGCAGCCACACGCAGG |
| 1360 | CCTGCGTGTGGCTGCCGGGCgccCACGATTTTCAACCGGCGATGGTC |
| 1388 | tctgttttatcagaccgcttctgcgttctgattcagtggtgatggtggtggtggtggtggTATCCATCCGAGTCCATATC |
| 1615 | aaattctgttttatcagaccgcttctgcgttctgatTCAGTATCCGGCGGTGTCGTAGTC |
| 1616 | attgtgagcggataacaatttcacacaggaaacagcttttATGTCACGCCATTCGAACAC |
| 1619 | aaattctgttttatcagaccgcttctgcgttctgatTCAGTAGCCTTCTGAAGCGGTATC |
| 1620 | gtgagcggataacaatttcacacaggaaacagcttttATGGAAAGCCTGTTAACCCTTCC |
| 1687 | ctgcgttctgattcagtggtgatggtggtggtggtggtgGTATCCGGCGGTGTCGTAGTC |
| 1689 | tgcgttctgattcagtggtgatggtggtggtggtggtgGTAGCCTTCTGAAGCGGTATC |
| 2172 | cagtgccaagcttgcatgcctgcaggtcgactctaGCTATGTTAGCGCTGACGATAGACG |
| 2173 | gtgcaggtagagcagcgtcgaaggcatcaATGGTGTCCTTTGTTATTTCAGTCTGTC |
| 2175 | gacagactgaaataacaaaggacaccatTGATGCCTTCGACGCTGCTCTACCTGCAC |
| 2176 | gctggagaaatgggaaagcatggcgcatCTGGAAAAACACCTGGCCATTGAGC |
| 2178 | gctcaatggccaggtgtttttccagATGCGCCATGCTTTCCCATTTCTCCAGC |
| 2179 | gataacaatttcacacaggaaacagctatgaccatgattCCGTTAACGCTAGCGCCAACG |
Lower case base pairs target recombination and upper case base pairs direct amplification.
Biofilm and twitching assays
S. marcescens biofilms were formed on glass test tubes at high shear force as previously described [35]. Single colonies were added to 20 mm glass test tubes with 5 ml of LB and incubated with aeration on a TC-7 tissue culture rotor (New Brunswick Scientific) overnight at 30°C, a temperature that supports robust biofilm formation. Resulting biofilms adhering to the glass tubes were rinsed with tap water and stained using 5 ml of 0.1% crystal violet. The relative amount of biofilm was determined by solubilization of the crystal violet with 33% glacial acetic acid, and measurement of absorbance at 590 nm with a Synergy 2 plate reader (Biotek).
Viability of biofilms and planktonic cells was determined by staining cells with SYTO-9 and propidium iodide using a commercial kit and the manufacturer's specifications (BacLight L7012, Invitrogen). Static biofilms were formed on glass cover-slips that were incubated upright in a 12-well dish with 3 ml of LB inoculated with bacteria. The coverslip was dipped thrice in PBS to remove non-adherent cells and stained using the BacLight kit. Planktonic cells were from cultures grown overnight in LB with shaking, these were incubated and stained for viability and rinsed with PBS. A 10 µl aliquot was placed on the bottom of a glass-bottomed 6-well dish (Matek) followed by the addition of 37°C low melt agarose (25 µl of 0.5% agarose) to prevent movement of the cells during micrsocopy. Images were obtained using epifluorescent microscopy and experiments were performed on two different days with similar results.
Molecular techniques
Plasmids were generated using yeast in vivo cloning [37]. To clone the SMA3506 (cpdS) open reading frame (ORF) from S. marcescens, primers (1140 and 1141) were designed to amplify SMA3506 with a change in the predicted start codon from “TTG” to “ATG”, for SMA3506 and all subsequent cAMP-PDE genes, to enhance translation efficiency. These primers also direct recombination into pMQ131 and pMQ132, which were linearized with SmaI. The recombination event places the SMA3506 ORF under control of the E. coli Plac promoter and the resulting plasmids were dubbed pMQ171 and pMQ172 respectively. The same strategy was used to clone cpdA (PA4969) from P. aeruginosa strain PA01 except that it was cloned into shuttle vectors pMQ132 using primers 1615 and 1616. Clones that conferred elevated PDE activity to crude lysates of E. coli, tested with bis(p-nitrophenyl) phosphate (bis-pNPP) as described below, were analyzed by PCR, sequenced and used for further analysis.
To make pMQ176 in which a N94A mutation was introduced into the SMA3506 ORF, the gene was amplified in two halves with overlapping regions into which the N94A mutation was engineered using primer sets 1141 with 1360, and 1140 with 1359. The two linear replicons were simultaneously recombined into pMQ131 using in vivo cloning. The His8-tagged versions of SMA3506 and SMA3506 -N94A were generated using 1388 and 1141. His8-tagged versions of the cpdA genes from E. coli and P. aeruginosa were generated with primer sets 1689 and 1620, and 1687 and 1616, respectively. Plasmids were sequenced to verify that unwanted mutations were not introduced during the PCR or recombination processes.
Mutagenesis
In-frame deletion of the cpdS gene was accomplished by using the allelic replacement vector pMQ236 [38]. Briefly, primers 2172 and 2173 amplified a 1468 base pair amplicon upstream of cpdS, and primer sets 2175 and 2176 and 2178 and 2179 were used to amplify 3646 and 3468 base pair amplicons with an overlapping region of DNA (54 base pairs) downstream of cpdS. This large region was cloned in case the integration of pMQ236 during the first step of allelic replacement caused a polar effect on the likely essential gyrase genes downstream of cpdS. The pMQ236 was linearized by digestion with SmaI, and combined with the PCR amplicons in a Saccharomyces cerevisiae transformation reaction. Plasmids were isolated from the resulting Ura+ colonies and were introduced into E. coli strain S17-1 λpir. PCR was used to screen for plasmids with DNA upstream and downstream of cpdS, and the cpdS deletion was verified by sequencing. The resulting plasmid pMQ360 included 1468 base pairs of DNA upstream of cpdS and 7060 base pairs of DNA downstream of cpdS.
To delete the chromosomal copy of cpdS, pMQ360 was introduced into WT S. marcescens (CMS376) by conjugation and selection with kanamycin. Plasmid insertion at the cpdS locus was verified by PCR. The I-SceI expressing plasmid pMQ240 was introduced into the merodiploid strain to introduce a double strand break in the pMQ360 backbone. Kanamycin sensitive single colonies were isolated, and the cpdS gene status was assayed by PCR among the kanamycin susceptible candidates. Roughly half had an amplicon corresponding to the mutant allele and the rest to the expected size of the wild-type cpdS gene. The diagnostic amplicons were sequenced to verify the chromosomal cpdS deletion.
