

Abstract
The nearly neutral theory, which proposes that most mutations are deleterious or close to neutral, predicts that the ratio of nonsynonymous over synonymous substitution rates (dN/dS), and potentially also the ratio of radical over conservative amino acid replacement rates (Kr/Kc), are negatively correlated with effective population size. Previous empirical tests, using life-history traits (LHT) such as body-size or generation-time as proxies for population size, have been consistent with these predictions. This suggests that large-scale phylogenetic reconstructions of dN/dS or Kr/Kc might reveal interesting macroevolutionary patterns in the variation in effective population size among lineages. In this work, we further develop an integrative probabilistic framework for phylogenetic covariance analysis introduced previously, so as to estimate the correlation patterns between dN/dS, Kr/Kc, and three LHT, in mitochondrial genomes of birds and mammals. Kr/Kc displays stronger and more stable correlations with LHT than does dN/dS, which we interpret as a greater robustness of Kr/Kc, compared with dN/dS, the latter being confounded by the high saturation of the synonymous substitution rate in mitochondrial genomes. The correlation of Kr/Kc with LHT was robust when controlling for the potentially confounding effects of nucleotide compositional variation between taxa. The positive correlation of the mitochondrial Kr/Kc with LHT is compatible with previous reports, and with a nearly neutral interpretation, although alternative explanations are also possible. The Kr/Kc model was finally used for reconstructing life-history evolution in birds and mammals. This analysis suggests a fairly large-bodied ancestor in both groups. In birds, life-history evolution seems to have occurred mainly through size reduction in Neoavian birds, whereas in placental mammals, body mass evolution shows disparate trends across subclades. Altogether, our work represents a further step toward a more comprehensive phylogenetic reconstruction of the evolution of life-history and of the population-genetics environment.
Keywords: effective population size, life-history evolution, Markov chain Monte Carlo, Bayesian statistics, mitochondrial genome, nearly neutral model
Introduction
Effective population size is a fundamental parameter of population genetics, shaping the structure of polymorphism within populations, and influencing patterns of divergence. Within populations, effective population size determines the force of genetic drift, and therefore influences the level of genetic diversity (Kimura 1983). In the long time scale, population size influences the probability of fixation of nonneutral mutations with small fitness effects (Ohta 1992). In particular, slightly deleterious mutations may behave as effectively neutral in small populations and, in contrast, be efficiently eliminated in large populations. This differential fixation probability could lead to variation in the substitution rate as a function of long-term effective population size (Ne; Ohta 1992).
Historically, the class of slightly deleterious mutations was introduced in the context of the nearly neutral theory (Ohta 1973), itself proposed as an extension of the strict neutral theory (Kimura 1968) including mutations with small fitness effects (Ohta 1992). The nearly neutral theory was originally invoked to explain why the correlation between substitution rate and generation time was more pronounced for synonymous substitutions than for nonsynonymous substitutions. The rationale was that an inverse relationship between population size and generation time could lead to a mutual compensation between the decrease in the mutation rate, and the increase in the fixation probability of slightly deleterious nonsynonymous mutations, in species with long generation-times but small effective population sizes (Chao and Carr 1993; Ohta 1993). The presence of slightly deleterious mutations was subsequently identified as the likely reason for the excess of nonsynonymous polymorphism, compared with divergence, in mitochondrial protein-coding genes (Nachman 1998; Rand and Kann 1998; Gerber et al. 2001). Similarly, by comparing levels of polymorphism and divergence between species, Eyre-Walker et al. (2002) and Keightley et al. (2005) suggested that Hominidae may have experienced reduced selective constraints on their protein-coding genes compared with Muroidea. These results were interpreted in the light of the nearly neutral theory, based on the fact that hominids are believed to have a smaller long-term effective population size (Ne) than murids (Eyre-Walker et al. 2002; Keightley et al. 2005).
At larger evolutionary scale, several studies have attempted to explain between-species differences in the ratio of nonsynonymous (dN) over synonymous (dS) substitution rates by variation in population size. In particular, Johnson and Seger (2001) and Woolfit and Bromham (2005) have found that island birds species have, on average, a higher dN/dS ratio than their mainland relatives. Woolfit and Bromham (2003) reported an increase in the substitution rate in the 16S rRNA gene of endosymbiotic organisms, compared with free living species. Domesticated populations, which probably experienced a severe bottleneck during the domestication process, frequently show an increased dN/dS compared with wild progenitor species (Björnerfeldt et al. 2006; Wang et al. 2011). Finally, Popadin et al. (2007), Nikolaev et al. (2007), Lartillot and Delsuc (2012), and Romiguier et al. (2013) have found a positive correlation between body mass (or age at sexual maturity) and the dN/dS ratio in nuclear and mitochondrial protein-coding genes of placental mammals. Relying on the well-known negative relationship between body mass and population density in mammals (Damuth 1987; White et al. 2007), they interpreted this correlation in terms of a difference in long-term effective population size between small and large-bodied mammals and, therefore, as empirical evidence in support of the nearly neutral theory. These results are particularly interesting because they suggest that long-term trends in life-history evolution across mammalian orders result in correlated changes in selection efficacy between groups and leave a distinctive trace at large evolutionary scale.
Currently, however, this result is yet to be replicated in another group of animals. Although less obvious, a relationship between population density and body mass also exists in birds (e.g., Nee et al. 1991) and, therefore, a relationship between life-history traits (LHT) and the efficacy of selection is also expected in this group. To date, evidence that the efficacy of selection could vary between birds is equivocal. Analyzing approximately 8,400 protein-coding genes, Nam et al. (2010) have failed to identify any difference in the average dN/dS estimated in the chicken (Gallus gallus) lineage (which leads to the relatively large birds of the Galloanserae clade), compared with the zebra finch (Taeniopygia guttata) lineage (leading to the relatively small birds of the Passeriformes clade). Similarly, there is some debate as to whether island bird populations are more prone to accumulating slightly deleterious substitutions, compared with their mainland relatives (Johnson and Seger 2001; Woolfit and Bromham 2005; Wright et al. 2009).
The correlation between LHT and dN/dS also suggests that large-scale phylogenetic reconstruction of dN/dS might reveal long-term trends in effective population size, which in turn could be interpreted in terms of macroevolutionary patterns in life-history evolution across large phylogenetic groups. Currently, methods for reconstructing life-history or quantitative trait evolution along phylogenies (e.g., Venditti et al. 2011) do not directly use the substitution process. Yet, the correlation between substitution patterns and LHT suggests that potentially useful information about ancestral LHT is contained in molecular sequences (Lanfear et al. 2010) and can be integrated into methods for quantitative trait reconstruction (Lartillot and Delsuc 2012).
In practice, however, such integrated reconstruction methods require accurate estimation of ancestral substitution rates, both synonymous and nonsynonymous. Yet, this becomes increasingly difficult with increasing phylogenetic divergence. The accumulation of multiple substitutions at the same site over time, a process called mutational saturation, can easily erase the phylogenetic signal (Springer et al. 2001; Galewski et al. 2006) and lead to an underestimation of substitution rates. As synonymous substitutions accumulate more rapidly than nonsynonymous substitutions, this will lead to a differential saturation of dS and dN, thus potentially resulting in biased estimates of their ratio, particularly along branches corresponding to long periods of time, ancient evolutionary lineages or fast evolving species. This problem is especially pronounced in the case of the fast evolving mitochondrial genomes (Ballard and Whitlock 2004; Galtier et al. 2006; Nabholz et al. 2008). On the other hand, using mitochondrial genomes might be interesting, because, as opposed to nuclear genomic sequences, the animals mitochondrial genome is largely free from recombination (Elson and Lightowlers 2006; Galtier, Nabholz, et al. 2009). In nuclear genomes, recombination induces biased-gene conversion phenomena (Duret and Galtier 2009), which can potentially impact dN/dS (Berglund et al. 2009; Galtier, Duret, et al. 2009) and therefore potentially confound the relation between the nuclear dN/dS and effective population size (Lartillot 2013). Mitochondrial genomes, in contrast, because they are nonrecombining, should be immune from biased gene conversion. In certain regimes (in particular, under recurrent positive selection and large effective population sizes), lack of recombination may result in Hill–Robertson interference disturbing the simple relation between the substitution process and effective population size predicted by the nearly neutral theory (Bazin et al. 2006). However, mitochondrial genomes of mammals and birds appear to be in a globally nearly neutral regime (Mulligan et al. 2006; Nabholz, Mauffrey, et al. 2008, 2009) and should therefore more faithfully reflect variation in selection efficacy and intensity among lineages than nuclear protein-coding genes.
To overcome the problems caused by mutational saturation, one possibility would be to rely entirely on the less saturated amino acid substitutions. Amino acids substitutions can be classified as either radical or conservative, depending of the degree of similarity of the physicochemical properties of the initial and final amino acids (Zhang 2000; Sainudiin et al. 2005). Assuming that radical substitutions are more prone to be deleterious than conservative substitutions, the ratio between radical substitution rate (Kr) and conservative substitution rate (Kc) could be used in the same way as the dN/dS (Zhang 2000). Although the response of Kr/Kc to changes in effective population size may in principle depend on the exact shape of the distribution of selective effects across conservative and radical mutations, about which not much is currently known, previous analyses have nevertheless suggested that species with smaller effective population sizes are characterized by larger values of Kr/Kc (Eyre-Walker et al. 2002). Likewise, in mammals, the Kr/Kc ratio correlates positively with body mass or age at sexual maturity (Nikolaev et al. 2007; Popadin et al. 2007), a result that has been interpreted as a nearly neutral effect.
