

Abstract
Presenilins (PS1 and PS2) are multi-functional proteins involved in a diverse array of molecular and cellular functions, including proteolysis, development, neurogenesis, synaptic plasticity, ion channel regulation and phospholipid metabolism. Mutations in presenilin genes are responsible for the majority of Familial Alzheimer disease (FAD). Consequently, FAD-associated mutations in genes encoding PS1 or PS2 lead to several key cellular phenotypes, including alterations in proteolysis of β-amyloid precursor protein (APP) and Ca2+ entry. The mechanism underlying presenilin (PS)-mediated modulation of Ca2+ entry remains to be determined. Our previous studies showed that the PS-dependent down-regulation of phosphatidylinositol-4,5-bisphosphate (PIP2) is attributable to the observed Ca2+ deficits. In this study, we attempted to identify the ion channel that is subject to the PIP2 and PS-dependent modulation. We found that Ca2+ or Zn2+ entry via the transient receptor potential melastatin 7 (TRPM7) channel was attenuated by the presence of FAD-associated PS1 mutants, such as ΔE9 and L286V. TRPM7 has been implicated in Mg2+ homeostasis and embryonic development. The intracellular delivery of PIP2 restored TRPM7-mediated Ca2+ influx, indicating that the observed deficits in Ca2+ entry are due to down-regulation of PIP2. Conversely, PS1 and PS2 deficiency, previously shown to up-regulate PIP2 levels, potentiated TRPM7-mediated Ca2+ influx. PS-dependent changes in Ca2+ influx could be neutralized by a TRPM7 channel blocker. Collectively, these results indicate that TRPM7 may underlie the Ca2+ entry deficits observed in FAD-associated PS mutants and suggest that the normal function of PS involves regulation of TRPM7 through a PIP2-dependent mechanism.
INTRODUCTION
Alzheimer disease (AD) is a progressive and irreversible neurodegenerative disorder that leads to cognitive, memory and behavioral impairments. In the pathogenesis of AD, cerebral elevation and accumulation of the amyloid β-peptide (Aβ) are necessary steps (Hardy and Selkoe 2002). Most cases of AD are idiopathic, and advanced age serves as a major risk factor. In contrast, approximately 5% of AD cases are familial (FAD), and some cases are attributable to autosomal dominant mutations in presenilin (PS1 and PS2) genes (Tanzi and Bertram 2005). PSs constitute one component of a high-molecular-weight γ-secretase complex, which consists of at least three other transmembrane proteins: nicastrin, PEN-2 and APH-1 (De Strooper 2003; O’Brien and Wong 2011). Consistent with the role of PS as catalytic subunit of γ-secretase, FAD mutations in PSs affect APP processing increasing the ratio of Aβ42 to Aβ40 (Scheuner et al., 1996).
In addition to the role for APP processing, PSs are intimately linked to cellular Ca2+ homeostasis (Mattson 2001; LaFerla 2002; Stutzmann 2007). Capacitative Ca2+ entry (CCE) is a refilling mechanism that regulates the coupled process of inositol-1,4,5-trisphosphate (IP3)-mediated release of Ca2+ from endoplasmic reticulum (ER) and the replenishment of intracellular Ca2+ through plasma membrane channels (Berridge 2002). Cells that lack PS1 or express a dominant negative PS1 mutant show potentiation of CCE, whereas FAD-linked PS mutants attenuate CCE (Yoo et al., 2000). The ER of PS mutant cells is overloaded with Ca2+ causing potentiation of IP3-mediated Ca2+ signaling (Leissring et al., 2000; Akbari et al., 2004). Consistent with this result, one study reported PSs function as Ca2+ leak channels in the ER membrane regulating intracellular Ca2+ homeostasis (Tu et al., 2006). The same study reported that FAD-linked PS mutants exhibit decreased leak channel activity, which may explain the potentiated IP3-mediated Ca2+ signaling. Thus, cellular Ca2+ signaling in response to agonist stimulation could be exaggerated in FAD-associated PS mutations.
The transient receptor potential melastatin 7 (TRPM7) channel is widely expressed in various tissues including the brain. It is associated with anoxic neuronal death, neurodegenerative disease (Aarts et al., 2003; Hermosura et al., 2005; Wei et al., 2007), and has been implicated in the regulation of Mg2+ and Ca2+−homeostasis (Nadler et al., 2001; Monteilh-Zoller et al., 2003; Schmitz et al., 2003). TRPM7 channel is also permeable to Zn2+, playing an important role for Zn2+-mediated neuronal injury (Inoue et al., 2010). TRPM7 has also been implicated in embryonic development and thymopoiesis (Jin et al., 2008). Recently, we reported that activation of Mg2+ inhibitory cation (MIC) currents are chronically suppressed by the presence of FAD-linked PS mutants via down-regulation of phosphatidylinositol-(4,5)-bisphosphate (PIP2) levels (Landman et al., 2006). Since the TRPM7 channel has been implicated in mediating the MIC current (Penner and Fleig 2007), and requires PIP2 for channel activation (Runnels et al. 2002), we tested whether the TRPM7 channel is the targeted ion channel subject to PIP2 and PS-dependent modulation.
METHODS
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) except PIP2-carrier system (Echelon Biosciences, Salt Lake City, UT).
Cell Culture
Human embryonic kidney (HEK) cells (HEK293) and Chinese hamster ovarian (CHO) cells were stably transfected with 10 μg of each plasmid (PS1 WT, PS1 ΔE9, L286V) using SuperFect (Qiagen, Valencia, CA) transfection reagent according to the manufacturer’s protocol. Individual Zeocin-resistant colonies were isolated and screened for PS1 expression by Western blotting. Stable cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin in the presence of 250 μg/ml Zeocin (Invitrogen, Carlsbad, CA) and 400 μg/ml G418 (Calbiochem, Gibbstown, NJ) at 37°C. PS1/2-deficient MEFs were kindly provided by B. De Strooper (Center for Human Genetics/VIB04, Belgium).
Cytoplasmic Calcium Measurements
Cytoplasmic free Ca2+ concentration ([Ca2+]i) was measured using fura-2AM. Freshly prepared cells were incubated in DMEM containing 4 μM fura2-AM at 22–25°C for 40 min and washed twice with HEPES Ringer solution containing 145 mM NaCl, 3.6 mM KCl, 10 mM HEPES, 1.3 mM CaCl2, 1 mM MgCl2, 5 mM D-glucose and pH was titrated to 7.4 with NaOH. The fluorescence was monitored in a stirred quartz-microcuvette (1 ml) in a cell holder of a model CAF-110 fluorescence spectrophotometer (Jasco, Tokyo, Japan) at the wavelengths of 340 and 380 nm (excitation), and 510 nm (emission). The results were calibrated by adding 10 μM ionomycin with 10 mM CaCl2, which produced the maximum value of fluorescence ratio (340 nm/380 nm, Rmax), and 35 μM EGTA which produced the minimum value of fluorescence ratio (Rmin). [Ca2+]i was calculated by assuming a dissociation constant of 150 nM for Ca2+-fura-2 at 22–25°C. Single-cell fluorescence for measuring [Ca2+]i was carried out using a model IX70 inverted microscope (Olympus, Tokyo, Japan) equipped with 40 × or 60 × water-immersion objectives attached to a frame-transfer and back-illuminated charge-coupled device (CCD) camera (Quantix, Tucson, AZ) and Metafluor software (Universal Imaging, Ypsilanti, MI). Cells were cultured on cover glasses, then loaded with 2 μM Fluo-4-AM (Invitrogen, Carlsbad, CA) in serum-free medium at 22–25°C for 45 min. After washing three times with serum-free medium, cells were transferred to recording chambers, and allowed to accommodate for 20 min. Dual excitation wavelengths of 340 nm/380 nm with a 400 nm dichroic mirror were used, and emitted light was collected using a high pass filter of 450 nm. Cytosolic Ca2+ changes are presented as ratio values (340 nm/380 nm).