Biochemical applications
Protein purification was performed using E. coli (S17-1 λpir) bearing plasmids with poly-histidine tagged versions of cpdA, PA4969, SMA3506, SMA3506-N94A, and the empty vector, pMQ131, for a mock-purification control. Cultures of bacteria (50–200 ml) were grown overnight in LB with kanamycin. Bacteria were pelleted by centrifugation and frozen at −80°C. Pellets were thawed on ice, disrupted in 15 ml lysis solution consisting of B-per lysis solution (Pierce), Halt-protease inhibitor, and DNase I using sonication. Lysates were clarified by centrifugation and aqueous fractions were passed over nickel-affinity columns using the manufacturers protocols (Pierce product no. 78100). PAGE analysis of eluted protein fractions indicate the presence of a predominant band that migrates at the expected size. Western blots using anti-histidine antibodies indicate the presence of a single band in the SMA3506-His8 and SMA3506-N94A-His8 samples that was absent in those made from the empty vector mock purification samples. Western blots were performed as previously described [39].
Generation of a cpdS cyaA double mutant
Based on the prediction that a cpdS cyaA double mutant would behave as a cyaA mutant with respect to prodigiosin pigment production, we screened the cpdS strain that had been randomly mutagenized with a transposon for isolates with a precocious red colony phenotype. These were noted as precocious red colony mutants (PRC). A mariner-based transposon from plasmid pSC189 was introduced into S. marcescens strain CMS2910 (ΔcpdS mutant) as previously described to initiate mutagenesis. Ten independent conjugation pools were plated onto LB agar plates selective for S. marcescens that had experienced a transposon insertion, and these were incubated at 30°C. After 16–20 hours, plates were visually inspected for hyper-red colonies, and one was chosen from nine of the conjugation plates from a total of ∼4000 mutant colonies. Four of the nine PRC mutant isolates were chosen based upon their enhanced pigmentation, increased hemolysis on blood agar plates and increased biofilm production – phenotypes exhibited by cyaA mutants. A medium-copy plasmid bearing the wild-type cyaA gene (pMQ158) was introduced into the four candidates. Three of the four mutant strains were complemented for their pigment and biofilm phenotypes (hemolysis was not tested). When the cyaA plasmid was lost through serial passage without selective antibiotic, the cyaA mutant-like phenotypes were restored (data not shown). The transposon insertion site for the cyaA-plasmid complemented candidates was performed as previously described [40] using SacII to digest the genome and T4 DNA ligase. The circularized genome fragments containing the plasmid were selected with kanamycin in E. coli strain EC100D pir-116 (Epicentre), plasmid minipreps were prepared and sequenced. Two of the mutants had transposons in the cyaA gene located at base pair 102 and 632 respectively; the other insertion site was not determined.
In vitro cAMP phosphodiesterase assays
Phosphodiesterase activity conferred by cAMP-PDE genes was measured using the method of Kuchma, et al. [41] with minor alterations. E. coli cpdA mutant strain JM3000 was used. JM3000 with the empty vector or test plasmid was grown for 16–18 h in LB medium with selective antibiotic, 1 ml of culture was obtained and the bacteria were pelleted by centrifugation. Bacterial cells were washed with 1 ml of reaction buffer (5 mM MgCl2, 50 mM Tris-HCl pH 9.3, and 50 mM NaCl), suspended in 0.75 ml reaction buffer and sonicated on ice until the lysate cleared. Lysates were clarified by centrifugation (16,000×g for 15 m at 4°C) and the protein concentration of the supernatant was determined by Bradford analysis. Lysates were normalized to 100 µg/ml in 0.1 ml reaction buffer and mixed with 0.1 ml of 10 mM bis-pNPP (Sigma product number N3002). The release of p-nitrophenol was measured in 96 well plates using a plate reader (A = 410 nm). Experiments were performed with triplicate biological replicates on two different days.
A thin layer chromatography (TLC) based cAMP-PDE assay was used to separate cAMP from 5′-AMP. Briefly 15 µl of reaction mixture (10 µl of 30 mM cAMP in enzyme buffer (1 mM MnCl2, 1 mM FeCl2, 50 mM Tris-Cl, 50 mM NaCl, pH 8), 10 µl of protein or protein elution buffer as a negative control), and 5 µl of water were incubated for 1 hour at 37°C. Aliquots (5 µl) were spotted and dried on a silica gel HLF TLC plate (Analtech product no. 47021) and separated using an ethanol/water/ammonium bicarbonate mixture (70:30:0.2 M) as a solvent [42]. Separated nucleotides were visualized with short wave UV light and photographed with a digital camera. Heat inactivation of protein was performed by incubation of the reaction mixture at 95°C for 20 minutes. The experiment was performed at least three times with each protein on different days and with at least two independent preparations of each protein.
An in vitro cAMP-PDE assay, based on the method of Hosono [43], was developed in which cAMP was mixed with either a positive control cAMP-PDE or SMA3506 or SMA3506-N94A and with alkaline phosphatase (AP). The basis of the assay being that cAMP-PDE activity will generate 5′-AMP from cAMP which can then be cleaved by AP to generate phosphate, whereas AP does not generate free phosphate from cAMP (confirmed in pilot assays). Available phosphate can be measured with the ascorbic acid-molybdate method, which generates a colorometric output measurable at A820. A reaction mixture composed of 500 µl of solution A (50 mM Tric-Cl, 5 mM MnCl2, 5 mM FeCl2, 50 mM NaCl, pH8), 100 µl of solution B (20 mM MgSO4, 100 µl of 2 mM cAMP, 100 µl of alkaline phosphatase (Sigma P5931 –28 units/ml)), and 200 µl of test protein or mock control (mock purified protein from E. coli with empty vector). This reaction was incubated for 2 hours at 38°C, at which time 100 µl of 55% TCA was added to stop the reaction. An equal volume of coloring reagent (1.2 N H2SO4, 0.5% ammonium molybdate, and 0.2% ascorbic acid) was then used to measure phosphate concentration indicating cAMP-PDE activity. The mixture was incubated at 38°C for 90 minutes and absorbance was read at 820 nm. A standard curve of phosphate was generated and used to measure phosphate release by the purified proteins and control samples.
Intracellular cAMP analysis
Cyclic-AMP concentrations were determined using an Enzyme Immunoassay (EIA) (Caymen Chemical-product number 581001) according to the manufacturers specifications using the acetylation protocol. S. marcescens strains were grown in LB to an OD600 of 4.3–5.0 and normalized to lowest culture OD600. Five ml of culture was collected and pelleted by centrifugation at 16,000×g for 1 minute. Bacterial pellets were washed three times in charcoal filtered phosphate-buffered saline (PBS) to remove extracellular cAMP. Pellets were lysed by sonication in 250 μl of charcoal filtered PBS (Fisher Scientific Sonic Dismembrator model number 100) on ice until lysates became visually clear.