In this study, we pursue three goals: 1) We incorporated a model based on amino acid substitutions in the framework recently developed by Lartillot and Poujol (2011) so as to evaluate the correlation between variation in Kr/Kc and variation in quantitative traits along phylogenetic trees; 2) we reanalyzed the correlation between Kr/Kc and LHT in placental mammals, using a dense taxon sampling and the complete mitochondrial proteome, and explored the same question in birds, so as to assess the degree of generality of the correlation patterns of Kr/Kc with LHT across the two groups. Finally, 3) we relied on the correlation between LHT and amino acid substitution patterns to perform a phylogenetic reconstruction of LHT evolution, thereby offering a synthetic picture of what an integrated comparative analysis of complete mitochondrial genomes can say about life-history evolution in birds and placental mammals.
Materials and Methods
The Amino Acid Model
The framework introduced in Lartillot and Poujol (2011) is based on a codon model specified according to the formalism of Muse and Gaut (1994). Specifically, the rate of substitution between codons c1 and c2, differing at only one position, and with respective nucleotides n1 and n2 at that position, is given by the following equation:
| (1) |
where R is a 4 × 4, normalized, general time-reversible nucleotide substitution process. Substitution rates between nonnearest neighbor codons are equal to 0. This model is called MGdNdS in the following. The parameter λ represents the synonymous substitution rate, whereas ω = dN/dS is the ratio of nonsynonymous over synonymous substitution rates. The R matrix is assumed to be constant in time, while λ and ω vary among lineages in correlation with LHT (here, body size, female maturity, and maximum longevity) and, in some cases, other quantitative traits describing nucleotide compositional variation between taxa (discussed later). Note that, although the codon model is formally time-reversible for any fixed value of ω, the overall substitution process is not time-reversible, due to the variation in ω through time. In the general case, a total of L quantitative traits are considered, jointly with K = 2 substitution parameters (λ and ω), summing up to a total of M = K + L parameters, assumed to follow a M-dimensional log-normal Brownian process running along the lineages of the phylogeny and parameterized by a M × M covariance matrix Σ. Using a log-normal Brownian model amounts to assuming linear correlations between variations in the logarithm of the quantitative traits and the substitution parameters. This is in turn equivalent to making the hypothesis that all LHT and substitution parameters scale allometrically with body size. The parameters of the model (the matrix R, the reconstruction of dS, dN/dS, and quantitative traits along the phylogeny, the covariance matrix Σ and divergence times) are jointly estimated using Bayesian MCMC methods (see Lartillot and Poujol 2011 for more details).
Based on this formalism, we now develop a model, which we call RadConsAA, directly specified at the amino acid level and capturing the variation among lineages in the ratio Kr/Kc of radical over conservative amino acid replacement rates. Following an analogy with equation (1), instead of a codon model, we propose a 20 × 20 amino acid replacement process, such that the rate of substitution between amino acids a and b is given as follows:
| (2) |
where R is now a 20 × 20, normalized, general-time reversible amino acid replacement matrix parameterized as follows: 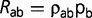 , where
, where 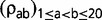 is a set of 190 relative exchangeabilities, and
is a set of 190 relative exchangeabilities, and 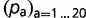 are the 20 equilibrium frequencies of the amino acid replacement process. Both ρ and π are free parameters of the model. The parameter λ is now the conservative substitution rate, and η is proportional to the ratio of conservative over radical amino acid replacement rates (Kr/Kc). As in the MGdNdS model, we let λ and η vary among lineages, jointly with L other quantitative traits, whereas ρ and π are assumed to be constant across the entire phylogeny. Note that, because the relative exchangeabilities are left unconstrained and are empirically estimated, unless additional constraints are specified, the parameters of the model are not identifiable. Specifically, defining, for any positive constant A, η' = ηA,
are the 20 equilibrium frequencies of the amino acid replacement process. Both ρ and π are free parameters of the model. The parameter λ is now the conservative substitution rate, and η is proportional to the ratio of conservative over radical amino acid replacement rates (Kr/Kc). As in the MGdNdS model, we let λ and η vary among lineages, jointly with L other quantitative traits, whereas ρ and π are assumed to be constant across the entire phylogeny. Note that, because the relative exchangeabilities are left unconstrained and are empirically estimated, unless additional constraints are specified, the parameters of the model are not identifiable. Specifically, defining, for any positive constant A, η' = ηA, 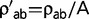 , for all radical pairs of amino acids (a,b) and
, for all radical pairs of amino acids (a,b) and 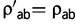 for all conservative pairs will result in the same substitution process Q. To restore identifiability, we impose the arbitrary convention that η = 1 at the root of the tree. Therefore, as a measure of Kr/Kc, η is defined only up to a proportionality constant. In a second step, reconstructions of Kr/Kc along the tree are scaled by displaying Kr/Kc =
for all conservative pairs will result in the same substitution process Q. To restore identifiability, we impose the arbitrary convention that η = 1 at the root of the tree. Therefore, as a measure of Kr/Kc, η is defined only up to a proportionality constant. In a second step, reconstructions of Kr/Kc along the tree are scaled by displaying Kr/Kc = 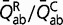 = η
= η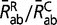 , where
, where 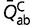 and
and 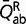 are the mean rates (such as defined by the entries of the 20 × 20 Q matrix) over conservative and radical amino acid pairs, respectively, and
are the mean rates (such as defined by the entries of the 20 × 20 Q matrix) over conservative and radical amino acid pairs, respectively, and 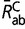 and
and 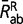 are similarly defined based on the R matrix. This rescaling is done separately for each parameter configuration obtained by MCMC, before averaging over the sample approximately from the posterior distribution. With this rescaling, the displayed reconstructions of Kr/Kc effectively represent the instant mean ratio of relative rates between radical and conservative amino acid pairs at any point of time along the phylogeny.
are similarly defined based on the R matrix. This rescaling is done separately for each parameter configuration obtained by MCMC, before averaging over the sample approximately from the posterior distribution. With this rescaling, the displayed reconstructions of Kr/Kc effectively represent the instant mean ratio of relative rates between radical and conservative amino acid pairs at any point of time along the phylogeny.
To classify amino acid substitutions as radical or conservative, we follow Sainudiin et al. (2005). First, amino acids are partitioned according to three criteria: volume (large amino acids: L, I, F, M, Y, W, H, K, R, E, and Q), polarity (polar amino acids: Y, W, H, K, R, E, Q, T, D, N, S, and C), or charge (positively charged amino acid: H, K, R; negatively charged amino acids: D, E and neutrally charged amino acids: A, N, C, Q, G, I, L, M, F, P, S, T, W, Y, and V). Next, any combination of criteria defines a model in which substitutions that do not conserve all of these criteria are considered as radical. In what follows, we consider four distinct models, corresponding to charge alone (RadConsAA-Chg), volume alone (RadConsAA-Vol) or polarity alone (RadConsAA-Pol), as well as the combination of volume and polarity (RadConsAA-PolVol).
To control for nucleotide compositional variation in the context of a RadConsAA analysis, we computed the empirical frequencies of A, C, G, and T at the third coding positions of the alignment (denoted as πA, πc, πG, and πT, respectively). As most compositional variation seen in mammalian mitochondrial genomes is due to variation in πc and πT (Gibson et al. 2005), we considered these two log-transformed variables as additional quantitative traits, to be analyzed along with LHT (i.e., as two additional quantitative traits in the model). Then, multiple regression (as described later) allows estimation of the correlation between η and the three life-history variables, while controlling for compositional variation in C and T. Similarly, we also controlled for GC bias, GC skew, and AT skew, where GC bias is defined as (πG + πC)/(1 − [πG + πC]); GC skew as exp([πG − πC]/[πG + πC]) and AT skew as exp([πA − πT]/[πA + πT]). Finally, we also performed the analyses excluding NADH dehydrogenase 6 (ND6). In birds and mammals, ND6 is the only protein-coding gene located on the cytosine-rich light strand of the mitochondrial genome and therefore has a different nucleotide composition, compared with other mitochondrial protein-coding genes.
We used the following priors: a uniform prior on divergence times, with fossil calibrations (hard constraints), a fixed topology (see “Phylogenetic trees” section) and an inverse-Wishart prior on the covariance matrix, of parameter Σ0 = Diag (κ1 … κM), where M is the dimension of the Brownian process. For each m = 1 … M, the prior on κm is a truncated log-uniform prior between 10−3 and 103 (equivalently, each κm has a density proportional to 1/ κm over [10−3, 103]). Truncation ensures normalization of the posterior density. A uniform Dirichlet distribution is used for the priors on the relative exchange rates and the equilibrium frequencies of the general time-reversible matrix R. Concerning the prior on the value of the Brownian process at the root of the tree, we use a uniform prior between −100 and 100 for the entries corresponding to the logarithm of the substitution parameters λ and ω, and a normal distribution of mean and variance determined empirically for the LHT in mammals (mean and standard deviation of 8.4 for the logarithm of the body mass in grams, of 5.8 for the logarithm of the age at sexual maturity in days, and 5.2 for the logarithm of the longevity in months) and in birds (mean and standard deviation of 4.6 for the logarithm of the body mass, 2.3 for the logarithm of the maturity and 5.9 for the logarithm of the longevity). These values were obtained using the mean of the logarithm of the current life-histories traits (discussed later). Finally, we used the linear approximation for branch-specific averages of λ, ω, and η (as defined in Lartillot and Poujol 2011).
Monte Carlo
Under all settings, and for all data sets, MCMC runs were duplicated. Convergence was first checked visually. The results obtained from the two independent runs were very similar in all cases, except for the large placental data set (with 273 taxa), for which one run was stuck in a local optimum. Exceptionally, we ran two additional chains for this particular case. Convergence was then more quantitatively assessed using the tracecomp program in the PhyloBayes package (Lartillot et al. 2009), for estimating the effective sample size and the overlap between two independent runs, based on the following summary statistics: the logarithm of the likelihood and of the prior density, the mean substitution rate over the tree, the mean dN/dS or mean Kr/Kc over the tree, the entries of the covariance matrix and the age of the root.