Zinc Imaging
The cytoplasmic zinc level ([Zn2+]i) was imaged using zinc-sensitive fluorescence dye, FluoZin-3 (Invitrogen, Carlsbad, CA). Cells were incubated with 5 μM FluoZin-3-AM in extracellular bath solution for 30 min at 37°C, which was followed by de-esterification for another 30 min at 22–25°C. The dye-loaded cells were held in a recording chamber under an inverted microscope as for measuring [Ca2+]i. Excitation wavelength was 490 nm, and emission wavelength was 520 nm. Fluorescence intensities (ΔF) were normalized to the resting values (F, average of five-data points recorded immediately before the change of solutions).
Silencing TRPM7 Expression Using Small Interference RNA (shRNA)
A TRPM7-specitic shRNA (TRPM7-shRNA) based on human TRPM7 corresponding to coding region 2204–2228 (AAC AAT GGC TAA AGC ATT AGT TGC, GenBank accession number NM_017672.3) were obtained from Integrated DNA Technologies (Coralville, IA). The control shRNA (control-shRNA) was nonsilencing RNA construct that did not affect any endogenous genes. The shRNAs were transfected into HEK293 cells using RNAiFect transfection reagent (Qiagen). Briefly, the shRNAs (20 pmoles) were mixed with 15 μl RNAiFect and 100 μl serum-free DMEM. After the liposome/RNA mix was incubated for 15 min at room temperature, the mixture was added to cells with 1900 μl growth medium in a 60 mm-diameter plate. Forty-eight hours later, cells were harvested and Western blotting was performed to confirm transfection efficiencies. For single-cell calcium and zinc imaging experiments, HEK293 cells were transfected with silencing of the same sequence for the human TRPM7-shRNA tagged Cy5 and thereby the transfected cells could be identified during measurement of [Ca2+]i and [Zn2+]i. Electrophysiology-Patch pipettes were fabricated from borosilicate glass (TW150–4; World Precision Instrument, Sarasota, FL) using a model P-97 Flaming/Brown micropipette puller (Sutter Instrument, Novato, CA). Patch-clamp experiments were conducted in a standard whole-cell recording configuration at room temperature. TRPM7 currents were recorded as described previously (Jeong et al., 2006). The extracellular Na+-based divalent cation-free (DVF) recording solution contained 130 mM Na methanesulfonate, 5 mM NaCl, 10 mM HEPES, and 12 mM HEDTA, titrated to pH 7.2 with NaOH. The pipette solution contained: 135 mM Cs•methanesulfonate, 10 mM BAPTA, 10 mM HEPES, and 3.17 mM CaCl2, titrated to pH 7.2 with CsOH. The internal-free [Ca2+] was buffered at 100 nM, calculated using Maxchelator software (Stanford University). Currents were amplified using an Axopatch 1D patch-clamp amplifier (Axon Instruments, Foster City, CA). Voltage stimuli were delivered to the cell as ramp pulses, and cells were held at 0 mV between each voltage stimulus. Data were analyzed using pClamp7 (Axon Instruments) and the Origin 6.0 program (OriginLab, Northampton, MA).
Cell Surface Biotinylation
HEK293 cells in poly-L-lysine coated 10 cm-diameter dishes were treated with 1 mM H2O2 for 10 min at 37°C. Cell surface labeling with NHS-LC-LCbiotin (Pierce, Etten-Leur, Netherlands) was performed. One hour after homogenizing, biotinylated proteins were precipitated using streptavidin-agarose beads (Sigma-Aldrich, St. Louis, MO). TRPM7 expression was analyzed by immunoblot for the precipitates (plasma membrane fraction).
Western Blot Analysis
Proteins were resolved by 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto nitrocellulose membranes. After transfer, each nitrocellulose membrane was blocked in Tris-buffered saline/Tween 20 containing membrane-blocking agent for overnight at 4°C, and then incubated with goat anti-TRPM7 antibody (Abcam, Cambridge, MA) overnight at 4°C. After washing, membranes were incubated for 1 h at 22–25°C with horseradish peroxidase-conjugated anti-goat IgG antibody (Zymed, Carlsbad, CA) and washed. Peroxidase activity was visualized with enhanced chemiluminescence. Blots were quantified with the Multi Gauge software using a LAS-3000 system (Fuji Film, Japan).
PIP2 Assay
Steady state level of PIP2 in membrane was assayed using an enzyme-linked immunosorbent assay (ELISA) kit (Echelon Biosciences Inc.). CHO cells were plated at 70–80% confluence in a 60 mm-diameter dish and cells were collected by removing the medium by gentle aspiration and immediately adding 1 ml ice cold 0.5 M trichloroacetic acid. The extraction of neutral lipids was done according to the manufacturer’s protocol using methanol and chloroform.
Statistical Analysis
Values are expressed as mean ± SE. Statistical significances between the means of two groups were analyzed by an unpaired Student’s t test. A p < 0.05 was considered statistically significant.
RESULTS
TRPM7 Channel Mediates MIC Current
The Mg2+ inhibitory cation (MIC) current has been shown to be suppressed in cells expressing FAD-linked PS mutants (Landman et al., 2006). Since TRPM7 is suggested to play a role in the MIC current (Monteilh-Zoller et al., 2003; Jeong et al., 2006), we characterized currents mediated by the TRPM7 channel. We first down-regulated the expression of the TRPM7 channel by using shRNAs targeting TRPM7 (TRPM7-shRNA). Western blot analysis showed that the expression level of endogenous TRPM7 was significantly reduced in HEK293 cells transfected with TRPM7-shRNA compared to cells transfected with control-shRNA [Fig. 1(A)]. We next performed whole-cell patch clamp recordings using these TRPM7-shRNA-transfected HEK293 cells. We used divalent-free extracellular solution, and ramp voltage pulses. As shown in Figure 1(B), the non-selective MIC currents from control-shRNA transfected cells showed slight inward rectification as described previously (Landman et al., 2006). The presence of the TRPM7 channel blocker, Gd3+, in the extracellular solution blocked TRPM7 current. The MIC current densities measured at −100 mV were −110.0 ± 17.2 pA/pF (n = 7) for control-shRNA transfected cells, and −39.8 ± 10.0 pA/pF (n = 9) for TRPM7-shRNA transfected cells. Thus, the reduction of MIC current density was evident in cells transfected with TRPM7-shRNA, suggesting that the TRPM7 channel mediates the MIC current.
Figure 1.