Cell lysate protein concentrations were determined by Bradford Assay and normalized to the least concentrated sample (0.7–6.2 µg/ml of protein). 50 μl of lysate was used per well for EIA. cAMP concentrations (pmol/ml) were determined using a standard curve and online tools from Caymen chemical (www.myassays.com). Experiments were performed with at least three biological replicates per assay and repeated on at least two different days.
Transmission Electron Microscopy (TEM)
Performed as previously described using overnight cultures of cells grown in LB broth, washed with PBS, spotted on formvar coated grids, strained with uranyl acetate (1%) and viewed with a JEM-1210 electron microscope. The experiment was performed at least two times per genotype with independent biological replicates. At least 50 images were taken at 10,000× magnification per genotype based on the presence of non-clumped bacteria. Cell length was determined using Image J Software.
Yeast agglutination assay
These assays were performed as previously described [35], [44]. For the type I fimbriae-specific yeast agglutination assay bacteria were grown overnight in LB, washed in PBS and adjusted to OD600 = 1.0 in PBS. Yeast (Sigma product no. YSC2) was added to PBS at a concentration of 0.2 g/10 ml. To a 1 cm cuvette, PBS (1.5 ml), yeast (0.5 ml) and bacteria (0.4 ml) were added and shaken. Cuvettes were placed in a spectrophotometer and the OD600 was taken at time intervals up to 10 minutes. Experiments were done with triplicate to quadruplicate biological replicates and performed on at least two different days.
Statistical Analysis
One-way ANOVA with Tukey's multiple comparison test was done for experiments with more than two groups. A two-tailed unpaired Student's T-test was performed for experiments with two groups. Fisher's exact test was used for experiments with categorical variables. Analysis was performed using Graphpad Prism 5.
Results
Multicopy expression of the E. coli cpdA gene stimulates S. marcescens biofilm formation
Based on previous studies, we developed a simple model for the role of cAMP as a regulator of biofilm formation by S. marcescens (Figure 1A). The primary goal of this study was to examine the contributions of cAMP-PDE activity on biofilm formation by S. marcescens. To test this, we cloned a known 3′,5′-cAMP-PDE gene (cpdA) from E. coli and placed it under control of the Plac promoter on a multicopy plasmid. The plasmid was introduced into S. marcescens and it conferred a dramatic increase in biofilm formation (Figure 1B). This result provided impetus to seek a native cAMP-PDE gene and to test the importance of this putative gene in biofilm formation.
Figure 1. Role of cAMP-phosphodiesterase activity in biofilm formation and identification of a cAMP-phosphodiesterase gene in the S. marcescens genome.

A. Model for cAMP metabolism and inhibitory effect on biofilm production. Adenylate cyclase (AC) catalyzes synthesis of cAMP from ATP, whereas cyclic-AMP phosphodiesterase (cAMP-PDE) catalyzes hydrolysis of cAMP to 5′-AMP. B. Crystal violet stained biofilms on the side of test tubes formed under high-sheer conditions. Shown is a wild-type S. marcescens strain with either the empty vector or the vector with a wild-type copy of the E. coli cAMP-PDE gene, cpdA. C. Genomic context of the S. marcescens cpdS gene, a candidate cAMP-PDE gene.
Identification of a candidate cAMP-PDE in S. marcescens
An open reading frame (ORF), SMA3506, was identified by blasting the cpdA gene from E. coli against a sequenced strain of Serratia marcescens (Sanger Center). The SMA3506 ORF will be referred to as cpdS for cyclic-AMP phosphodiesterase from S. marcescens as described below. This gene is the fourth open reading frame in a group of nine genes that are oriented in the same direction and may be in an operon (Figure 1C). This genomic context is similar to that of cpdA from E. coli, with inclusion of DNA gyrase, gyrA, in the putative operon [45]. The cpdS ORF was cloned from S. marcescens strain CMS376 to test if its protein product functions as a PDE and to determine whether this activity has a role in surface attachment. cpdS was placed under control of the Plac promoter on a pBBR1-based plasmid (pMQ171). cpdS from laboratory strain CMS376 was sequenced (GenBank number EU925585) and the DNA sequence was 96.3% identical to cpdS from the sequenced S. marcescens strain, Db11. Compared to five previously characterized class III 3′,5′-cAMP-PDEs, the CpdS predicted protein is 73% identical to the CpdA protein from E. coli, 70% identical to the CpdA protein from K. pneumoniae, 58% identical to the Icc protein from Haemophilus influenza, 42% identical to the Rv0805 gene from Mycobacterium tuberculosis, and 40% identical to CpdA from Pseudomonas aeruginosa (Figure S1). All of these proteins contain conserved residues for catalytic activity and metal binding and contain a conserved sequence for the active site from purple acid phosphatases (D-(X)n-GD-(X)n-GNH[E/D]-(X)n-H-(X)n-GHXH) that is necessary for PDE activity [46] (Figure S1).
CpdS can hydrolyze cAMP in vitro
To determine whether the protein encoded by cpdS could hydrolyze cAMP in vitro, we generated poly-histidine-tagged recombinant CpdS (Figure S2A). For a negative control, a mutant version (cpdS-N94A) with an alanine substitute for a highly conserved asparagine residue that is necessary for cAMP-PDE activity in CpdA of P. aeruginosa and E. coli was made (Figure S2A). Immunobots of crude lysates from E. coli and S. marcescens support that the recombinant CpdS and CpdS-N94A were stable (Figure S2B). CpdS-His8 and CpdS-N94A-His8 were incubated with cAMP and the resulting products were separated with thin layer chromatography (TLC). The production of 5′-AMP was observed when cAMP was mixed with the purified CpdA-His8 positive control protein from E. coli and CpdS-His8 (Figure 2A). Heat-killed cAMP-PDE protein, the no protein control, and the mutant CpdS-N94A-His8 protein were unable to hydrolyze cAMP (Figure 2A).
Figure 2. CpdS exhibits cAMP-PDE activity in vitro.

A. Thin layer chromatography to examine cAMP-PDE activity from purified CpdA from E. coli, CpdS, and CpdS-N94A mixed with cAMP. HK = indicates heat killed protein before being mixed with cAMP. B. cAMP-PDE assay shows dose responsive activity by CpdS and positive controls CpdA and PA4969 (CpdA from P. aeruginosa), but not by the CpdS-N94A mutant or a mock purified protein.
A second method was used to verify cAMP hydrolysis in which cAMP was placed in a reaction mixture with both test protein (CpdA, CpdS, or CpdS-N94A) and alkaline phosphatase (AP). In this reaction, if cAMP is hydrolyzed into 5′-AMP, AP can release inorganic phosphate from 5′-AMP, which can be measured colorometrically using the ascorbic acid-molybdate method [43]. The positive controls CpdA from E. coli and P. aeruginosa (PA4969) released phosphate, indicating cAMP-PDE activity (Figure 2B). CpdS-His8, unlike CpdS-N94A-His8 and the mock-purified protein control (Mock), exhibited dose-dependent cAMP-PDE activity (Figure 2B, Figure S2C).