Covariance Analysis
Covariance analyses were conducted as in Lartillot and Poujol (2011), reporting mean posterior correlation coefficients, and assessing the strength of the statistical support by estimating the posterior probability (PP) of a positive or a negative correlation between the two traits of interest. Specifically, the marginal correlation coefficient between two entries k and l of the multivariate process (which can correspond to either substitution parameters or quantitative traits) is defined as follows:
| (3) |
where Σ is the covariance matrix. Equation (3) is averaged over the sample obtained by MCMC, yielding the point estimate of the correlation coefficient. Statistical support is indicated by always reporting the estimated PP of a positive correlation. Therefore, a PP close to 1 indicates a high PP of a positive correlation, whereas a PP close to 0 indicates, by symmetry, a high PP of a negative correlation. By convention, a correlation with a PP greater than 0.975 or smaller than 0.025 is deemed supported, whereas a PP greater than 0.95 or smaller than 0.05 is called marginally supported. Note that this is not formally equivalent to a classical Bayesian point-hypothesis testing procedure. Testing the point hypothesis of no correlation would be done by associating a positive prior probability mass with the parameter configuration corresponding to a correlation coefficient exactly equal to 0, and then calculating the PP of this configuration. On the other hand, the present test is able to detect situations in which there is a strong support for a positive or negative correlation between any pairs of variables (Lartillot and Poujol 2011).
The correlation coefficient defined by equation (3) corresponds to the marginal correlation between the two variables k and l. As such, it may include any indirect correlation of the two variables with other entries of the multivariate process. Unless otherwise specified, multiple regression was done as in Lartillot and Delsuc (2012), whereby correlations are systematically controlled for all other variables. This is done by computing the precision matrix Ω (which is the inverse of the covariance matrix Σ). The partial correlation coefficient between variables k and l is then defined as follows:
| (4) |
In the particular case of base composition, we estimated the correlation between variables controlling only for base composition using the method introduced by Lartillot and Poujol (2011):
| (5) |
where k and l are the variables of interest and b is the variable (related to base composition) to be controlled for. When several such variables need to be controlled for, equation (5) is applied recursively. The Kr/Kc models introduced here have been implemented in the Coevol software program, available from www.phylobayes.org.
Sequence Data, Multiple Sequence Alignments
In this study, we analyzed two mitochondrial data sets composed of the 13 protein-coding genes representing 11,442 bp for 92 bird species and 11,529 bp for 273 placental mammals. The bird data set includes the 80 species of Neoaves and Galloanserae used in Pacheco et al. (2011), combined with eight species of Ratites and Tinamiformes (Härlid et al. 1998; Haddrath and Baker 2001) and four near-complete passerines mitochondrial genomes (Nabholz et al. 2010). The placental data set included all the placentals species for which the complete mitochondrial genomes was available as of September 2011 in NCBI/GenBank database (http://www.ncbi.nlm.nih.gov/genbank/). To perform the time consuming MGdNdS analysis, this data set was reduced to 194 species by randomly selecting one species per genus.
Sequences were downloaded from the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov/, last accessed September 2011). For each protein-coding gene, amino acid sequences were aligned using MUSCLE (Edgar 2004). The alignments were checked by eye and manually corrected if needed, and then used as a template for aligning nucleotide sequences while respecting the underlying coding structure. All the species names, taxonomic information, gene accession numbers, and the alignments are available on Dryad (http://datadryad.org, doi:10.5061/dryad.72594).
Phylogenetic Trees
For placental mammals, we followed Murphy et al. (2007) and grouped Afrotheria and Xenarthra in the so-called Atlantogenata, as the monophyletic sister group of Boreoeutheria (Laurasiatheria and Euarchontoglires; Madsen et al. 2001; Murphy et al. 2001). Within these clades, we respected the topology proposed by Delsuc et al. (2002) for Xenarthra; Dubey et al. (2007), Flynn et al. (2005), Teeling et al. (2005), and Springer et al. (2004) for Laurasiatheria; and Blanga-Kanfi et al. (2009), Fabre et al. (2009), Huchon et al. (2002), and Perelman et al. (2011) for Euarchontoglires.
In the case of birds, some uncertainties remain regarding the relationship at the base of Neoavian clade (i.e., all the extant birds excluding Galloanserae and Palaeognathae, see e.g., Hackett et al. 2008; Pacheco et al. 2011; Pratt et al. 2009). Here, we chose to apply a pragmatic approach by testing two alternative topologies: first, a phylogeny was estimated using a maximum likelihood method (RAxML with GTR + G substitution model; version 7.0.4, Stamatakis 2006). Second, we used the topology of Hackett et al. (2008), which represents the most up-to-date phylogeny obtained with nuclear markers (this topology is presented in supplementary fig. S1, Supplementary Material online). The results obtained with the two topologies being very similar, we only present the results obtain with the maximum likelihood topology, except if stated otherwise.
Fossil Calibrations
In the case of birds, following Benton et al. (2009), we constrained the Paleognathae/Neognathae divergence to lie between 66 and 86.5 Ma and the crown Passeriformes (Acanthisitta/other Passeriformes) to be younger than 65 Myr. The oldest known penguins (Sphenisciformes) were estimated at 60–61 Myr (Slack et al. 2006), which allows us to constrain the split between Sphenisciformes (penguins) and Procellariiformes (the “tube-nosed” seabirds: albatrosses and petrels) to be older than 62 Myr.
For placentals, we used 13 fossil calibrations, all obtained from Benton et al. (2009), including calibrations of the crown Hominoidea, Hylobatidae (used only for the mitochondrial data set), Catarrhini, Primates, Lagomorpha, Glires, Rodentia, Rodentia (minus sciurids), Muridae, Carnivora, Laurasiatheria, Laurasiatheria (minus Lipotyphla), and Boreoeutheria (see corresponding paragraphs in Benton et al. 2009 for details). All the calibrations are presented in supplementary table S1, Supplementary Material online.
Life-History Traits
Body mass for all the bird species analyzed here were obtained from Dunning (2007). Maximum recorded lifespan (a proxy for longevity) and female age at sexual maturity were extracted from the AnAge database (de Magalhães and Costa 2009). In the case of birds, where longevity and age at sexual maturity were recorded, we used the mean traits at the genus level. In the case of placentals, data for the three LHT were obtained from the PANTheria database (Jones et al. 2009). For birds, body mass was available for all the 92 species whereas maximum longevity and age at sexual maturity were available for 60 and 44 species, respectively. For placental mammals, body mass was available for 230 species, maximum longevity for 189 and age at sexual maturity for 183 species. The distribution of these traits is shown in supplementary figure S2, Supplementary Material online.
Results
Using a previously developed phylogenetic covariance model (Lartillot and Poujol 2011), and extending so as to estimate variation in the ratio of radical over conservative amino acid replacement rates (Kr/Kc), we analyzed the relationship between dN/dS, Kr/Kc, and three LHT (body mass, female age at sexual maturity, and maximum recorded lifespan, as a proxy for longevity) in the mitochondrial proteome (13 protein-coding genes) of 92 birds and 273 placental mammals.
The Relationship between dN/dS and LHT
In placentals, we found a positive correlation between mitochondrial dN/dS and the three LHT (PP of a positive correlation > 0.96, table 1). This is well in line with previous studies using partially overlapping data sets (Popadin et al. 2007; Lartillot and Poujol 2011). Statistical support is lost for all LHT in partial correlations (i.e., controlling for dS and the other two LHT) although the effect of longevity remains moderately strong (PP = 0.94; table 1).
Table 1.
Covariance between dN/dS and dS and LHT Using the Nucleotide Data Sets
| Data Set | Body Mass | Longevity | Age at Sexual Maturity |
|---|---|---|---|
| Marginal correlationa | |||
| Placentalia194 | 0.47 (>0.99)** | 0.51 (>0.99)** | 0.38 (0.98)** |
| Birds | −0.30 (0.07) | −0.63 (<0.01)** | −0.18 (0.28) |
| Partial correlationb | |||
| Placentalia 194 | 0.12 (0.83) | 0.22 (0.94) | −0.08 (0.30) |
| Birds | 0.04 (0.56) | −0.60 (0.01)** | 0.17 (0.63) |
aCorrelation coefficients corresponding to the marginal correlation between each pair of variables.
bCorrelation coefficients corresponding to the partial correlations.
**PP < 0.025 or >0.975.
Unexpectedly, an opposite result was obtained in birds. In this group, the mitochondrial dN/dS displays a negative correlation with LHT. The correlation is supported in the case of longevity (PP = 0.01; table 1) and remains so in partial correlation (i.e., controlling for dS, body mass and maturity; PP = 0.01; table 1).
The Relationship between Mitochondrial Kr/Kc and LHT
The high mutation rate experienced by the mitochondrial genome (Ballard and Whitlock 2004; Galtier et al. 2006; Nabholz, Glémin, et al. 2008) may prevent accurate estimation of dN/dS. In our data set, there is a marked saturation visible when uncorrected pairwise divergence is plotted against phylogenetic distance (fig. 1). As is clear from figure 1, saturation is more pronounced on synonymous than on nonsynonymous substitutions.
Fig. 1.—

Pairwise uncorrected divergence against phylogenetic distance estimated on the bird (A) and on the placental (B) data sets. On both panels, gray dots correspond to measures based on amino acids and black triangles to measures based on nucleotides. Phylogenetic distances are estimated by maximum likelihood using CODEML (Yang 2007; mtmam.dat substitution matrix) for the amino acid data, and BASEML (GTR+GAMMA substitution model) for nucleotides.