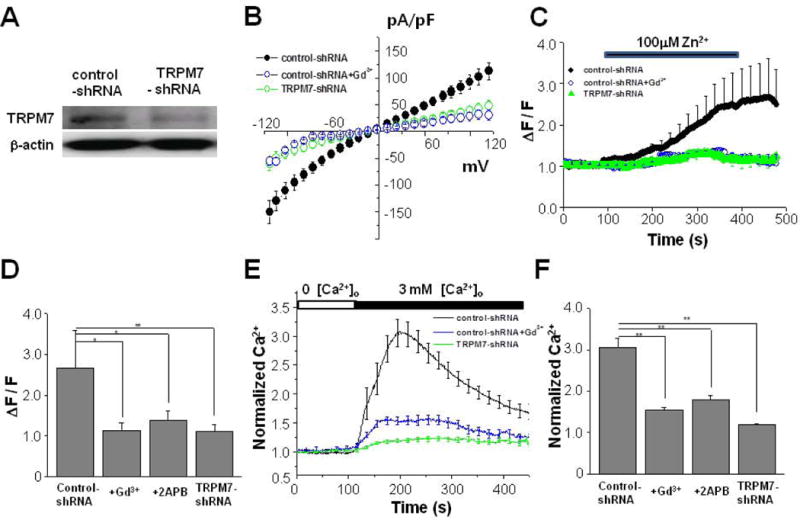
Zn2+-permeable TRPM7 channel underlies Ca2+ influx following depletion of extracellular Ca2+. (A) Western blotting result showed that shRNA against TRPM7 down-regulates the expression of endogenous TRPM7 channels from HEK293 cells. Cells were transfected either with control-shRNA or TRPM7-shRNA as described in Methods. Similar results were obtained from three independent experiments. The β-actin signal represents the internal loading control. (B) MIC currents were measured using whole-cell mode of patch clamp method from either control-shRNA (n = 7) or TRPM7-shRNA (n = 9) transfected cells. Na+-based divalent cation-free (DVF) recording solution was used. Ramp voltage was clamped from −120 mV to +120 mV during 100 ms from 0 mV holding voltage. As a TRPM7 channel blocker, 10 μM Gd3+ was added to the extracellular solution when measuring from control-shRNA transfected cells (n = 7). (C) Zinc-sensitive fluorescence dye, FluoZin-3, was used to measured the cytoplasmic zinc level ([Zn2+]i). Cy5-tagged transfected cells were identified before recordings from single HEK293 cells. [Zn2+]i was imaged from control-shRNA (n = 8) or TRPM7-shRNA (n = 9) transfected cells. In some control-shRNA transfected cells, 10 μM Gd3+ (n = 7) was added to the extracellular solution. (D) The normalized fluorescence intensities (ΔF) to the resting values (F) were compared from different conditions as in (C). In some control-shRNA transfected cells, 25 μM 2APB (n = 9) was added to the extracellular solution. (E) The cytoplasmic Ca2+ level ([Ca2+]i) was measured using Fluo-4 from single HEK293 cells. Cy5-tagged transfected cells were identified before recordings. [Ca2+]i was imaged from control-shRNA (n = 16) or TRPM7-shRNA (n = 16) transfected cells. In some control-shRNA transfected cells, 10 μM Gd3+ (n = 19) was added to the extracellular solution. (F) The normalized fluorescence intensities were compared from different conditions as in (E). In some control-shRNA transfected cells, 25 μM 2APB (n = 7) was added to the extracellular solution.
To further characterize TRPM7, we next examined the Zn2+ permeability, which is unique to TRPM7 but not store-operated Ca2+ entry (Monteilh-Zoller et al., 2003; Inoue et al., 2010). While the TRPM7 channel is a Ca2+-permeable non-selective cation channel, it is the only known Zn2+-permeable channel among TRP family members (Monteilh-Zoller et al., 2003; Inoue et al., 2010). We determined if down-regulation of TRPM7 channel affected the influx of Zn2+ by performing single-cell Zn2+ imaging to measure [Zn2+]i based on a published protocol (Inoue et al., 2010). When 100 μM Zn2+ was added to the extracellular solution, Zn2+ tracer FluoZin-3 intensity gradually increased in control-shRNA transfected cells (n = 8), indicating a gradual influx of Zn2+ [Fig. 1(C)]. The increased fluorescence level was maintained even after washing out Zn2+ from the extracellular solution. The increase of FluoZin-3 intensity was much smaller in TRPM7-shRNA transfected cells (n = 9) as compared to the control. The normalized fluorescence intensity values (ΔF/F) are compared in Fig. 1D. The addition of Gd3+ reduced [Zn2+]i influx (n = 7), suggesting that the influx of Zn2+ is via Gd3+-sensitive for the TRPM7 channel. Similar to Gd3+, the presence of 25 μM 2-aminoethoxydiphenyl borate (2APB), a known blocker of the TRPM7 channel, reduced [Zn2+]i (n = 9). Thus, these results indicate that TRPM7 channel is likely to mediate Zn2+ influx.
TRPM7 Channel Underlies Ca2+ Influx Following the Removal of Extracellular Ca2+
The TRPM7 channel is known to be responsible for the underlying current for spontaneously activated Ca2+ influx (Hanano et al., 2004). Decreasing extracellular divalent cations stimulates this current which is distinctively different from Ca2+-release activated Ca2+ current (CRAC) (Wei et al., 2007). During the experiment, the amount of Ca2+ released from the endoplasmic reticulum (ER) by thapsigargin, an ER Ca2+-depleting reagent, was not diminished (data not shown). Thus, during the experiment, Ca2+ was not depleted from ER when cells were exposed to a Ca2+-free extracellular solution.
To test whether the down-regulation of TRPM7 channel affected the spontaneously activated influx of Ca2+, [Ca2+]i was monitored by single-cell imaging using Fluo-4. After cells were exposed to a Ca2+-free extracellular solution, the addition of 3 mM extracellular Ca2+ induced increase of [Ca2+]i as shown in Figure 1(E). Compared to control-shRNA transfected cells (n = 16), TRPM7-shRNA transfected cells displayed reduced Ca2+ influx following the depletion of extracellular Ca2+ (n = 16). For comparison, the normalized Ca2+ levels are shown in Figure 1(F). The presence of Gd3+ blocked Ca2+ influx from control-shRNA transfected cells (n = 19). Similarly, the presence of 25 μM 2APB reduced [Ca2+]i (n = 7). These results indicate that Ca2+ influx following the depletion of extracellular Ca2+ was via TRPM7 channels. During the experiment, Ca2+ depletion from the endoplasmic reticulum (ER) was not observed when cells were exposed to a Ca2+-free extracellular solution. Also, during the experiments, the amount of Ca2+ released from ER was not diminished when cells were treated with thapsigargin, an ER Ca2+-depleting reagent (data not shown). Thus, only Ca2+ influx from the extracellular solution was measured, which was most likely mediated by TRPM7 channels without the activation of CCE.