We observed that the phosphodiesterase inhibitor IBMX inhibited CpdS-His8 in a dose dependent manner using the general phosphodiesterase substrate bis-pNPP to assess PDE activity [47], with 4.8 mM (4.1 to 5.6–95% confidence interval) of IBMX conferring a 50% reduction in the activity of 1 µg of CpdS-His8 (Figure S2D). These data indicate that the cpdS gene from S. marcescens codes for a phosphodiesterase able to hydrolyze cAMP in vitro.
CpdS exhibits in vivo cAMP-PDE activity
We performed assays to test the hypothesis that CpdS codes for a functional cAMP-PDE in vivo. E. coli with a cpdA mutation bearing the vector alone, the vector with cpdS, and the vector with cpdS-N94A were tested for PDE activity. PDE activity was observed in lysates from cells with multicopy expression of cpdS (pcpdS plasmid) or the positive control E. coli plasmid (pcpdA), but not from lysates from the CpdS-N94A strains (pcpdS-N94A plasmid) (Figure 3A). Complementation of the cpdA mutation in E. coli with the cpdS gene demonstrated that multicopy expression of cpdS was sufficient to confer PDE activity in vivo.
Figure 3. CpdS exhibits cAMP-PDE activity in vivo.

A. Phosphodiesterase activity measured from protein lysates of an E. coli cpdA mutant strain bearing the negative control empty vector (pMQ131), pcpdS (pMQ171), or positive control pcpdA (pEJK3). Bis-pNPP was used as a substrate to measure PDE activity. B–C. Intracellular cAMP levels measured using an EIA assay was measured from S. marcescens cells of the indicated genotypes. Vector = pMQ131, pcpdS = pMQ171, pcpdA = pEJK3, pcpdS-N94A = pMQ176, pcpdA-His8 = pEJK7, pcpdS-His8 = pMQ198. Asterisk indicates significant increase compared to cpdA + vector (A) or WT (B), p<0.05 by ANOVA with Tukey's post-test. The number sign indicates a significant reduction from cpdS + vector, p<0.05 by ANOVA with Tukey's post-test.
To test whether CpdS has a role in vivo in S. marcescens, we mutated the cpdS gene by deletion of the cpdS open reading frame and measured intracellular levels of cAMP. The cAMP levels from the cpdS mutant were ∼50-fold higher in the cpdS deletion mutant strain than the WT, whereas cAMP levels from an adenylate cyclase mutant, CMS524, were near the minimum detection level of the assay and 17-fold lower than the WT (Figure 3 B–C). Episomal expression of E. coli cpdA or S. marcescens cpdS on a plasmid reversed the elevated cAMP levels of the cpdS mutant, whereas the cpdS-N94A mutant gene did not (Figure 3C). Together these results indicate that CpdS is a functional cAMP-PDE in vivo.
CpdS mediates biofilm formation through type I fimbriae
To determine whether physiological levels of CpdS mediate S. marcescens biofilm formation, we assessed biofilm formation in the cpdS deletion strain. A significant 66% reduction in biofilm formation was observed in the cpdS mutant compared to the WT (Figure 4A–B). As with cpdA (Figure 1B), multicopy expression of cpdS enhanced biofilm formation by both the WT and cpdS mutant strain (Figure 4A–B). Multicopy expression of the PDE defective cpdS-N94A mutant gene did not confer a hyper-biofilm phenotype (Figure 4B).
Figure 4. CpdS mediates S. marcescens biofilm formation.
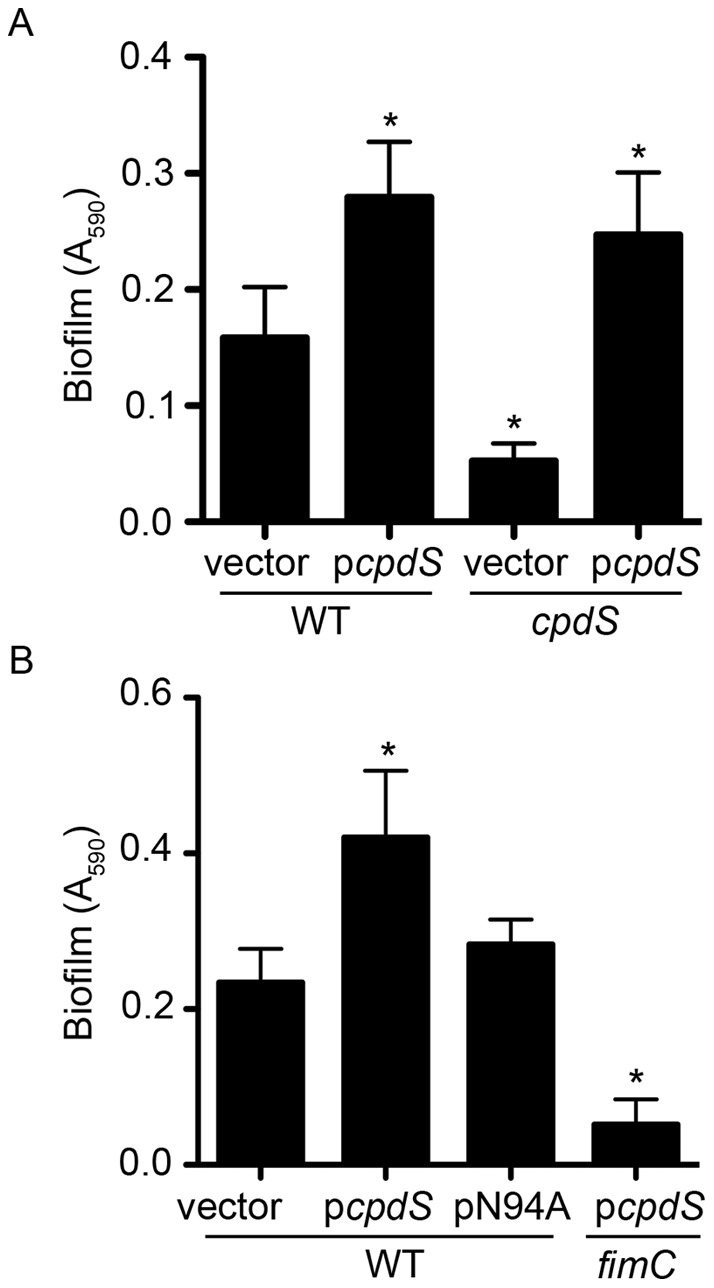
A–B. Quantification of crystal violet stained biofilms from the sides of glass test tubes grown under high-sheer conditions. Vector = pMQ131; pcpdS = pMQ171; pN94A = pMQ176. The asterisk indicates statistical difference from wild-type levels (n = 9, p<0.05, ANOVA with Tukey's post-test).