Differential saturation between dS and dN is likely to result in important distortions in the estimated dN/dS, raising doubts about the reliability of the correlation patterns displayed by dN/dS (table 1). Distortions caused by differential saturation may also explain some of the unexpected correlations observed between dN/dS and LHT, in particular in birds. Specifically, although smaller birds are expected to have smaller effective population sizes and, therefore, lower dN/dS ratios, in the presence of saturation, their higher overall rate of substitution (Nabholz et al. 2009) will only weakly impact dS but more strongly impact dN. As a result, the apparent dN/dS will increase, whereas in fact the real dN/dS is undergoing a decrease. This effect is also predicted in the case of mammals, although in that case, the better taxonomic sampling, combined with a wider overall range of variation in body size, may be sufficient for ensuring a robust positive correlation between dN/dS and body mass.
Measures of the strength of selection exclusively relying on the less saturated nonsynonymous substitutions may be more reliable than dN/dS in the context of mitochondrial genome evolution. With this in mind, we constructed several models based on the ratio of radical over conservative amino acid substitution rates. Biochemical conservativeness can be defined in several different ways, depending on whether amino acid replacements resulting in a change in charge, volume, or polarity were considered as radical. Accordingly, we defined four different versions of the model taking into account the three different changes mentioned above plus the combination of polarity and volume (see Materials and Methods).
In placentals, whichever definition of biochemical conservativeness is used, in all cases, Kr/Kc correlates positively with LHT, thus mirroring the correlation patterns observed for dN/dS (Popadin et al. 2007). Among the four models, the one considering a change in polarity or in volume as a radical change (RadConsAA-PolVol) gives a consistently stronger support for a positive correlation between LHT and Kr/Kc (table 2). Using this model, the correlation with LHT is stronger than using dN/dS (table 3). In contrast, the model using charge as the criterion for discriminating radical and conservative amino acid substitutions (RadConsAA-Chg) results in a weaker covariance, with a PP above 0.95 obtained only for longevity (table 2). Using a different method, Popadin et al. (2007) also found that a definition of radical substitutions based on charge results in a weaker correlation between body-mass and Kr/Kc, compared with volume or polarity. In contrast to our study, however, Popadin et al. (2007) reported that the model based on polarity or on volume alone were slightly better than the model based on polarity and volume in combination. In partial correlation analyses, Kr/Kc is found to correlate primarily with age at sexual maturity for the complete data set, and with longevity for the reduced data set (with 194 species, table 3).
Table 2.
Marginal Covariance between Kr/Kc and LHT According to Alternative Definitions of Biochemical Conservativeness
| Data Set | Classification | Body Mass | Longevity | Age at Sexual Maturity |
|---|---|---|---|---|
| Mammals (All) | Charge | 0.19 (0.91) | 0.43 (0.99)** | 0.17 (0.83) |
| Polarity | 0.29 (>0.99)** | 0.23 (0.94) | 0.49 (>0.99)** | |
| Volume | 0.30 (>0.99)** | 0.25 (0.95)* | 0.47 (>0.99)** | |
| Polarity and volume | 0.37 (>0.99)** | 0.43 (>0.99)** | 0.64 (>0.99)** | |
| Birds | Charge | −0.05 (0.42) | −0.28 (0.24) | −0.07 (0.41) |
| Polarity | 0.24 (0.90) | 0.06 (0.62) | −0.03 (0.44) | |
| Volume | 0.26 (0.80) | 0.33 (0.82) | 0.18 (0.69) | |
| Polarity and Volume | 0.37 (0.98)** | 0.23 (0.84) | 0.09 (0.59) | |
*PP > 0.95 or <0.05.
**PP <0.025 or >0.975.
Table 3.
Covariance between Kr/Kc, Kc, and LHT
| Data Set | Body Mass | Longevity | Age at Sexual Maturity |
|---|---|---|---|
| Marginal correlationa | |||
| Placentalia (All) | 0.37 (>0.99)** | 0.43 (>0.99)** | 0.64 (>0.99)** |
| Placentalia (N = 194) | 0.46 (>0.99)** | 0.60 (>0.99)** | 0.54 (>0.99)** |
| Birds | 0.37 (0.98)** | 0.23 (0.84) | 0.09 (0.59) |
| Partial correlationb | |||
| Placentalia (All) | −0.03 (0.39) | 0.21 (0.87) | 0.42 (>0.99)** |
| Placentalia (N = 194) | 0.04 (0.57) | 0.37 (0.97)* | 0.22 (0.88) |
| Birds | 0.30 (0.92) | 0.03 (0.53) | −0.12 (0.36) |
Note.—Change in polarity and volume are considered as radical substitutions.
aCorrelation coefficients corresponding to the marginal correlation between each pair of variables.
bCorrelation coefficients corresponding to the partial correlations.
*PP >0.95 or <0.05.
**PP < 0.025 or >0.975.
Interestingly, the results obtained on birds are at odds with the dN/dS analyses. Apart from the model based on charge, for which Kr/Kc displays negative but not supported correlations with LHT, all other models indicate a positive correlation between LHT and Kr/Kc (table 2). Under the model based on polarity and volume, support for a positive marginal correlation is found between body mass and Kr/Kc (PP = 0.98, table 2). The marginal correlation with the other LHT is not significant, which may seem surprising given that all LHT are strongly correlated with each other. However, this could be explained by the fact that a substantial fraction of values is missing for these two LHT (35% and 48% of the entries for longevity and maturity, respectively). The effect of body mass is lost in partial correlations (table 3). Similar results were obtained using the alternative topology for the bird phylogeny (PP = 0.99, supplementary table S2, Supplementary Material online).
The Influence of Base Composition and Mutation Bias
Mitochondrial nucleotide composition, especially in Cytosine (C) and in Thymine (T), varies substantially between mammalian species (Gibson et al. 2005). In birds, we observed a similar pattern, with the frequency of C (πC) being highly variable between species (ranging from 0.32 to 0.51), whereas A and G show moderate variation (from 0.29 to 0.43 and from 0.03 to 0.10, respectively). As in the case of mammals, in birds, the frequency of C (πC) appears strongly negatively correlated with the frequency of T (πT), suggesting that the joint variation in the two frequencies, in C and in T, is primarily the consequence of an underlying variation in the relative rate of cytosine deamination (Reyes et al. 1998).
Nucleotide compositional variation between species could potentially affect our estimation of dN/dS and Kr/Kc. Variation in nucleotide composition may differentially influence synonymous and nonsynonymous rates, owing to the fact that the nucleotide content is more variable on synonymous than on nonsynonymous sites. For Kr/Kc, the potential impact of compositional variation is less obvious, but could occur in several subtle ways. For instance, codons for polar amino acids have a tendency to be GC-poor compared with codons encoding nonpolar amino acids, and thus, a shift in nucleotide composition could potentially result in a higher rate of radical amino acid changes. In this context, the existence of a correlation between nucleotide composition and LHT, such as longevity (Jobson et al. 2010), raises the possibility that nucleotide compositional variation may be responsible for at least part of the correlations observed between LHT and dN/dS or Kr/Kc (tables 1 and 2), thus calling for a more detailed investigation of the impact of nucleotide composition on our estimation.
To do so, for the analyses using the MGdNdS model, we allow the equilibrium GC (GC*) of the codon substitution model to vary continuously along the tree (see Materials and Methods and Lartillot 2013). Variation in GC* among lineages is then treated like variation in dS and dN/dS, such that the overall covariance analysis includes a total of three substitution parameters and three LHT. Multiple regression then allows controlling for GC* when estimating the correlation between dN/dS and LHT. More specifically, we controlled for both the equilibrium GC composition at all codon position (GC*) and the equilibrium GC at the third-codon position (GC3*), the latter being markedly more variable than the former (Gibson et al. 2005). Both models leads to very similar results; therefore, we present only the GC3* results hereafter.
The GC3* appears positively correlated with dN/dS in birds (R = 0.57, PP > 0.99) and in placental mammals (R = 0.22, PP > 0.99). In placental mammals, GC3* is positively correlated with all LHT (PP > 0.99). In birds, GC3* correlates negatively with maximal longevity (R = −0.36, PP = 0.03), but not with body mass and age at sexual maturity. When controlling for GC3*, the correlation between dN/dS and LHT remains largely unchanged for birds (table 4). In placentals, the strength of the correlation is overall reduced, with body mass remaining the only correlate of dN/dS receiving a strong statistical support (PP > 0.99; table 4).
Table 4.
Partial Correlations between dN/dS and LHT Controlling for GC3 Content
| Data Set | Body Mass | Longevity | Age at Sexual Maturity |
|---|---|---|---|
| Placentalia (N = 194) | 0.38 (>0.99)** | 0.45 (0.91) | 0.16 (0.82) |
| Birds | −0.21 (0.15) | −0.54 (0.01)** | −0.25 (0.20) |
**PP <0.025 or >0.975.
To control for nucleotide composition in the case of the RadConsAA models, we opted for a pragmatic method circumventing the fact that nucleotide frequencies are not explicit parameters of the substitution model. Specifically, for each taxon, we computed the frequencies of C and T (πC and πT) at the third positions, and considered these (log-it transformed) frequencies as quantitative traits, to be included in our covariance analyses along with the three LHT. We then performed multiple regression analyses, so as to estimate the correlation of LHT with Kr/Kc while controlling for composition in C and T. Alternatively, we devised a control using GC bias, AT skew and GC skew (all log-transformed) instead of πC and πT (see Materials and Methods). These controls were conducted both on the original sequence alignments and on alignments from which the ND6 gene was excluded, as this gene, being located on the light strand, experiences a distinct mutation bias compared to other mitochondrial protein coding genes.
