
Table 1. Refinement and final model statistics for dsHCAII.
Values in parentheses are for the highest resolution bin.
| PDB code | 4hba |
| Wavelength (Å) | 1.5418 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 42.3, b = 41.2, c = 71.6, β = 104.2 |
| Total No. of measured reflections | 96044 |
| Total No. of unique reflections | 23307 |
| Resolution (Å) | 20.0–1.77 (1.83–1.77) |
| R merge † (%) | 7.0 (50.0) |
| 〈I/σ(I)〉 | 13.5 (2.7) |
| Completeness (%) | 98.2 (98.9) |
| Multiplicity | 4.1 (4.2) |
| R cryst ‡ (%) | 15.0 |
| R free § (%) | 18.9 |
| No. of residues | 257 |
| No. of protein atoms (including alternate conformations) | 2301 |
| No. of water molecules | 203 |
| R.m.s.d. | |
| Bond lengths (Å) | 0.010 |
| Angles (°) | 1.297 |
| Ramachandran statistics (%) | |
| Allowed | 100.0 |
| Most favored | 96.8 |
| Outliers | 0.0 |
| Average B factors (Å2) | |
| All | 19.5 |
| Main chain | 18.7 |
| Side chain | 20.7 |
| Solvent | 29.2 |
| Cα r.m.s.d.¶ (Å) | 0.2 |
†
R
merge = 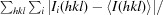
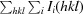 × 100.
× 100.
‡
R
cryst = 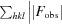 −
− 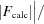
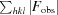 × 100.
× 100.
§
R free is calculated in the same way as R cryst except that it is calculated for data that were omitted from refinement (5% of reflections).
¶
Root-mean-square deviation of the Cα backbone compared with HCA II (PDB entry 3ks3).