The viability of cpdS mutant and WT cells was assessed using fluorescence-based viability assay to test the prediction that altered cell viability contributed to the reduced biofilm exhibited by the cpdS mutant. No obvious difference was observed between planktonic or biofilm cells (Figure S3), indicating that another mechanism underlied the differential ability of these strains to form biofilms.
Because it was previously noted that S. marcescens cyaA and crp mutants develop hyper-biofilms and elevated production of type I fimbriae [35], we tested whether CpdS mediates biofilm formation through differential biosynthesis of fimbriae. Multicopy expression of cpdS in a fimC mutant, which is defective in the fimbrial usher component and unable to make surface fimbriae [35], did not confer a hyper-biofilm (Figure 4B). This shows that fimbriae are required for the hyper-biofilm phenotype conferred by multicopy cpdS expression.
A type I fimbriae yeast agglutination assay [34] was used to assess the defect of fimbriae production by the cpdS mutant. We observed a significant (p<0.05) decrease in the percent of yeast agglutination by the ΔcpdS mutant (23.6 ± 5.1%) compared to the WT (32.6 ± 4.7%) (Figure 5A). Whereas a negative control fimC mutant strain deficient for measurable fimbriae [35] produced no measurable agglutination, a positive control hyper-fimbriated cyaA mutant strain generated 55.9 ± 4.6% agglutination (Figure 5A).
Figure 5. The cpdS mutant has reduced surface fimbriae.
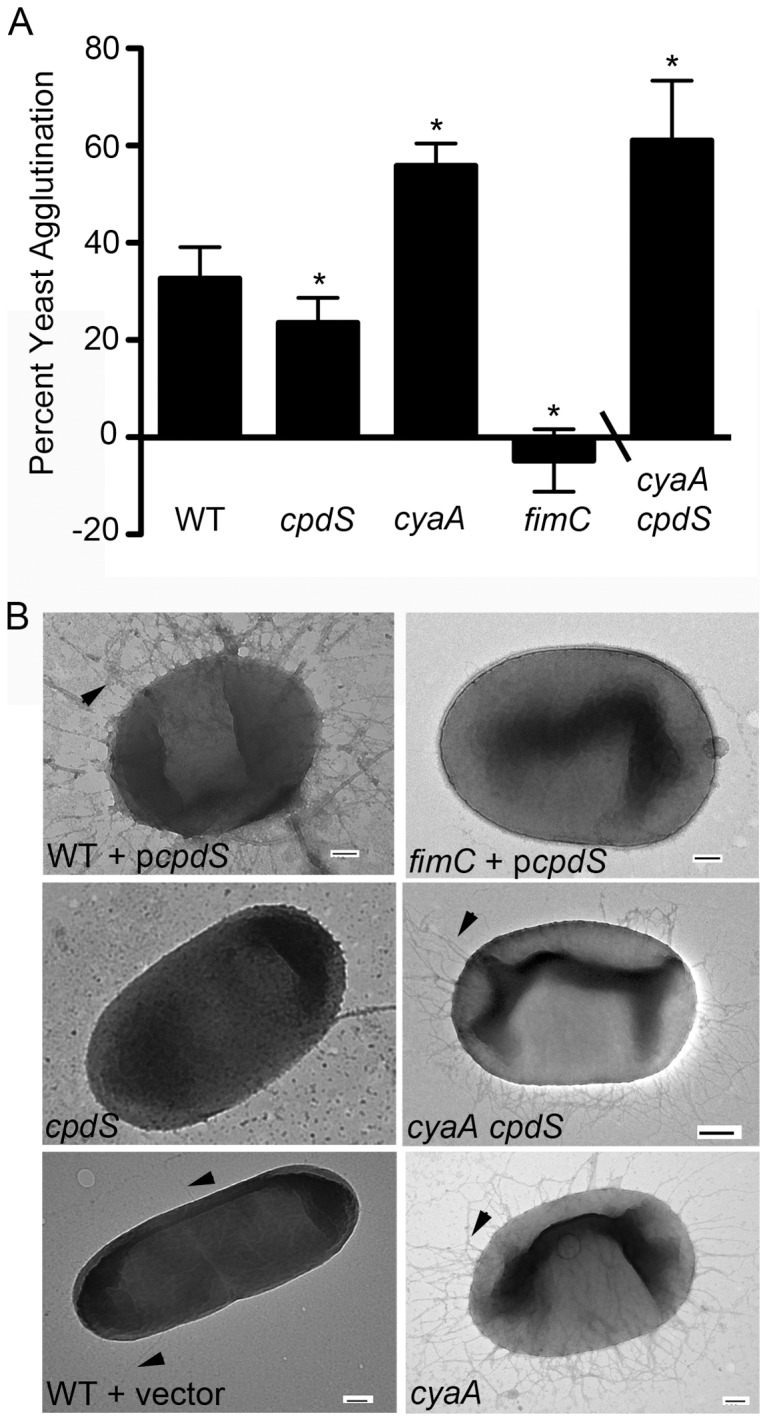
A. A yeast-agglutination assay was used to quantitate relative fimbriae expression. The asterisk indicates statistical difference from wild-type levels (n = 9, p<0.05, ANOVA with Tukey's post-test or Student's T-test). The cyaA cpdS double mutant data was from a different experiment (n = 6). B. Representative TEM micrographs of relevant strains. Black arrow-heads indicate fimbriae. Bar = 100 µm.
A cpdS cyaA double mutant was isolated, see materials and methods, for epistasis analysis. The cpdS cyaA double mutant was highly proficient at agglutination of yeast (63.8 ± 5.6), similar to the cyaA mutant, and unlike the cpdS mutant. This result suggests that the cyaA mutation is epistatic to the cpdS mutation for fimbriae production.
Transmission electron microscopy (TEM) was used to directly observe the fimbriae on the surface of the WT and cpdS mutant (Figure 5B). Although it was difficult to differentiate between individual cells, on a population basis it was clear that the cpdS mutant produced fewer fimbria-like appendages (Figure 5B). Whereas 58.6% of WT cells exhibited at least one surface fimbria, only 32.1% of ΔcpdS cells had a clear fimbria (n≥80 cells per genotype, p<0.01 Fisher's Exact Test).
A dramatic hyper-fimbria phenotype was observed from the WT with multicopy expression of cpdS (pcpdS) similar to the cyaA mutant and cpdS cyaA double mutant (Figure 5B). Significantly more (p<0.01, Fisher's Exact Test) WT cells with the cpdS gene on a plasmid (88%, n = 479) had at least one surface fimbria compared to the WT cell with the vector alone (60.3%, n = 179). These structures were confirmed as fimbriae, and the hyper-fimbria phenotype conferred by cpdS on a plasmid was absent in a fimC mutant, as FimC is the usher protein required for generation of fimbriae (Figure 5B).