We find that πC and πT are, respectively, positively and negatively correlated with longevity in mammals (PP > 0.99 for πC and PP < 0.01 for πT for all the LHT). These results are in agreement with previous analyses (Jobson et al. 2010). We also observe that Kr/Kc is positively correlated with πC (R = 0.35, PP > 0.99) and negatively with πT (R = −0.29, PP < 0.01). This correlation could be due in part to a joint covariation of Kr/Kc and composition in C and T with LHT. However, controlling for LHT does not eliminate the statistical support for the correlation of Kr/Kc with nucleotide composition (πC: R = 0.24, PP = 0.99; πT: R = −0.19, PP = 0.03), suggesting instead that compositional variation has a direct impact on Kr/Kc estimation. On the other hand, biases induced by compositional variation do not entirely explain the correlation of Kr/Kc with LHT, as controlling for πC and πT does not reduce the strength the correlation between LHT and Kr/Kc (table 5). Similarly, controlling for GC bias, AT skew and GC skew does not have any impact on the strength of the correlation (PP > 0.98 for all LHT; table 5) nor does the exclusion of ND6 (supplementary table S3, Supplementary Material online).
Table 5.
Covariance between Kr/Kc and LHT Controlling for Base Composition
| Data Set | Control | Body Mass | Longevity | Age at Sexual Maturity |
|---|---|---|---|---|
| Placentalia (All) | πC and πT | 0.33 (>0.99)** | 0.46 (>0.99)** | 0.53 (>0.99)** |
| Placentalia (All) | GC bias, AT/GC skew | 0.33 (>0.99)** | 0.38 (0.98)** | 0.54 (>0.99)** |
| Birds | πC and πT | 0.42 (0.99)** | 0.29 (0.88) | 0.19 (0.76) |
| Birds | GC bias, AT/GC skew | 0.41 (0.99)** | 0.27 (0.86) | 0.20 (0.77) |
Note.—Change in polarity and volume are considered as radical substitutions.
**PP < 0.025 or >0.975.
Interestingly, for birds, the relationship between πC and πT and LHT goes in the opposite direction, compared with what is observed in placental mammals: in birds, πC is negatively correlated with body mass although the correlation is not supported (R = −0.15, PP = 0.08) and πT is positively correlated with body mass, with marginal support (R = 0.17, PP = 0.95). As in mammals, controlling for πC and πT or for GC bias, AT skew and GC skew, with or without ND6, does not reduce the support for the correlation between body mass and Kr/Kc (PP ≥ 0.96; table 5 and supplementary tables S2 and S3, Supplementary Material online, for the results with the alternative topology and the correlation without ND6, respectively). Importantly, the fact that the correlation between Kr/Kc and composition goes in opposite directions in birds and in placental mammals, whereas in both cases Kr/Kc is positively correlated with body mass and/or longevity, is encouraging, as it confirms that compositional variation cannot entirely explain observed correlations between Kr/Kc and LHT.
Macroevolutionary Patterns of Kr/Kc and LHT
The reconstructions of dN/dS and Kr/Kc are displayed in supplementary figures S3 and S4, Supplementary Material online, for dN/dS and figures 2 and 3 for Kr/Kc. In the case of dN/dS (supplementary figs. S3 and S4, Supplementary Material online), branches with large values of dN/dS are mostly ancient branches. This is further indicated by the strong positive correlation between divergence times and dN/dS observed in placentals (fig. 4A), suggesting that dN/dS could indeed be overestimated because of an underestimation of dS in the more ancient branches of the phylogeny, itself due to the greater mutational saturation affecting synonymous substitutions. In the context of a nearly neutral interpretation of the variation in dN/dS, such differential saturation effects would result in artifactual inference of small ancestral effective population sizes. In addition, as the model uses the relationship between LHT and dN/dS to reconstruct ancestral LHT, the overestimation of dN/dS in ancient lineages may bias the estimation of ancestral LHT toward higher values.
Fig. 2.—

Reconstruction of Kr/Kc (as defined by change in polarity or volume) along the phylogeny of Placentalia. Color stands for the marginal posterior mean instant value of Kr/Kc at each node, with linear interpolation along branches.
Fig. 3.—

Reconstructionof Kr/Kc (as defined by change in polarity or volume) along the phylogeny of birds. Color stands for the marginal posterior mean instant value of Kr/Kc at each node, with linear interpolation along branches.
Fig. 4.—

Plot of dN/dS (A, C) and Kr/Kc (B, D) against divergence times in Placentalia (194 species; A, B) and in Birds (92 species; C, D). dN/dS, Kr/Kc (both in base 10 logarithm), and divergence times are estimated by their posterior means obtained under the MGdNdS and the RadConsAA-PolVol models, respectively. Black lines indicate lowest fit curves estimated using “loess” function in R (R Development Core Team 2004).
In contrast, patterns of Kr/Kc (fig. 2) are more consistent within orders, and across the tree: high values of Kr/Kc in orders such as Cetartiodactyla (especially cetaceans), Carnivora (particularly Ursidae and Pinnipedia), Primates (especially Catyrrhinii), and more isolated clades like elephants or sloths. Conversely, Glires (i.e., Rodentia + Lagomorpha), Eulipotyphla (i.e., insectivores within Laurasiatheria), or Afrosoricida and Macroscelidae (i.e., insectivores within Afrotheria) have low Kr/Kc values. Chiroptera have relatively low values, with large Pteropodidae having higher Kr/Kc ratios than other smaller Chiroptera. These patterns are globally consistent with Kr/Kc representing a more reliable molecular correlate of population size and LHT. Unlike what is observed with dN/dS, the early lineages of placental mammals do not have especially high values of Kr/Kc, and a negative, instead of a positive, correlation is observed between Kr/Kc and divergence times in placentals (Spearman’s rho = −0.67, P value < 0.01; fig. 4B), but note the increase of Kr/Kc toward the very base of the tree (fig. 4B). Nonetheless, the ancestor of placental mammals is estimated to have a bigger body mass using the MGdNdS model (between 2 and 40 kg) than under the RadConsAA model (between 0.6 and 7 kg). Similarly, the ancestors of each of the major groups of placental mammals are all inferred to be two to four times smaller using RadConsAA than using MGdNdS (table 6).
Table 6.
Body Mass Estimated under the MGdNdS and the RadConsAA Models for the Ancestors of the Major Mammalian Clades
| Clade | Body Mass Estimated under MGdNdS (kg) | Body Mass Estimated under RadConsAA (kg) |
|---|---|---|
| Xenarthra | 6.2 (1.1, 30.3) | 1.9 (0.3, 16.8) |
| Afrotheria | 11.7 (2.4, 47.5) | 1.7 (0.5, 11.9) |
| Laurasiatheria | 8.9 (2.3, 38.1) | 2.0 (0.6, 8.4) |
| Euarchontoglires | 6.0 (1.7, 22.4) | 0.9 (0.2, 4.4) |
Note.—Values are expressed as median (CI).
The saturation detected in nucleotide sequences may also explain why the relationship between LHT and dN/dS is weaker than the relationship between LHT and Kr/Kc. Based on all of these observations, we choose to favor the reconstruction of the ancestral LHT using the RadConsAA model over MGdNdS, and we will only refer to the results obtained using the RadConsAA model hereafter.
The ancestor of extant placental mammals was estimated to weigh 2 kg (95% credibility interval [CI]: 0.6–7 kg), reach its sexual maturity after 440 days (CI: 277–696 days) and have a life span of 15 years (CI: 10–23 years). This estimation is compatible with estimates obtained with a nuclear data set (mean = 1.6 kg; CI: 0.2–7 kg; Lartillot and Delsuc 2012). Comparing estimates obtained under the covariant or the uncorrelated models suggests that Kr/Kc brings information in favor of a slight downward revision of the ancestral LHT of a certain number of ancestors. For example, all the ancestor of major orders such as Primates, Rodentia, Chiroptera, Cetartiodactyla, and Carnivora are inferred to be heavier using the uncorrelated model. The largest discrepancy was observed for Cetartiodactyla where the ancestor is estimated to be almost twice as large with the uncorrelated model (median = 33 kg; CI: 9–113 kg) compared with what is observed under the covariant model (median = 15 kg; CI: 4–53 kg). The estimation for the ancestor of placental mammals is, however, nearly identical between the uncorrelated (median = 1.7 kg; CI: 0.5–6 kg) and the uncorrelated models.
The negative correlation between Kr/Kc and divergence times alluded to above, combined with the positive correlation between Kr/Kc and LHT, would seem to suggest the existence of a global trend in increasing body size in placental mammals, akin to a Cope’s rule. On the other hand, early placentals do not have especially low Kr/Kc ratios: Kr/Kc is larger in early branches connecting placental orders, than within Rodentia or Eulipotyphla (mean Kr/Kc = 0.85; CI: 0.69–1.1 and mean = 0.85; CI: 0.70–0.99 for Laurasiatheria and Euarchontoglires ancestor vs. mean = 0.93; CI = 0.71 and 1.22 for terminal branches of Rodentia and mean = 0.72; CI: 0.63–0.96 for terminal branches of Eulipotyphla). Such an intermediate Kr/Kc ratio suggests that early placentals were not especially small: indeed, even when taking into account the covariance between body mass and Kr/Kc (which leads to a smaller inferred ancestral body mass), the estimated body mass of the last common ancestor of placentals is bigger than the mass of a majority of extant mammals. The median body mass of extant mammals is 100 g, and only 23 % of the extant species weigh more than 2 kg (N = 3,913 species available in PanTheria database, Jones et al. 2009). Therefore, our estimate for the ancestor represents a fairly large animal compared with a typical extant placental mammal, and this, fundamentally because species-rich group of mammals such as Rodentia, Chiroptera, and Eulipotyphla are all small animals.
To further explore the trend in body mass during placental evolution, we computed the difference between the median ancestral body mass and median body mass of extant species for the 14 orders containing at least two species in our data set. Exactly one-half of the orders experienced an increase in size (the largest increase being observed in Perissodactyla; fig. 5), whereas the other half shows a decrease in body size (particularly in Eulipotyphla and Chiroptera; fig. 5), and thus not suggesting any systematic trend in body mass evolution globally across placental mammals.
Fig. 5.—

Difference between the median ancestral body mass estimated using the RadConsAA-PolVol model and the median of the body mass of extant placental mammals species (all in base 10 logarithm). A positive (respectively negative) value indicates a decrease (respectively an increase) in body mass. Taxonomy is as in PANTheria (Jones et al. 2009).