FAD-linked PS1 ΔE9 Mutant Attenuates Ca2+ Influx Mediated by Zn2+-permeable TRPM7 Channel
Based on the Zn2+ permeability of TRPM7, we next measured Zn2+ influx in HEK293 cells stably transfected with PS1 WT or PS1 ΔE9. When 100 μM Zn2+ was added to the extracellular solution, the intensity of FluoZin-3 gradually increased in PS1 WT cells (n = 7; [Fig. 2(A)]). The addition of a membrane-permeable chelator for Zn2+, TPEN, reduced the fluorescence intensity indicating reduced Zn2+ influx. Compared to PS1 WT-transfected cells, the increase of FluoZin-3 intensity was much smaller in PS1 ΔE9-transfected cells (n = 8). FluoZin-3 intensities were compared in Figure 2(B). Similar to PS1 ΔE9, another FAD-linked PS mutant PS1 L286V, showed reduced Zn2+ influx (n = 8). Addition of Gd3+ reduced [Zn2+]i most significantly in PS1 WT cells [Fig. 2(B)]. These results suggest that the activity of Zn2+-permeable TRPM7 channel is down-regulated in FAD-linked mutant PS1.
Figure 2.
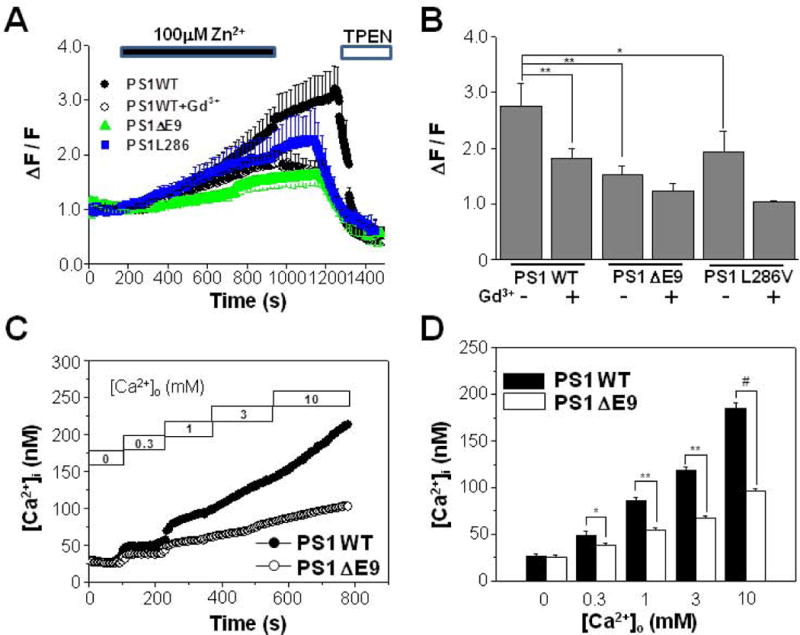
FAD-linked PS1 ΔE9 mutant attenuates Ca2+ influx mediated by Zn2+-permeable TRPM7. (A) Zn2+ influx was measured from HEK293 cells stably transfected with PS1 WT (n = 7), PS1 ΔE9 (n = 8), or PS1 L286V (n = 8). Also Zn2+ influx was measured from PS1 WT cells (n = 7) in the presence of 10 μM Gd3+. The intensity of Fluo Zin-3 gradually increased by the addition of 100 μM Zn2+ into the extracellular solution. The addition of membrane permeant chelator for Zn2+, TPEN, reduced the fluorescence intensity in all of cells tested. (B) The normalized fluorescence intensities were compared from different conditions as in (A). Zn2+ influx was also measured from PS1 ΔE9 (n = 7), or PS1 L286V (n = 7) cells in the presence of 10 μM Gd3+. (C) Cytoplasmic free Ca2+ concentration, [Ca2+]i, were compared between HEK293 cells transfected with either PS1 WT or PS1 ΔE9 using fura-2. Typical recordings are shown. Ca2+ influx were measured by adding back Ca2+ to the extracellular solution to obtain the indicated free Ca2+ concentrations. (D) When Ca2+ influx were compared they were larger in PS1 WT cells (n = 5) than in PS1 ΔE9 cells (n = 5). *, **, # represents p < 0.05, p < 0.01, p < 0.005 from paired t-tests, respectively.
We next tested whether Ca2+ influx following the removal of extracellular Ca2+ in the absence of intracellular Ca2+ store depletion could also be affected in FAD-linked mutant PS1 cells. When internal calcium ([Ca2+]i), was monitored using fura-2, the resting [Ca2+]i in the presence of physiological external calcium (calcium out, [Ca2+]o) was similar in PS1 WT (69.2 ± 2.1 nM, n = 24) and PS1 ΔE9 cells (62.3 ± 3.0 nM, n = 23) (data not shown). Also, [Ca2+]i in Ca2+-free extracellular solution did not differ between these cells [Fig. 2(C) and 2(D)]. To measure TRPM7-mediated Ca2+ influx, cells were first exposed to a Ca2+-free extracellular solution and subsequently the extracellular Ca2+ concentration, [Ca2+]o, was elevated stepwise as previously described (Chun et al., 2010). Typically, [Ca2+]i increased when Ca2+ was added stepwise to obtain the indicated [Ca2+]o [Fig. 2(C)]. At all of the tested [Ca2+]o, [Ca2+]i levels were greater in PS1 WT cells than in PS1 ΔE9 cells [Fig. 2(D)]. The discrepancy in [Ca2+]i between these cells increased as [Ca2+]o was elevated. For example, when [Ca2+]o was 0.3 mM, [Ca2+]i in PS1 WT cells and PS1 ΔE9 cells were 48.5 ± 4.3 nM and 37.8 ± 2.13 nM (n = 5), respectively. When [Ca2+]o was increased to 10 mM, [Ca2+]i in PS1 WT cells and PS1 ΔE9 cells were 185 ± 5 nM and 96.3 ± 2.2 nM (n = 5), respectively. These results suggest that PS1 plays an important role in regulating the Ca2+ influx following the depletion of extracellular Ca2+ and that the PS1 ΔE9 mutation down-regulates the Ca2+ entry mediated by TRPM7.
To further characterize the mechanism underlying the attenuated Ca2+ influx in PS1 ΔE9 cells, the effect of 2APB was tested. Cells were pretreated with 25 μM 2APB for 100 s in Ca2+-free solution, and then Ca2+ influx was measured by elevating the Ca2+ concentration of extracelluar solution to 3 mM. When Ca2+ was measured at 500 s, pretreating PS1 WT cells with 2APB inhibited Ca2+ influx by about 20% [Fig. 3(A)]. [Ca2+]i was 176 ± 7 nM (n = 11) in PS1 WT control cells, and 130 ± 6 nM (n = 7) in 2APB-treated cells. Thus, 2APB was able to block some component of the Ca2+ influx pathway following depletion of extracellular Ca2+ in PS1WT cells. In contrast, pretreating PS1 ΔE9 cells with 2APB did not influence Ca2+ in flux [Fig. 3(B)]. [Ca2+]i was 124 ± 8 nM (n = 10) in control and 111 ± 4 nM (n = 7) in 2APB-treated cells, suggesting that diminished 2APB-sensitive Ca2+ influx activity in PS1 ΔE9 cells. When the effects of 2APB on Ca2+ influx were compared, it was clear that the discrepancy in [Ca2+]i between PS1 WT and PS1 ΔE9 cells was significant in the absence of 2APB (filled bars), and that [Ca2+]i levels showed no difference in the presence of 2APB (open bars) [Fig. 3(C)]. These results are consistent with the down-regulation of the 2APB-sensitive TRPM7 current in PS1 ΔE9 cells, leading to the attenuation of Ca2+ influx.