Yeast agglutination analysis confirmed that multicopy expression of cpdS induced fimbriae production (Figure S4). The WT strain with the vector alone exhibited significantly less yeast agglutination than the WT strain with pMQ171 (pcpdS). The WT strain with the same plasmid but with the N94A mutation (pMQ176) induced a similar amount of yeast agglutination as the WT with the empty vector supporting that cAMP-PDE activity was required for the increased surface fimbriae phenotype.
Discussion
This study has demonstrated that S. marcescens cpdS (SMA3506) encodes a functional cAMP-PDE, although S. marcescens cAMP-PDE activity has been previously detected in the soluble fraction of crude cell extracts [48], [49]. Our results provided evidence that i) CpdS hydrolyzes cAMP to 5′-AMP (Figure 2A–B); ii) CpdS exhibits similar in vitro cAMP-PDE activity to CpdA homologs of E. coli and P. aeruginosa (Figure 2); iii) CpdS requires a conserved metal ion binding site through mutational analysis of cpdS-N94A (Figure 2–3); and iv) CpdS contains highly conserved amino acid residues and metal ion binding sites among class III cAMP-PDEs through multiple sequence alignment analysis (Figure S1). In addition, in vivo cAMP-PDE assays exhibited a 50-fold increase in intracellular cAMP levels in a cpdS deletion strain (Figure 3). We conclude that these genetic and biochemical data establish CpdS as a bona fide cAMP-PDE.
It is intriguing that isogenic cpdA mutant strains in E. coli [45] S. typhimurium [50], V. vulnificus [51], Klebsiella pneumonia [52], and P. aeruginosa [53] possess a wide range (∼2–30 fold increase) of intracellular cAMP compared to wild-type strains. This suggests that bacteria have adopted divergent cAMP-PDE-independent strategies for maintenance of cAMP concentrations. Indeed, there is compelling evidence that E. coli alternatively maintains intracellular cAMP through TolC-mediated cAMP export [54].
The biological relevance of bacterial cAMP-PDEs is inadequately understood, for which little functional significance has been identified except for cAMP homeostasis, sugar fermentation and transformation competence [45], [50], [53], [55]. Notably, CpdA homologs of V. cholerae [56] and P. aeruginosa are required for colonization efficiency and regulation of twitching-motility and virulence factors [53], [57], [58], respectively. The stringent control of cAMP levels is important in a wide variety of bacterial behaviors, thus we hypothesized that cAMP-PDEs could have an uncharacterized role in mediating other pathogenesis-associated phenotypes.
Through mutational, phenotypic, and microscopic analyses, we determined that CpdS is involved in the production of type I fimbrial adhesins and biofilm formation (Figure 4–5). These conclusions are supported by our findings that a cpdS deletion mutant is defective for biofilm formation but can be restored to wild-type levels with multicopy expression of cpdS (Figure 4). Furthermore, multicopy expression of cpdS in a wild-type background displays significantly elevated biofilm formation, however, biofilms were unaffected by over-expression of PDE-defective cpdS-N94A (Figure 4). We found that biofilm phenotypes were dependent on fimC, encoding a chaperone usher protein of the fimbrial operon fimABCD, similar to previous S. marcescens biofilm studies (Figure 5) [35]. Consistently, we observed a decrease in type I fimbriation for cpdS deletion mutants as determined through indirect (yeast agglutination) and direct (TEM) assays (Figure 5). Considering the importance of ACs and cAMP-CRP in type I fimbriae production and biofilm formation from prior work with E. coli and S. marcescens [24], [34], [35], it should be expected that cAMP levels are necessarily fine-tuned through cAMP-PDEs. It is plausible that the reciprocal interplay of ACs and cAMP-PDEs precisely modulates cAMP to permit flexibility in adjusting cAMP-regulated biofilm formation. To our knowledge, this report is the first description of a bacterial cAMP-PDE with the role of directing biofilm formation.
Ultimately, it will be important to identify the specific environmental signals, growth conditions, and/or regulatory proteins that modify cAMP-PDE effects on surface adhesins and biofilm formation. For instance, the potential influence of cAMP-CRP-dependent phase variation on type I fimbriation was not ascertained in this study as has been detailed in E. coli [23]. The orientation of the fimA promoter defines the phase as “on” or “off” which corresponds to the presence or absence of fimbrial transcription, respectively, with dramatic effects on early virulence and fimbriae-dependent bacterial colonization [59]. Although fimbrial phase-variation has not been described in S. marcescens, phase-related determinants such as the alarmone (p)ppGpp or sigma factor RpoS could feasibly have affected our observations of type I fimbriae [60], [61]. It is has been demonstrated through mutational analyses of E. coli catabolite repression machinery that cAMP-CRP is a negative regulator of rpoS expression [62], [63], and decreased rpoS expression has been shown in a V. vulnificus cpdA mutant [64]. Given the central role of RpoS in mediating the general stress response that contributes to antibiotic tolerance within biofilms [18], it is enticing to consider the possibly broader function of cAMP-dependent pathways in linking phase-dependent fimbriation with biofilm-mediated drug tolerance. Interestingly, it has been observed that cAMP-PDE mutants of S. typhimurium have increased antibiotic sensitivity, in which antibiotic and carbon sources were coupled through cAMP-dependent transporters [65], [66].
Bacterial signaling molecules and carbon source availability have a profound impact on the dispersal or detachment of biofilms [67]. A recent report by Huynh and colleagues highlighted the involvement of cAMP in glucose starvation-induced P. aeruginosa biofilm dispersal [68]. Because surface adhesins are largely involved in the primary attachment stage of surface colonization, we speculate that the observed importance of cAMP-PDEs on biofilm formation in our study was mediated at this early step. However, cAMP is emerging as a key signal in the later stages of biofilm development, namely biofilm dispersal, suggesting that cAMP flux may have a more important role in biofilm dynamics than formerly appreciated.
Supporting Information
Alignment of type III cAMP-Phosphodiesterase proteins.
(DOCX)
Purification and cAMP-PDE activity of recombinant CpdS.
(TIFF)
Live-Dead staining of WT and cpdS planktonic and biofilm cells.
(TIF)
Fimbriae-dependent yeast agglutination was induced by multicopy expression of cpdS .
(TIFF)
Acknowledgments
The authors thank Dr. Nanette Fulcher for instrumental help with charcoal purification, the University of Pittsburgh Center for Biological Imaging for the use of the electron microscope, and Denise Polaski for critical reading of the manuscript.