In birds, saturation is expected to have the same negative impact on dN/dS and, as in mammals, we note the absence of a positive correlation between dN/dS and divergence time when using Kr/Kc (compare figs. 3 and 4C and D with supplementary fig. S4, Supplementary Material online). Using the same rationale as above, we again favor the ancestral reconstruction obtain with RadConsAA model, over that obtained under the MGdNdS model. We obtain an estimate of approximately 640 g (CI: 0.2–1.9 kg) for the ancestral body mass of Neornithes. Compared with extant species, this represent a fairly large bird, that is, about the size of a common buzzard Buteo buteo. From this ancestor, diversification of modern birds mainly occurred through size reduction. The ancestor of Neoaves (all the extant birds excluding Galloanserae and Palaeognathae) was already estimated to be smaller than the Neornithes ancestor (median = 341 g; CI: 142–820 g) and the great majority of modern birds (6,556 species out of 8,177 Neoaves) have today a body mass lower than 150 g (Dunning 2007). This trend is, however, not followed by Palaeognathae (Ratites + Tinamiformes), nor by Galloanserae. The ancestor of Palaeognathae was estimated to be heavier than the Neornithes ancestor (median = 2.1 kg and 95% CI: 530 g to 8.7 kg), after which body mass still increases toward the very large extant Ratites (but not in Tinamiformes). In Galloanserae, body mass remains fairly stable from approximately 800 g (95% CI: 210 g to 3.1 kg) for the ancestral species to a median of 705 g (95% CI: 102 g to 4.2 kg) for the extant species. Using the same method as for Placentalia, we computed the difference between the median ancestral body mass and the median body mass of extant species for the 11 orders (or clades) (fig. 6). In 7 out of the 11 clades, body mass has decreased during Avian evolution (fig. 6). This might indicate that, in contrast to placental mammals, life-history evolution preferentially occurred through size reduction in birds. Finally, most ancestral body mass estimates under the uncorrelated model are slightly larger than under the covariant model with Kr/Kc.
Fig. 6.—

Difference between the median ancestral body mass estimated using the RadConsAA-PolVol and the median of the body mass of extant bird species (all in base 10 logarithm). The “water-birds clade” includes the orders Ciconiiformes, Gaviiformes, Pelecaniformes, Sphenisciformes, and Procellariiformes. A positive (respectively negative) value indicates a decrease (respectively an increase) in body mass. Taxonomy as in Dunning (2007).
Discussion
Extending several recent methodological developments concerning the analysis of correlations between substitution patterns and LHT, and adapting them so as to deal with the specific features of the evolutionary regime of mitochondrial DNA (in particular the high rate of synonymous substitution), the present article proposes a synthetic view of what the comparative analysis of mitochondrial genomes can tell us about the evolution of LHT, and population genetics parameters in birds and mammals.
Mutational Saturation and the Relation between Substitution Patterns and LHT
Assuming that body mass represent a proxy of long-term effective population size (discussed later), the nearly neutral theory predicts a positive correlation of dN/dS and Kr/Kc with body size. Globally, placental mitochondrial genomes agree with this expectation. Birds, on the other hand, display contradictory patterns: negative correlation between dN/dS and body mass, but positive correlation between Kr/Kc and body mass (albeit not robust to controlling for other traits and dS). We argue that the conflicting results observed in the case of birds reflect the poor estimation of dN/dS in mitochondrial data sets, rather than an absence of relationship between body mass and the efficacy of selection in birds. This interpretation relies on two points. First, synonymous substitutions appear to be highly saturated in mitochondrial sequences (see Results section and fig. 1). Despite the fact that the mitochondrial mutation rate is lower in birds than in mammals (Hickey 2008; Nabholz et al. 2009), saturation may be more pronounced in birds because, in our data sets, terminal branch lengths are often longer in the case of birds than for placentals (median = 0.13 vs. 0.43 substitution per nucleotide sites, for placental mammals and birds, respectively, in both cases, using third-codon positions). This appears to be an indirect consequence of the taxonomic sampling of the bird data set, mainly designed to tackle the Neoaves phylogeny (Gibb et al. 2007; Pratt et al. 2009). Second, in placental mammals, the relationship between Kr/Kc and LHT is stronger that the relationship with dN/dS (compare tables 1 and 3), further suggesting that the RadConsAA model, despite its increased overall parameterization, is better at estimating the rate of fixation of slightly deleterious mutations in mitochondrial sequences. Finally, it is expected that any relationship between the substitution process and LHT would be more difficult to pinpoint in birds, because they have a markedly narrower range of variation in LHT compared with mammals.
In the case of mammals, owing to a better taxonomic sampling and a wider range of LHT variation in this group, mutational saturation, although strong, does not qualitatively change the correlation patterns of dN/dS with LHT. On the other hand, the reconstruction of dN/dS along the phylogeny (supplementary fig. S2, Supplementary Material online) clearly appears to be unreliable: the large values observed in ancient and fast evolving lineages are probably the result of a differential saturation of dS and dN, and are thus almost certainly artifactual. Such biases in the estimation of ancestral dN/dS are likely to have a strong impact on the marginal reconstruction of LHT evolution along the tree. Typically, given the positive correlation inferred between dN/dS and LHT, this might result in an upward bias in the estimated ancestral body sizes. In contrast, as far as ancestral LHT inference is concerned, Kr/Kc seems to represent a more reliable molecular marker than dN/dS.
Variation in Kr/Kc among Lineages: A Nearly Neutral Effect?
The nearly neutral theory predicts that both dN/dS and, under certain conditions, Kr/Kc, should correlate negatively with effective population size (Ne). Here, and elsewhere (Nikolaev et al. 2007; Lartillot and Delsuc 2012; Romiguier et al. 2013), what is more specifically observed is a correlation between dN/dS or Kr/Kc and body mass (and other LHT). Interpreting these observations in the light of the nearly neutral theory requires additional assumptions concerning the link between body mass and long-term Ne. In this direction, a negative correlation between population density and body size has been reported in a wide range of taxonomical groups (White et al. 2007). This correlation seems stronger in mammals than in birds (e.g., Silva et al. 1997). In turn, it is reasonable to assume that effective population size is correlated with population density. This correlation is expected to be weakened, although not suppressed, by additional covariates of Ne, such as unequal sex ratio, population structure, inbreeding, and variance in the number of offspring could influence Ne (Frankham 1995; Charlesworth 2009). These additional factors may in fact explain the relatively weak correlation between body size and Kr/Kc observed here, as well as the weak correlations previously reported between dN/dS and LHT. Note that the fact that the level of mitochondrial neutral diversity is only weakly negatively correlated with body mass could be taken as an argument against the link between body mass and Ne (Berlin et al. 2007; Nabholz et al. 2008 but see Piganeau and Eyre-Walker 2009). Nucleotide diversity is, however, very sensitive to recent drop in population size and therefore could be a very poor estimate of long term Ne. This sensitivity to population fluctuation could explain why, in birds, no correlation was found between census population size and the level of neutral polymorphism of one mitochondrial protein-coding gene (Nabholz et al. 2009). dN/dS or Kr/Kc, on the other hand, are not expected to be strongly impacted by recent population bottlenecks but instead reflect variation in long term Ne. By imposing an upper limit on the population size of a given species, body mass could influence long term Ne and therefore be a better predictor of variation in dN/dS (and Kr/Kc) than short term Ne such as estimated from genetic diversity or current census population size.
From a molecular evolutionary perspective, the most important question raised by the present analysis is whether, and to what extent, Kr/Kc reflects variation in effective population size. Although the correlation patterns observed in our analysis are globally compatible with a nearly neutral model, alternative interpretations should also be considered.
Among potentially confounding factors acting on Kr/Kc, positive selection represents one possible candidate. Positive selection has been suggested as a dominant force in the molecular evolutionary regime of mitochondrial genomes (Bazin et al. 2006), although mostly in invertebrates. In contrast, patterns of genetic diversity in mitochondrial genomes of birds and mammals suggest a regime dominated by purifying selection and random drift in these groups (Mulligan et al. 2006; Nabholz et al. 2008, 2009). Isolated cases of positive selection in mitochondrial protein sequences have been reported in mammals, and particularly in anthropoid primates (Grossman et al. 2004). In the specific case of anthropoids, we have checked that the correlation between Kr/Kc and LHT was robust to the removal of all sequences of this clade (not shown).
Mitochondrial nucleotide composition is correlated with LHT and particularly with longevity (Jobson et al. 2010), a relationship confirmed by our results. This raises the possibility that changes in nucleotide composition, and more generally in the mutation spectrum could influence the estimation of variation in dN/dS or Kr/Kc and confound the correlation patterns of these two variables with LHT. Here, we have checked that the correlation between Kr/Kc and LHT remains significant upon controlling for compositional variation (table 5). Similarly, the correlation between dN/dS and LHT is robust when controlling for equilibrium GC3 (table 4). Finally, the fact that Kr/Kc positively correlates with LHT in birds and mammals, despite of opposite compositional trends in the two groups, provides an internal negative control suggesting that compositional variation at the nucleotide level cannot entirely explain the correlation patterns of Kr/Kc with LHT. This said, our controls do not entirely rule out potential biases due to more general changes in the mutation spectrum (e.g., the transition–transversion ratio, Belle et al. 2005). An in depth analysis of the question would require explicit models of the mutation-selection balance at the codon level, and of how this balance may shift as a result of changes in either the mutation process or the efficacy of selection.