Figure 3.
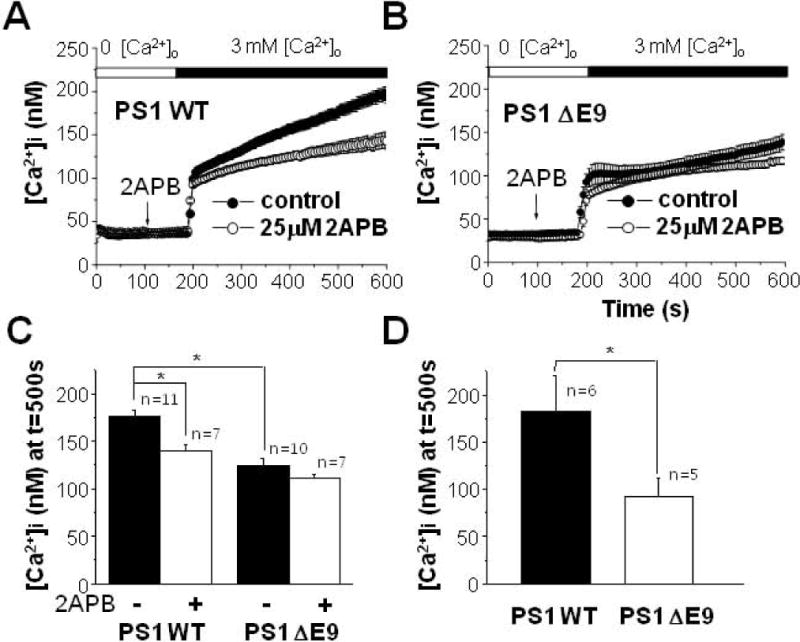
Effect of 2APB on Ca2+ influx following the depletion of extracellular Ca2+ in PS1 WT and PS1 ΔE9 cells. (A) PS1 WT cells were pre-treated either with (n = 7) or without (n = 11) 25 μM 2APB for 100 s in Ca2+-free solution. Then, [Ca2+]i was measured by adding back 3 mM Ca2+ into extracelluar solution. 2APB was able to block some component of Ca2+ influx only in PS1 WT cells. (B) Similarly, PS1 ΔE9 cells were pre-treated either with (n = 7) or without (n = 10) 25 μM 2APB before [Ca2+]i was measured by adding back 3 mM Ca2+ into extracelluar solution. 2APB was not able to block Ca2+ influx. (C) The effects of 2APB on Ca2+ influx were compared between PS1 WT and PS1 ΔE9 cells. (D) The effects of γ-secretase inhibitor on Ca2+ influx following the depletion of extracellular Ca2+. PS1 WT (n = 6) and PS1 ΔE9 cells (n = 5) were pre-treated with 100 nM compound E for 8 h before measuring Ca2+ influx by adding back 3 mM Ca2+ into extracelluar solution. * represents p < 0.05 from paired t-tests.
We next tested whether the difference in Ca2+ influx between PS1 WT and PS1 ΔE9 mutant was cell-type specific. For this purpose, PS1 WT- or PS1 ΔE9-transfected CHO cells were used. When [Ca2+]i was measured at 500 s after [Ca2+]o was elevated to 3 mM from 0 mM as described above, it was 428 ± 36 nM (n = 7) in PS1 WT cells and 251 ± 25 nM (n = 5) in PS1 ΔE9 cells. This result was consistent with our findings in HEK293 cells. We also tested whether another FAD-associated PS1 mutant could produce the same effect on Ca2+ influx as PS1 ΔE9 by measuring Ca2+ influx in CHO cells transfected with either PS1 WT or L286V. Adding 3 mM [Ca2+]o following the depletion of extracellular Ca2+ increased [Ca2+]i to 403 ± 47 nM (n = 6) in PS1 WT cells and 282 ± 26 nM (n = 7) in CHO cells transfected with PS1 L286V mutant. Thus, the PS1 L286V mutation showed the same down-regulation of Ca2+ influx as the PS1 ΔE9 mutation.
Attenuation of Ca2+ Influx By FAD-linked PS1 Mutation Is Not Dependent On γ-Secretase Activity
We next tested whether the down-regulation of Ca2+ influx in PS1 ΔE9 mutant cells was dependent on γ-secretase activity (Hardy and Selkoe 2002; O’Brien and Wong 2011). When both PS1 WT- and PS1 ΔE9-transfected HEK cells were pre-incubated for 8 h with a potent γ-secretase inhibitor (100 nM of compound E), the discrepancy in [Ca2+]i between two cells persisted [Fig. 3(D)], indicating that the Ca2+ influx pathway is not dependent on the γ-secretase activity.
PS Deficiency Leads To Increased Ca2+ Influx
To determine the role for PSs in TRPM7-mediated Ca2+ influx, we next examined the effects of PSKO on Ca2+ influx. The resting [Ca2+]i in PS1/2 double knockout (PSKO) mouse embryonic fibroblast (MEF) cells (Landman et al., 2006) increased in the presence of physiological [Ca2+]o compared to PSWT cells [Fig. 4(A)]. Also, [Ca2+]i of PSKO cells was higher than that of PSWT cells in the absence of [Ca2+]o. When [Ca2+]i was measured while Ca2+ was added stepwise to obtain the indicated [Ca2+]o as in Figure 2(C), [Ca2+]i exhibited graded increases in PSWT (n = 5) and PSKO cells (n = 5) in response to increased [Ca2+]o [Fig. 4(B)]. At all of the tested [Ca2+]o, [Ca2+]i levels obtained at different [Ca2+]o were larger in PSKO cells than in PSWT cells, and the discrepancy in [Ca2+]i between the two cell types increased as [Ca2+]o was elevated, which recapitulated results obtained from PS1 WT and PS1 ΔE9 cells in Figure 2(D). These results indicate that PS1/2 deficiency increased Ca2+ influx, confirming that the normal function of PSs includes regulating this Ca2+ influx pathway following the depletion of extracellular Ca2+.
Figure 4.