Funding Statement
This study was funded by NIH grant AI085570 and a Research to Prevent Blindness Career Development Award to RS and support from NIH grant EY08098, the Eye and Ear Foundation of Pittsburgh, and unrestricted funds from Research to Prevent Blindness. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Botsford JL, Harman JG (1992) Cyclic AMP in prokaryotes. Microbiol Rev 56: 100–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorke B, Stulke J (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6: 613–624. [DOI] [PubMed] [Google Scholar]
- 3. McDonough KA, Rodriguez A (2012) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat Rev Microbiol 10: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Postma PW, Lengeler JW, Jacobson GR (1993) Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57: 543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Botsford JL, Drexler M (1978) The cyclic 3′,5′-adenosine monophosphate receptor protein and regulation of cyclic 3′,5′-adenosine monophosphate synthesis in Escherichia coli . Mol Gen Genet 165: 47–56. [DOI] [PubMed] [Google Scholar]
- 6. Richter W (2002) 3′,5′ Cyclic nucleotide phosphodiesterases class III: members, structure, and catalytic mechanism. Proteins 46: 278–286. [DOI] [PubMed] [Google Scholar]
- 7. Rickman L, Scott C, Hunt DM, Hutchinson T, Menendez MC, et al. (2005) A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol Microbiol 56: 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith RS, Wolfgang MC, Lory S (2004) An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun 72: 1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curtiss R 3rd, Goldschmidt RM, Fletchall NB, Kelly SM (1988) Avirulent Salmonella typhimurium delta cya delta crp oral vaccine strains expressing a streptococcal colonization and virulence antigen. Vaccine 6: 155–160. [DOI] [PubMed] [Google Scholar]
- 10. Petersen S, Young GM (2002) Essential role for cyclic AMP and its receptor protein in Yersinia enterocolitica virulence. Infect Immun 70: 3665–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skorupski K, Taylor RK (1997) Cyclic AMP and its receptor protein negatively regulate the coordinate expression of cholera toxin and toxin-coregulated pilus in Vibrio cholerae . Proc Natl Acad Sci U S A 94: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shanks RM, Stella NA, Arena KE, Fender JE (2013) Mutation of crp mediates Serratia marcescens serralysin and global secreted protein production. Res Microbiol 164: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shanks RM, Stella NA, Lahr RM, Wang S, Veverka TI, et al. (2012) Serratamolide is a hemolytic factor produced by Serratia marcescens . PLoS One 7: e36398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stella NA, Kalivoda EJ, O'Dee DM, Nau GJ, Shanks RM (2008) Catabolite repression control of flagellum production by Serratia marcescens . Res Microbiol 159: 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fux CA, Costerton JW, Stewart PS, Stoodley P (2005) Survival strategies of infectious biofilms. TRENDS in Microbiology 13: 34–40. [DOI] [PubMed] [Google Scholar]
- 16. Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2: 95–108. [DOI] [PubMed] [Google Scholar]
- 17. Kolter R, Greenberg EP (2006) The superficial life of microbes. Nature 441: 300–302. [DOI] [PubMed] [Google Scholar]
- 18. Mah TF, O'Toole GA (2001) Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9: 34–39. [DOI] [PubMed] [Google Scholar]
- 19. Lewis K (2008) Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322: 107–131. [DOI] [PubMed] [Google Scholar]
- 20. Connell H, Agace W, Klemm P, Schembri MA, Marild S, et al. (1996) Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci U S A 93: 9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pratt LA, Kolter R (1998) Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol Microbiol 30: 285–293. [DOI] [PubMed] [Google Scholar]
- 22. Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A (2001) The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci U S A 98: 6911–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muller CM, Aberg A, Straseviciene J, Emody L, Uhlin BE, et al. (2009) Type 1 fimbriae, a colonization factor of uropathogenic Escherichia coli, are controlled by the metabolic sensor CRP-cAMP. PLoS Pathog 5: e1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jackson DW, Simecka JW, Romeo T (2002) Catabolite repression of Escherichia coli biofilm formation. J Bacteriol 184: 3406–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fong JC, Yildiz FH (2008) Interplay between cyclic AMP-cyclic AMP receptor protein and cyclic di-GMP signaling in Vibrio cholerae biofilm formation. J Bacteriol 190: 6646–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang W, Pascual-Montano A, Silva AJ, Benitez JA (2007) The cyclic AMP receptor protein modulates quorum sensing, motility and multiple genes that affect intestinal colonization in Vibrio cholerae . Microbiology 153: 2964–2975. [DOI] [PubMed] [Google Scholar]
- 27. Houot L, Watnick PI (2007) A novel role for Enzyme I of the Vibrio cholerae phosphoenol-pyruvate phosphotransferase system in regulation of growth in a biofilm. J Bacteriol 190: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mahlen SD (2011) Serratia infections: from military experiments to current practice. Clin Microbiol Rev 24: 755–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hejazi A, Falkiner FR (1997) Serratia marcescens . J Med Microbiol 46: 903–912. [DOI] [PubMed] [Google Scholar]
- 30. Rice SA, Koh KS, Queck SY, Labbate M, Lam KW, et al. (2005) Biofilm formation and sloughing in Serratia marcescens are controlled by quorum sensing and nutrient cues. J Bacteriol 187: 3477–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nichols WA, Clegg S, Brown MR (1990) Characterization of the type 1 fimbrial subunit gene (fimA) of Serratia marcescens . Mol Microbiol 4: 2119–2126. [DOI] [PubMed] [Google Scholar]
- 32. Adegbola RA, Old DC (1982) New fimbrial hemagglutinin in Serratia species. Infect Immun 38: 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Labbate M, Zhu H, Thung L, Bandara R, Larsen MR, et al. (2007) Quorum-sensing regulation of adhesion in Serratia marcescens MG1 is surface dependent. J Bacteriol 189: 2702–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shanks RM, Stella NA, Kalivoda EJ, Doe MR, O'Dee DM, et al. (2007) A Serratia marcescens OxyR homolog mediates surface attachment and biofilm formation. J Bacteriol 189: 7262–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalivoda EJ, Stella NA, O'Dee DM, Nau GJ, Shanks RM (2008) The cyclic AMP-dependent catabolite repression system of Serratia marcescens mediates biofilm formation through regulation of type 1 fimbriae. Appl Environ Microbiol 74: 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke D DD, Stearns T. (2000) Methods In Yeast Genetics, A Cold Spring Harbor Laboratory Course Manual. Plainview, NY: Cold Harbor laboratory Press.