Altogether some caution is therefore needed when interpreting variation in mitochondrial Kr/Kc among lineages. Explanations in terms of positive selection, or more subtle artifacts caused by changes in the mutation spectrum, cannot be entirely excluded. Nevertheless, when combining the observations made on mammals and on birds, a nearly neutral interpretation remains the best explanation currently available. Accordingly, figures 2 and 3 would represent a tentative (albeit uncalibrated) reconstruction of the evolutionary trends in long-term effective population size in mammals and birds. In the case of mammals, this can be compared with a previous reconstruction based on the nuclear dN/dS (restricted to GC conservative transversions, which are immune from GC-biased gene conversion, Lartillot 2013). Although effective population size is not expected to be the same for nuclear and mitochondrial sequences, owing to differences in the mode of transmission, and in the recombination and selection regimes, nevertheless, at the scale of placental mammals, the qualitative patterns of variation should be relatively concordant between the two genetic compartments. Indeed, the two reconstructions agree in many respects. In particular, the long-term effective population size is inferred to be small in anthropoids, Cetacea, Panungulata (except Procavia) and Pilosa, large in Eulipotyphla, Rodentia, and Chiroptera, and intermediate in the ancestor of Placentalia. There are also differences between the two reconstructions, in particular for Carnivora, inferred to have a smaller long-term effective population size based on mitochondrial Kr/Kc, than based on the nuclear GC-conservative dN/dS (although in both cases, Canidae are inferred to have larger effective population sizes than Ursidae, Pinnipeds, or Felidae). Such discrepancies suggest the presence of confounding factors (among which compositional variation or local episodes of positive selection). Ultimately, these reconstructions could be calibrated using data about polymorphism in extant species, although this would require specific assumptions about the correlation between short- and long-term effective population size.
Evolution of Body Mass in Birds and Mammals
The correlation between Kr/Kc and LHT implies that information about ancestral LHT can be gained from reconstructions of patterns of Kr/Kc along phylogenies. In this respect, our analysis provides interesting insights into the evolution of life-history, and particularly the evolution of body-size, in placentals and birds.
In placental mammals, instead of a global trend across the entire group, our reconstruction integrating information about ancestral Kr/Kc suggests that the ancestors of some specific orders (e.g., Carnivora, Perissodactyla, and Cetartiodactyla) might have been slightly smaller than what would be inferred based on the body mass of extant taxa. This in turn suggests the existence of some local trends in increasing body size in these specific orders. On the other hand, other orders clearly show a pattern of stasis, or even of a decrease, in body size (e.g., Chiroptera, Rodentia, and Eulipotyphla).
Our analysis also suggests that the ancestors of both placental mammals and birds were not small, compared with extant species. This is particularly obvious in birds where most of the diversification seems to have been accompanied by reduction in size, but the same is true for placental mammals: the last common ancestor of Placentalia is inferred to be between 0.6 and 7 kg. This is larger than a majority of extant placental mammals. Comparison of this estimate with the fossil record is not an easy task. Nevertheless, current paleontological knowledge suggests that, although Eutherians known from the Cretaceous had apparently undergone a large ecological diversification (Luo 2007; Wilson et al. 2012) they were, on average, rather small (between 0.1 and 1 kg), with the exception of the 12–14 kg carnivorous Repenomamus giganticus (Hue et al. 2005). Our estimation is, therefore, in the upper side of the range of body masses inferred for cretaceous fossils, even if some isolated larger fossils are documented. Interestingly, our estimate is more compatible with a recent study using nuclear protein-coding gene GC evolution (Romiguier et al. 2013), although on the lower side of the confidence interval reported in this study.
Like mammals, birds were also ecologically and morphologically fairly diverse in the cretaceous (Chiappe and Dyke 2002). Body mass of these birds range from a few dozen grams to approximately 1 kg (body mass extrapolated using limbs size and scaling equation from Hone et al. 2008), which is compatible with our estimation (0.2 to 1.9 kg). Small individuals often belong to Enantiornithes, whereas larger birds were found in Ornithurae (e.g., Hesperornis and Ichthyornis)—a clade including the modern Neornithes (Chiappe and Dyke 2002). In the Cretaceous, Neornithes fossils are unfortunately very fragmentary and can be difficult to identify (Mayr 2009). Within the Paleogene however, most modern orders are represented in the fossil record (Mayr 2009), however, body mass has not yet been estimated for these taxa.
The present reconstruction assumes that the evolution of body mass and Kr/Kc is correctly described by a nondirectional Brownian motion. In a similar analysis of correlations between body size, LHT, and substitution rates in nuclear sequences in placental mammals (Lartillot and Delsuc 2012), it was noted that the assumptions behind the Brownian model are probably violated, and that this may have an impact on ancestral reconstructions, even taking into account correlations between body mass and substitution patterns. In the long term, further elaborating on the present model, integrating several molecular markers, and accounting for departures from the undirected Brownian assumption, could result in more contrasted and more informative patterns of LHT evolution.
Supplementary Material
Supplementary figures S1–S4 and tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the ARCAD foundation project (http://www.arcad-project.org/) to B.N.; the Natural Science and Engineering Research Council of Canada and the Canadian Foundation for Innovation to N.U. and N.L.; and the French Agence Nationale pour La Recherche, Ancestrome project to N.L. This is ISEM publication number ISE-M 2013-068. Computational resources were provided by Calcul Québec, Compute Canada, and Montpellier MBB Platform.
Literature Cited
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Bazin E, Glémin S, Galtier N. Population size does not influence mitochondrial genetic diversity in animals. Science. 2006;312:570–572. doi: 10.1126/science.1122033. [DOI] [PubMed] [Google Scholar]
- Belle EMS, Piganeau G, Gardner M, Eyre-Walker A. An investigation of the variation in the transition bias among various animal mitochondrial DNA. Gene. 2005;355:58–66. doi: 10.1016/j.gene.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Benton MJ, Donoghue PCJ, Asher RJ. Calibrating and constraining molecular clocks. In: Hedges SB, Kumar S, editors. The timetree of life. Oxford: Oxford University Press; 2009. pp. 35–86. [Google Scholar]
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e1000026. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin S, Tomaras D, Charlesworth B. Low mitochondrial variability in birds may indicate Hill–Robertson effects on the W chromosome. Heredity. 2007;99:389–396. doi: 10.1038/sj.hdy.6801014. [DOI] [PubMed] [Google Scholar]
- Björnerfeldt S, Webster MT, Vilà C. Relaxation of selective constraint on dog mitochondrial DNA following domestication. Genome Res. 2006;16:990–994. doi: 10.1101/gr.5117706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanga-Kanfi S, et al. Rodent phylogeny revised: analysis of six nuclear genes from all major rodent clades. BMC Evol Biol. 2009;9:71. doi: 10.1186/1471-2148-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L, Carr DE. The molecular clock and the relationship between population size and generation time. Evolution. 1993;47:688–690. doi: 10.1111/j.1558-5646.1993.tb02124.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 2009;10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- Chiappe LM, Dyke GJ. The mesozoic radiation of birds. Annu Rev Ecol Syst. 2002;33:91–124. [Google Scholar]
- Damuth J. Interspecific allometry of population density in mammals and other animals: the independence of body mass and population energy-use. Biol J Linn Soc. 1987;31:193–246. [Google Scholar]
- Delsuc F, et al. Molecular phylogeny of living xenarthrans and the impact of character and taxon sampling on the placental tree rooting. Mol Biol Evol. 2002;19:1656–1671. doi: 10.1093/oxfordjournals.molbev.a003989. [DOI] [PubMed] [Google Scholar]
- de Magalhães JP, Costa J. A database of vertebrate longevity records and their relation to other life-history traits. J Evol Biol. 2009;22:1770–1774. doi: 10.1111/j.1420-9101.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- Dubey S, et al. Molecular phylogenetics of shrews (Mammalia: Soricidae) reveal timing of transcontinental colonizations. Mol Phylogenet Evol. 2007;44:126–137. doi: 10.1016/j.ympev.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Dunning JBJ. CRC handbook of avian body masses. 2nd ed. Boca Raton (FL): CRC Press; 2007. [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson JL, Lightowlers RN. Mitochondrial DNA clonality in the dock: can surveillance swing the case? Trends Genet. 2006;22:603–607. doi: 10.1016/j.tig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD, Smith NGC, Gaffney D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol. 2002;19:2142–2149. doi: 10.1093/oxfordjournals.molbev.a004039. [DOI] [PubMed] [Google Scholar]
- Fabre P-H, Rodrigues A, Douzery EJP. Patterns of macroevolution among primates inferred from a supermatrix of mitochondrial and nuclear DNA. Mol Phylogenet Evol. 2009;53:808–825. doi: 10.1016/j.ympev.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Flynn JJ, Finarelli JA, Zehr S, Hsu J, Nedbal MA. Molecular phylogeny of the carnivora (mammalia): assessing the impact of increased sampling on resolving enigmatic relationships. Syst Biol. 2005;54:317–337. doi: 10.1080/10635150590923326. [DOI] [PubMed] [Google Scholar]
- Frankham R. Effective population size/adult population size ratios in wildlife: a review. Genet Res. 1995;66:95–107. doi: 10.1017/S0016672308009695. [DOI] [PubMed] [Google Scholar]