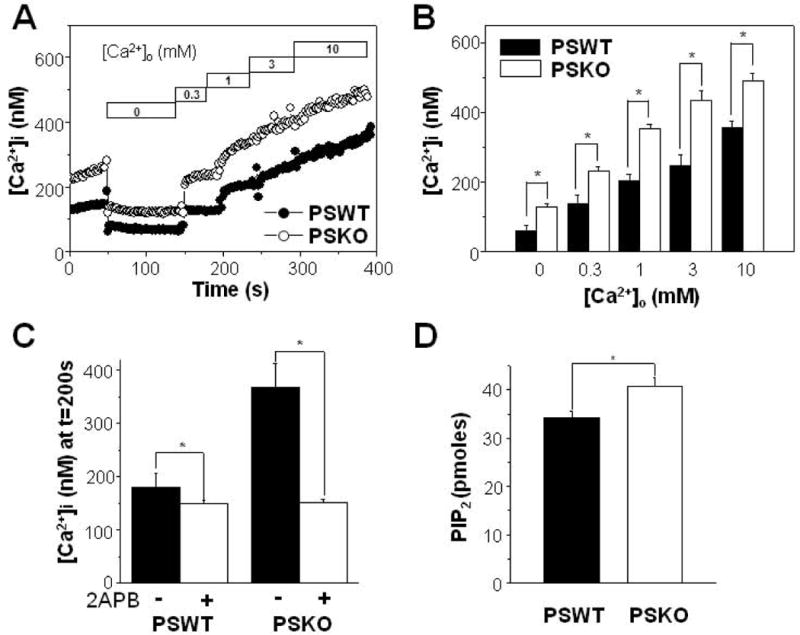
Increased Ca2+ influx following the depletion of extracellular Ca2+ in PS1/2 double knockout (PSKO) MEF cells. (A) From this typical recording, PSKO cells showed increased resting [Ca2+]i either in the presence or in the absence of physiological [Ca2+]o compared to wild type PS1/2-transfected (PSWT) cells. Ca2+ influx were measured as in Fig. 2C by adding back Ca2+ to the extracellular solution as indicated. (B) Ca2+ influx were larger in PSKO MEF cells (n = 5) compared to PSWT cells (n = 5). Ca2+ influx was measured as in (A). (C) The effects of 2APB on Ca2+ influx were compared between PSWT (n = 8) and PSKO MEF cells (n = 8) as described in Fig. 3A. 2APB was able to block larger amount of Ca2+ influx from PSKO cells. (D) Steady state levels of PIP2 in membrane were assayed by using and ELISA kit from PSKO MEF cells (n = 4) and to PSWT MEF cells (n = 4). * represents p < 0.05 from paired t-test.
Next, the effects of 2APB on Ca2+ influx were compared in PSWT and PSKO cells. Cells were pretreated with 25 μM 2APB for 100 s in a Ca2+-free solution, and then Ca2+ influx was measured by the addition of 3 mM Ca2+ to the extracelluar solution. Pretreating PSWT cells with 2APB resulted in about 13% inhibition in [Ca2+]i from 172 ± 21 nM (control) to 150 ± 5 nM (2APB-treated) [Fig. 4(C); n = 8]. In PSKO cells, 2APB inhibited Ca2+ influx by 61%. [Ca2+]i decreased from 370 ± 52 nM (control) to 146 ± 8 nM (2APB-treated) (n = 8). The discrepancy in [Ca2+]i between PSWT and PSKO cells was significant in the absence of 2APB [closed bars in Fig. 4(C)]. However, in the presence of 2APB, there was no difference in [Ca2+]i between them [open bars in Fig. 4(C)]. These results are consistent with our previous conclusion that 2APB-sensitive TRPM7 channels function as the Ca2+ influx pathway following the depletion of extracellular Ca2+.
TRPM7 Expression in PS1 Mutant ΔE9 Cells
One possible mechanism for the inhibition of Ca2+ influx in PS1 ΔE9 cells could be the down-regulation of the channel protein. For this reason, we tested whether PS1 ΔE9 alters the expression level of TRPM7 by comparing TRPM7 protein levels in PS1 WT- and PS1 ΔE9-transfected HEK293 cells using Western blot analysis. TRPM7 proteins were probed with a polyclonal antibody raised against goat TRPM7. Figure 5(A) shows an example of blots from 4 different experiments obtained from whole-cell homogenates. The intensities of the bands were similar between PS1 WT and PS1 ΔE9 cells, and densitometric analysis of the bands showed no difference in TRPM7 levels [Fig. 5(B)]. Similarly, the levels of TRPM7 on the cell surface were the same between PSWT and PSKO cells [n = 5; Fig. 5(C)]. Densitometric analysis of the bands is shown in Figure 5(D). These results indicate that changes in the expression levels of the TRPM7 channel are unlikely to be responsible for the decreased Ca2+ influx in PS1 ΔE9 cells, or the increased Ca2+ influx in PSKO cells.
Figure 5.
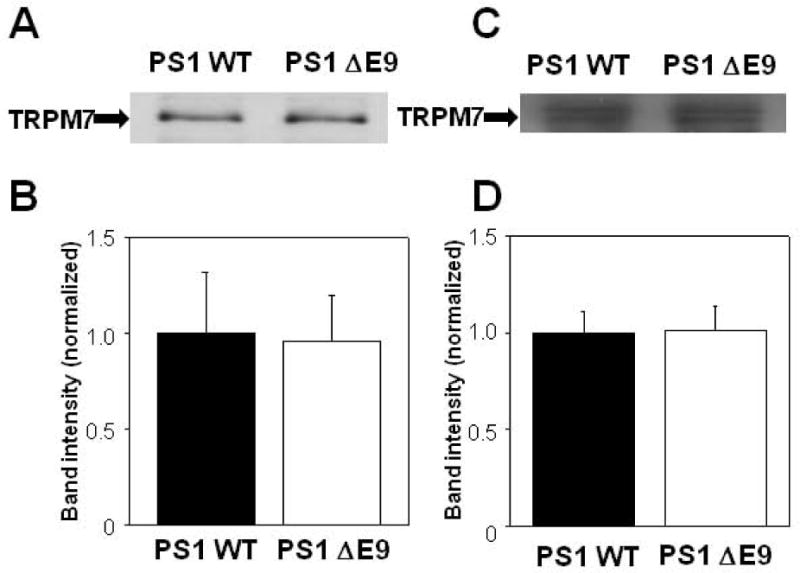
Expression levels of the endogenous TRPM7 channel in PS1 WT and PS1 ΔE9 HEK cells. (A) A typical Western blotting result and (B) densitometric analysis of the bands (n = 4) shows that the total expression levels of TRPM7 in membrane fractions were not different in PS1 WT and PS1 ΔE9 cells. (C) A typical Western blotting result and (D) densitometric analysis of the bands (n = 5) shows that the cell surface TRPM7 levels were not different in PS1 WT and PS1 ΔE9 cells.