- 37. Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA (2006) Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72: 5027–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shanks RM, Kadouri DE, MacEachran DP, O'Toole G A (2009) New yeast recombineering tools for bacteria. Plasmid 62: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shanks RM, Dashiff A, Alster JS, Kadouri DE (2012) Isolation and identification of a bacteriocin with antibacterial and antibiofilm activity from Citrobacter freundii . Arch Microbiol 194: 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chiang SL, Rubin EJ (2002) Construction of a mariner-based transposon for epitope-tagging and genomic targeting. Gene 296: 179–185. [DOI] [PubMed] [Google Scholar]
- 41. Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, et al. (2007) BifA, a c-di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol 189: 8165–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Higashida H, Hossain KZ, Takahagi H, Noda M (2002) Measurement of adenylyl cyclase by separating cyclic AMP on silica gel thin-layer chromatography. Anal Biochem 308: 106–111. [DOI] [PubMed] [Google Scholar]
- 43. Hosono K (1988) Acylpeptide inhibitors of phosphodiesterase produced by Bacillus subtilis . Methods Enzymol 159: 497–504. [DOI] [PubMed] [Google Scholar]
- 44. Kalivoda EJ, Stella NA, Aston MA, Fender JE, Thompson PP, et al. (2010) Cyclic AMP negatively regulates prodigiosin production by Serratia marcescens . Res Microbiol 161: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imamura R, Yamanaka K, Ogura T, Hiraga S, Fujita N, et al. (1996) Identification of the cpdA gene encoding cyclic 3′,5′-adenosine monophosphate phosphodiesterase in Escherichia coli . J Biol Chem 271: 25423–25429. [DOI] [PubMed] [Google Scholar]
- 46. Shenoy AR, Visweswariah SS (2006) New messages from old messengers: cAMP and mycobacteria. TRENDS in Microbiology 14: 543–550. [DOI] [PubMed] [Google Scholar]
- 47. Bobrov AG, Kirillina O, Perry RD (2005) The phosphodiesterase activity of the HmsP EAL domain is required for negative regulation of biofilm formation in Yersinia pestis . FEMS Microbiol Lett 247: 123–130. [DOI] [PubMed] [Google Scholar]
- 48. Okabayashi T, Ide M (1970) Cyclic 3′,5′-nucleotide phosphodiesterase of Serratia marcescens . Biochim Biophys Acta 220: 116–123. [DOI] [PubMed] [Google Scholar]
- 49. Winkler U, Scholle H, Bohne L (1975) Mutants of Serratia marcescens lacking cyclic nucleotide phosphodiesterase activity and requiring cyclic 3′,5′-AMP for the utilization of various carbohydrates. Arch Microbiol 104: 189–196. [DOI] [PubMed] [Google Scholar]
- 50. Botsford JL (1984) Cyclic AMP phosphodiesterase in Salmonella typhimurium: characteristics and physiological function. J Bacteriol 160: 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim HS, Kim SM, Lee HJ, Park SJ, Lee KH (2009) Expression of the cpdA gene, encoding a 3′,5′-cyclic AMP (cAMP) phosphodiesterase, is positively regulated by the cAMP-cAMP receptor protein complex. J Bacteriol 191: 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin CT, Chen YC, Jinn TR, Wu CC, Hong YM, et al. (2013) Role of the cAMP-dependent carbon catabolite repression in capsular polysaccharide biosynthesis in Klebsiella pneumoniae . PLoS One 8: e54430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fuchs EL, Brutinel ED, Klem ER, Fehr AR, Yahr TL, et al. (2010) In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J Bacteriol 192: 2779–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hantke K, Winkler K, Schultz JE (2011) Escherichia coli exports cyclic AMP via TolC. J Bacteriol 193: 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Macfadyen LP, Ma C, Redfield RJ (1998) A 3′,5′ cyclic AMP (cAMP) phosphodiesterase modulates cAMP levels and optimizes competence in Haemophilus influenzae Rd. J Bacteriol 180: 4401–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Merrell DS, Hava DL, Camilli A (2002) Identification of novel factors involved in colonization and acid tolerance of Vibrio cholerae . Mol Microbiol 43: 1471–1491. [DOI] [PubMed] [Google Scholar]
- 57. Inclan YF, Huseby MJ, Engel JN (2011) FimL regulates cAMP synthesis in Pseudomonas aeruginosa . PLoS One 6: e15867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nolan LM, Beatson SA, Croft L, Jones PM, George AM, et al. (2012) Extragenic suppressor mutations that restore twitching motility to fimL mutants of Pseudomonas aeruginosa are associated with elevated intracellular cyclic AMP levels. Microbiologyopen 1: 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gunther NWt, Snyder JA, Lockatell V, Blomfield I, Johnson DE, et al. (2002) Assessment of virulence of uropathogenic Escherichia coli type 1 fimbrial mutants in which the invertible element is phase-locked on or off. Infect Immun 70: 3344–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aberg A, Shingler V, Balsalobre C (2006) (p)ppGpp regulates type 1 fimbriation of Escherichia coli by modulating the expression of the site-specific recombinase FimB. Mol Microbiol 60: 1520–1533. [DOI] [PubMed] [Google Scholar]
- 61. Dove SL, Smith SG, Dorman CJ (1997) Control of Escherichia coli type 1 fimbrial gene expression in stationary phase: a negative role for RpoS. Mol Gen Genet 254: 13–20. [DOI] [PubMed] [Google Scholar]
- 62. Lange R, Hengge-Aronis R (1994) The cellular concentration of the sigma S subunit of RNA polymerase in Escherichia coli is controlled at the levels of transcription, translation, and protein stability. Genes Dev 8: 1600–1612. [DOI] [PubMed] [Google Scholar]
- 63. Ueguchi C, Misonou N, Mizuno T (2001) Negative control of rpoS expression by phosphoenolpyruvate: carbohydrate phosphotransferase system in Escherichia coli . J Bacteriol 183: 520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee HJ, Park SJ, Choi SH, Lee KH (2008) Vibrio vulnificus rpoS expression is repressed by direct binding of cAMP-cAMP receptor protein complex to its two promoter regions. J Biol Chem 283: 30438–30450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alper MD, Ames BN (1975) Cyclic 3′, 5′-adenosine monophosphate phosphodiesterase mutants of Salmonella typhimurium . J Bacteriol 122: 1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alper MD, Ames BN (1978) Transport of antibiotics and metabolite analogs by systems under cyclic AMP control: positive selection of Salmonella typhimurium cya and crp mutants. J Bacteriol 133: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McDougald D, Rice SA, Barraud N, Steinberg PD, Kjelleberg S (2012) Should we stay or should we go: mechanisms and ecological consequences for biofilm dispersal. Nat Rev Microbiol 10: 39–50. [DOI] [PubMed] [Google Scholar]
- 68. Huynh TT, McDougald D, Klebensberger J, Al Qarni B, Barraud N, et al. (2012) Glucose starvation-induced dispersal of Pseudomonas aeruginosa biofilms is cAMP and energy dependent. PLoS One 7: e42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miller VL, Mekalanos JJ (1988) A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR . J Bacteriol 170: 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of type III cAMP-Phosphodiesterase proteins.
(DOCX)
Purification and cAMP-PDE activity of recombinant CpdS.
(TIFF)
Live-Dead staining of WT and cpdS planktonic and biofilm cells.
(TIF)
Fimbriae-dependent yeast agglutination was induced by multicopy expression of cpdS .
(TIFF)