Elevation of PIP2 Levels Restores the Attenuated Ca2+ Influx from PS1 ΔE9 Cells
We previously showed that PIP2 levels are down-regulated in FAD-associated PS1 mutant HEK cells (Landman et al., 2006). Using an ELISA assay specific for PIP2, we found increased levels of PIP2 in PSKO cells compared to PSWT cells [Fig. 4(D)]. To explore the possibility that aberrant PIP2 metabolism may underlie the observed decrease of TRPM7-mediated Ca2+ influx, we tested whether the elevation of PIP2 levels could restore Ca2+ influx in PS1 ΔE9 cells. The PIPn-carrier system was utilized for the intracellular delivery of PIP2, since carrier compounds are “charge-neutralization” species that deliver anionic PIP2 into the cells (Ozaki et al., 2000). After carriers were added at a 1:1 molar ratio with PIP2, the complex was diluted to the final desired concentration into cell-containing solutions. After the carrier-PIP2 complex was incubated with PS1 ΔE9 cells for 15 min to allow cellular uptake, Ca2+ influx was measured by the addition of 3 mM Ca2+ to the extracellular solution. Carrier-PIP2 complex increased Ca2+ influx in a dose dependent manner [Fig. 6(A); n = 4]. The half maximum increase of Ca2+ influx was obtained at 5.3 ± 0.5 μM PIP2 [Fig. 6(B)]. This suggests that, the reduced Ca2+ influx in PS1 ΔE9 cells is due to reduced PIP2 level in PS1 ΔE9 cells. Next, the effects of 10 μM carrier-PIP2 complex were compared in PS1 ΔE9 and PS1 WT cells [Figs. 6(C) and 6(D)]. This concentration was near the saturating concentration for the maximal effect on Ca2+ influx. Without the carrier-PIP2 complex, [Ca2+]i levels measured at different [Ca2+]o were larger in PS1 WT cells [filled circles in Fig. 6(C)] than in PS1 ΔE9 cells [open circles in Fig. 6(C)] at all of the tested [Ca2+]o, confirming the previous observation [Fig. 2(C)]. However, in the presence of 10 μM carrier-PIP2 complex, the discrepancy in [Ca2+]i between the two cell types was abolished [filled and open squares in Fig. 6(C)]. Thus, there was no difference in [Ca2+]i between the two cell types at all of tested [Ca2+]o in the presence of PIP2 [Fig. 6(D)] indicating that the saturating concentration of PIP2 abolished the difference in [Ca2+]i between PS1 ΔE9 and PS1 WT cells. These results suggest that down-regulation of PIP2 levels is the underlying mechanism for the reduced TRPM7-mediated Ca2+ influx in PS1 ΔE9 cells.
Figure 6.
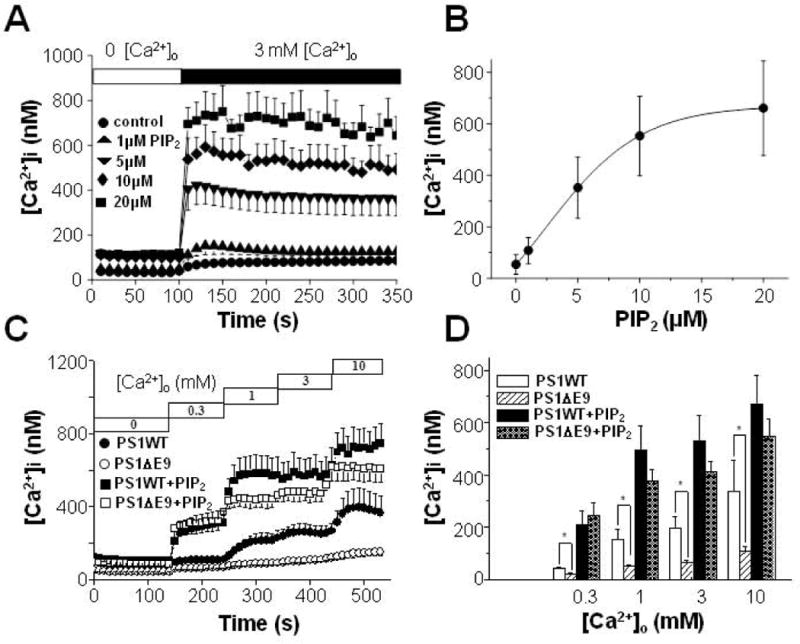
Carrier-delivered PIP2 abolishes the difference in Ca2+ influx following the depletion of extracellular Ca2+ between PS1 WT and PS1 ΔE9 HEK cells. (A) PS1 ΔE9-transfected HEK293 cells were incubated with carrier-PIP2 complex at indicated concentrations for 15 min before Ca2+ influx was measured by adding back 3 mM Ca2+ (n = 4). (B) Ca2+ influx was increased by carrier-PIP2 complex in a dose dependent manner. The half maximum increase of Ca2+ influx was obtained at 5.3 ± 0.5 μM. (C) The effect of 10 μM carrier-PIP2 complex on Ca2+ influx were compared between PS1 ΔE9 and PS1 WT cells. Without PIP2, PS1 WT cells (filled circles; n = 4) showed larger Ca2+ influx than in PS1 ΔE9 cells (open circles; n = 5). With PIP2, Ca2+ influx were not different between PS1 WT cells (filled squares; n = 6) and PS1 ΔE9 cells (open squares; n = 5). (D) The effect of 10 μM carrier-PIP2 complex on Ca2+ influx were compared between PS1 ΔE9 and PS1 WT. * represents p < 0.05 from paired t-tests.
DISCUSSION
In addition to functioning as the catalytic site of the APP processing γ-secretase, PSs are involved with diverse cell functions during the development as well as in the normal and diseased brain. In particular, PSs are intimately linked to cellular Ca2+ homeostasis by regulating CCE and Ca2+ signaling in the ER (Mattson 2001; LaFerla 2002; Yoo et al., 2000; Leissring et al., 2000; Akbari et al., 2004). The present study demonstrates that TRPM7 is a target ion channel that is regulated by PSs, since the abrogation of PSs greatly enhances Ca2+ influx through TRPM7 channels. We also show that TRPM7 activity is suppressed by FAD-associated PS1 mutations. These results suggest that a normal function of PS1 includes the regulation of Ca2+ permeability and FAD-associated mutations affect TRPM7-mediated Ca2+ influx.
The best studied function of PS on Ca2+ homeostasis is the modulation of ER Ca2+. PS1 or PS2 FAD mutations produce enhanced ER Ca2+ release, which may be due to the Ca2+ overload in this organelle (Leissring et al., 2000). APP intracellular domain (AICD) generated by γ-secretase was suggested as a mediator for the PS regulation of ER Ca2+ content, since a AICD protein complex may regulate the expression of genes required for Ca2+ metabolism (Leissring et al., 2002). For the regulation of Ca2+ influx by PSs, however, PS-associated proteolytic activity is not required, since we showed that pretreating the cells with a γ-secretase inhibitor did not influence Ca2+ influx in PS1 WT and PS1 ΔE9 cells. Thus, PSs regulates TRPM7 activity in a γ-secretase-independent mechanism.
Our studies also demonstrate that PS-dependent regulation of TRPM7 involves PIP2. One of FAD-linked PS mutants, PS1 ΔE9, showed decreased TRPM7-mediated Ca2+ influx. Western blotting analysis confirmed that the expression level of TRPM7 channel is not different between PS1 WT and PS1 ΔE9 cells. However, PIP2 enrichment restored the decreased resting Ca2+ permeability in PS1 ΔE9 in a dose-dependent manner. Thus, the present study identifies the involvement of PIP2 imbalance in impaired Ca2+ homeostasis. The mechanism by which PS1 mutations induce PIP2 imbalance is not yet known. Intriguingly, however, FAD-linked PS mutations have an elevated activity of phospholipase C, an enzyme that modulates PIP2 hydrolysis (Cedazo-Minguez et al., 2002; Cowburn et al., 2007). PIP2 is a minor component in the plasma membrane, but is an important signaling molecule, regulating a variety of ion channels and transporters (Suh and Hille 2005). Previously, we showed that the down-regulated PIP2 levels are observed in FAD mutant PS cells (Landman et al., 2006). In this study, we showed that TRPM7 mediated Ca2+ influx was recovered from PS1 ΔE9 cells when PIP2 was supplied. Taken together, these results indicate that reduced PIP2 level is the cause of reduced Ca2+ influx observed in PS1 ΔE9 cells, and that the TRPM7 channel is the underlying Ca2+ influx pathway. Based on our results, one possibility is that altered PIP2 metabolism may directly influence the activity of channel. As shown in our previous study (Landman et al., 2006), reduced availability of PIP2 in FAD mutant cells may directly lead to the suppression of Ca2+ through the TRPM7 channel. Conversely, elevated levels of PIP2 in PSKO cells leads to increased availability of the PIP2 pool that may result in enhanced Ca2+ entry. Since changes in PIP2 levels could give rise to other cellular changes, it is also conceivable that reduced or PIP2 levels in PS FAD cells and increased PIP2 PSKO cells, may influence Ca2+ entry via an indirect mechanism, such as cell surface targeting or endosomal trafficking of TRPM7, without affecting the levels of channel.
TRPM7 channel has been implicated in the control of cellular proliferation and viability by transporting various trace metal ions and by the regulation of Mg2+ and Ca2+ homeostasis (Nadler et al., 2001; Monteilh-Zoller et al., 2003; Schmitz et al., 2003). TRPM7 has also been implicated in embryonic development (Jin et al., 2008). The channel is widely expressed in various tissues including the brain, and is associated with anoxic neuronal death (Aarts et al., 2003). TRPM7 channels are activated by detecting lowered extracellular Ca2+ and Mg2+ levels in hippocampal neurons, providing a means by which this channel contributes to neuronal death during transient brain ischemia (Wei et al., 2007). Interestingly, a missense mutation in TRPM7 channel causes Guamanian amyotrophic lateral sclerosis and Parkinsonism dementia, probably due to an increased sensitivity to inhibition by intracellular Mg2+ (Hermosura et al., 2005). Thus, down-regulation of TRPM7 channel may confer a susceptibility to these neurodegenerative diseases in environments severely deficient in Ca2+ and Mg2+. Similarly, the deficit of TRPM7 currents and the resulting down-regulation of Ca2+ influx observed in FAD-linked PS mutants may contribute to neurodegenerative changes associated with AD. Consistent with this, changes in intracellular Ca2+ were suggested as a critical component of AD (Mattson 2003; Stutzmann 2007). On the other hand, the down-regulation of PIP2 level per se in FAD-linked PS mutations may be the direct cause of accumulation of Aβ levels in the brain. Consistent with this possibility, the lipid composition has been suggested as an important regulator for γ-secretase (Osenkowski et al., 2008; Osawa et al., 2008). Specifically, PIP2 potentially regulates γ-secretase activity by suppressing its association with the substrate to reduce the production of Aβ (Osawa et al., 2008). Alternatively, a recent report has shown that the oligomeric form of Aβ decreases the level of PIP2, resulting in synaptic dysfunction (Berman et al., 2008). Considering the growing evidence connecting lipid metabolism with AD pathogenesis (Di Paolo and Kim 2011), further research is required to determine the role for signaling lipids, such as PIP2, in PS-dependent regulation of TRPM7.
Acknowledgments
The authors thank L.B.J. McIntire for critical comments on the manuscript.
Footnotes
Contract grant sponsors: Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0072220) to S.C; US National Institute Heath grant NS074536 to T.-W. K.
References
- Akbari Y, Hitt BD, Murphy MP, Dagher NN, Tseng BP, Green KN, Golde TE, LaFerla FM. Presenilin regulates capacitative calcium entry dependently and independently of gamma-secretase activity. Biochem Biophys Res Commun. 2004;322:1145–1152. doi: 10.1016/j.bbrc.2004.07.136. [DOI] [PubMed] [Google Scholar]
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dall’Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci. 2008;11:547–554. doi: 10.1038/nn.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Popescu BO, Ankarcrona M, Nishimura T, Cowburn RF. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J Biol Chem. 2002;277:36646–36655. doi: 10.1074/jbc.M112117200. [DOI] [PubMed] [Google Scholar]
- Chun YS, Shin S, Kim Y, Cho H, Park MK, Kim T-W, Voronov S, Di Paolo G, Suh B-C, Chung S. Cholesterol modulates ion channels via downregulation of phosphatidylinositol 4,5-bisphosphate. J Neurochem. 2010;112:1286–1294. doi: 10.1111/j.1471-4159.2009.06545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowburn RF, Popescu BO, Ankarcrona M, Dehvari N, Cedazo-Minguez A. Presenilin-mediated signal transduction. Physiol Behav. 2007;92:93–97. doi: 10.1016/j.physbeh.2007.05.053. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, Sumimoto H, Ito Y, Mori Y, Inoue R. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J Pharmacol Sci. 2004;95:403–419. doi: 10.1254/jphs.fp0040273. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hermosura MC, Nayakanti H, Dorovkov MV, Calderon FR, Ryazanov AG, Haymer DS, Garruto RM. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci USA. 2005;102:11510–11515. doi: 10.1073/pnas.0505149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Branigan D, Xiong ZG. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J Biol Chem. 2010;185:7430–7439. doi: 10.1074/jbc.M109.040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Shin SY, Kim HS, Bae CD, Uhm DY, Park MK, Chung S. Regulation of magnesium-inhibited cation current by actin cytoskeleton rearrangement. Biochem Biophys Res Commun. 2006;339:810–815. doi: 10.1016/j.bbrc.2005.11.093. [DOI] [PubMed] [Google Scholar]
- Landman N, Serban G, Shin SY, Kang MS, Chung S, Kim TW. Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc Natl Acad Sci USA. 2006;103:19524–19529. doi: 10.1073/pnas.0604954103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Müller U, Saftig P, De Strooper B, Wolfe MS, Golde TE, LaFerla FM. A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002;99:4697–4702. doi: 10.1073/pnas.072033799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL, Camandola S. Presenilin mutations and calcium signaling defects in the nervous and immune systems. Bioessays. 2001;23:733–744. doi: 10.1002/bies.1103. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg. ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa S, Funamoto S, Nobuhara M, Wada-Kakuda S, Shimojo M, Yagishita S, Ihara Y. Phosphoinositides suppress gamma-secretase in both the detergent-soluble and -insoluble states. J Biol Chem. 2008;283:19283–19292. doi: 10.1074/jbc.M705954200. [DOI] [PubMed] [Google Scholar]
- Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki S, DeWald DB, Shope JC, Chen J, Prestwich GD. Intracellular delivery of phosphoinositides and inositol phosphates using polyamine carriers. Proc Natl Acad Sci USA. 2000;97:11286–11291. doi: 10.1073/pnas.210197897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penner R, Fleig A. The Mg2+ and Mg(2+)-nucleotide-regulated channel-kinase TRPM7. Handb Exp Pharmacol. 2007;179:313–328. doi: 10.1007/978-3-540-34891-7_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WL, Sun HS, Olah ME, Sun X, Czerwinska E, Czerwinski W, Mori Y, Orser BA, Xiong ZG, Jackson MF, Tymianski M, MacDonald JF. TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc Natl Acad Sci USA. 2007;104:16323–16328. doi: 10.1073/pnas.0701149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim T-W. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]