- Galewski T, et al. The evolutionary radiation of Arvicolinae rodents (voles and lemmings): relative contribution of nuclear and mitochondrial DNA phylogenies. BMC Evol Biol. 2006;6:80. doi: 10.1186/1471-2148-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glémin S, Ranwez V. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 2009;25:1–5. doi: 10.1016/j.tig.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Galtier N, Enard D, Radondy Y, Bazin E, Belkhir K. Mutation hot spots in mammalian mitochondrial DNA. Genome Res. 2006;16:215–222. doi: 10.1101/gr.4305906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Nabholz B, Glémin S, Hurst GDD. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol. 2009;18:4541–4550. doi: 10.1111/j.1365-294X.2009.04380.x. [DOI] [PubMed] [Google Scholar]
- Gerber AS, Loggins R, Kumar S, Dowling TE. Does nonneutral evolution shape observed patterns of DNA variation in animal mitochondrial genomes? Annu Rev Genet. 2001;35:539–566. doi: 10.1146/annurev.genet.35.102401.091106. [DOI] [PubMed] [Google Scholar]
- Gibb GC, Kardailsky O, Kimball RT, Braun EL, Penny D. Mitochondrial genomes and avian phylogeny: complex characters and resolvability without explosive radiations. Mol Biol Evol. 2007;24:269–280. doi: 10.1093/molbev/msl158. [DOI] [PubMed] [Google Scholar]
- Gibson A, Gowri-Shankar V, Higgs PG, Rattray M. A comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol Biol Evol. 2005;22:251–264. doi: 10.1093/molbev/msi012. [DOI] [PubMed] [Google Scholar]
- Grossman LI, Wildman DE, Schmidt TR, Goodman M. Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet. 2004;20:578–585. doi: 10.1016/j.tig.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Hackett SJ, Kimball RT, Reddy S, et al. A phylogenomic study of birds reveals their evolutionary history. Science. 2008;320:1763–1768. doi: 10.1126/science.1157704. [DOI] [PubMed] [Google Scholar]
- Haddrath O, Baker AJ. Complete mitochondrial DNA genome sequences of extinct birds: ratite phylogenetics and the vicariance biogeography hypothesis. Proc Biol Sci. 2001;268:939–945. doi: 10.1098/rspb.2001.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härlid A, Janke A, Arnason U. The complete mitochondrial genome of Rhea americana and early avian divergences. J Mol Evol. 1998;46:669–679. doi: 10.1007/pl00006347. [DOI] [PubMed] [Google Scholar]
- Hickey AJR. Avian mtDNA diversity? An alternate explanation for low mtDNA diversity in birds: an age-old solution? Heredity. 2008;100:443. doi: 10.1038/hdy.2008.6. [DOI] [PubMed] [Google Scholar]
- Hone DWE, Dyke GJ, Haden M, Benton MJ. Body size evolution in Mesozoic birds. J Evol Biol. 2008;21:618–624. doi: 10.1111/j.1420-9101.2007.01483.x. [DOI] [PubMed] [Google Scholar]
- Hu Y, Meng J, Wang Y, Li C. Large Mesozoic mammals fed on young dinosaurs. Nature. 2005;433:149–152. doi: 10.1038/nature03102. [DOI] [PubMed] [Google Scholar]
- Huchon D, et al. Rodent phylogeny and a timescale for the evolution of Glires: evidence from an extensive taxon sampling using three nuclear genes. Mol Biol Evol. 2002;19:1053–1065. doi: 10.1093/oxfordjournals.molbev.a004164. [DOI] [PubMed] [Google Scholar]
- Jobson RW, Dehne-Garcia A, Galtier N. Apparent longevity-related adaptation of mitochondrial amino acid content is due to nucleotide compositional shifts. Mitochondrion. 2010;10:540–547. doi: 10.1016/j.mito.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Johnson KP, Seger J. Elevated rates of non-synonymous substitution in island birds. Mol Biol Evol. 2001;18:874–881. doi: 10.1093/oxfordjournals.molbev.a003869. [DOI] [PubMed] [Google Scholar]
- Jones KE, et al. PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology. 2009;90:2648–2648. [Google Scholar]
- Keightley PD, Lercher MJ, Eyre-Walker A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 2005;3:e42. doi: 10.1371/journal.pbio.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge (UK): Cambridge University Press; 1983. [Google Scholar]
- Lanfear R, Welch JJ, Bromham L. Watching the clock: studying variation in rates of molecular evolution between species. Trends Ecol Evol. 2010;25:495–503. doi: 10.1016/j.tree.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Lartillot N. Interaction between selection and biased gene conversion in mammalian protein coding sequence evolution revealed by a phylogenetic covariance analysis. Mol Biol Evol. 2013;30:356–368. doi: 10.1093/molbev/mss231. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Delsuc F. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution. 2012;66:1773–1787. doi: 10.1111/j.1558-5646.2011.01558.x. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011;28:729–744. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- Luo Z-X. Transformation and diversification in early mammal evolution. Nature. 2007;450:1011–1019. doi: 10.1038/nature06277. [DOI] [PubMed] [Google Scholar]
- Madsen O, et al. Parallel adaptive radiations in two major clades of placental mammals. Nature. 2001;409:610–614. doi: 10.1038/35054544. [DOI] [PubMed] [Google Scholar]
- Mayr G. Paleogene fossil birds. Berlin, Heidelberg (Germany): Springer-Verlag; 2009. [Google Scholar]
- Mulligan CJ, Kitchen A, Miyamoto MM. Comment on “Population size does not influence mitochondrial genetic diversity in animals.”. Science. 2006;314:1390. doi: 10.1126/science.1132585. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, et al. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17:413–421. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Glémin S, Galtier N. Strong variations of mitochondrial mutation rate across mammals—the longevity hypothesis. Mol Biol Evol. 2008;25:120–130. doi: 10.1093/molbev/msm248. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Glémin S, Galtier N. The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol Biol. 2009;9:54. doi: 10.1186/1471-2148-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Jarvis ED, Ellegren H. Obtaining mtDNA genomes from next-generation transcriptome sequencing: a case study on the basal Passerida (Aves: Passeriformes) phylogeny. Mol Phylogenet Evol. 2010;57:466–470. doi: 10.1016/j.ympev.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Mauffrey J-F, Bazin E, Galtier N, Glemin S. Determination of mitochondrial genetic diversity in mammals. Genetics. 2008;178:351–361. doi: 10.1534/genetics.107.073346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW. Deleterious mutations in animal mitochondrial DNA. Genetica. 1998;102-103:61–69. [PubMed] [Google Scholar]
- Nam K, et al. Molecular evolution of genes in avian genomes. Genome Biol. 2010;11:R68. doi: 10.1186/gb-2010-11-6-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nee S, Read AF, Greenwood JJD, Harvey PH. The relationship between abundance and body size in British birds. Nature. 1991;351:312–313. [Google Scholar]
- Nikolaev SI, et al. Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc Natl Acad Sci U S A. 2007;104:20443–20448. doi: 10.1073/pnas.0705658104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Ohta T. The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst. 1992;23:263–286. [Google Scholar]
- Ohta T. An examination of the generation-time effect on molecular evolution. Proc Natl Acad Sci U S A. 1993;90:10676–10680. doi: 10.1073/pnas.90.22.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco MA, et al. Evolution of modern birds revealed by mitogenomics: timing the radiation and origin of major orders. Mol Biol Evol. 2011;28:1927–1942. doi: 10.1093/molbev/msr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau G, Eyre-Walker A. Evidence for variation in the effective population size of animal mitochondrial DNA. PLoS One. 2009;4:e4396. doi: 10.1371/journal.pone.0004396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popadin K, et al. Accumulation of slightly deleterious mutations in mitochondrial protein-coding genes of large versus small mammals. Proc Natl Acad Sci U S A. 2007;104:13390–13395. doi: 10.1073/pnas.0701256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt RC, et al. Toward resolving deep neoaves phylogeny: data, signal enhancement, and priors. Mol Biol Evol. 2009;26:313–326. doi: 10.1093/molbev/msn248. [DOI] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Mutation and selection at silent and replacement sites in the evolution of animal mitochondrial DNA. Genetica. 1998;102-103:393–407. [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2004. [Google Scholar]
- Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15:957–966. doi: 10.1093/oxfordjournals.molbev.a026011. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJP, Galtier N. Genomic evidence for large, long-lived ancestors to placental mammals. Mol Biol Evol. 2013;30:5–13. doi: 10.1093/molbev/mss211. [DOI] [PubMed] [Google Scholar]
- Sainudiin R, et al. Detecting site-specific physicochemical selective pressures: applications to the Class I HLA of the human major histocompatibility complex and the SRK of the plant sporophytic self-incompatibility system. J Mol Evol. 2005;60:315–326. doi: 10.1007/s00239-004-0153-1. [DOI] [PubMed] [Google Scholar]
- Silva M, Brown JH, Downing JA. Differences in population density and energy use between birds and mammals: a macroecological perspective. J Anim Ecol. 1997;66:327–340. [Google Scholar]
- Slack KE, et al. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol Biol Evol. 2006;23:1144–1155. doi: 10.1093/molbev/msj124. [DOI] [PubMed] [Google Scholar]
- Springer MS, et al. Mitochondrial versus nuclear gene sequences in deep-level mammalian phylogeny reconstruction. Mol Biol Evol. 2001;18:132–143. doi: 10.1093/oxfordjournals.molbev.a003787. [DOI] [PubMed] [Google Scholar]
- Springer MS, Stanhope MJ, Madsen O, de Jong WW. Molecules consolidate the placental mammal tree. Trends Ecol Evol. 2004;19:430–438. doi: 10.1016/j.tree.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Teeling EC, et al. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307:580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- Venditti C, Meade A, Pagel M. Multiple routes to mammalian diversity. Nature. 2011;479:393–396. doi: 10.1038/nature10516. [DOI] [PubMed] [Google Scholar]
- Wang Z, et al. Domestication relaxed selective constraints on the yak mitochondrial genome. Mol Biol Evol. 2011;28:1553–1556. doi: 10.1093/molbev/msq336. [DOI] [PubMed] [Google Scholar]
- White EP, Ernest SKM, Kerkhoff AJ, Enquist BJ. Relationships between body size and abundance in ecology. Trends Ecol Evol. 2007;22:323–330. doi: 10.1016/j.tree.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Wilson GP, et al. Adaptive radiation of multituberculate mammals before the extinction of dinosaurs. Nature. 2012;483:457–460. doi: 10.1038/nature10880. [DOI] [PubMed] [Google Scholar]
- Woolfit M, Bromham L. Increased rates of sequence evolution in endosymbiotic bacteria and fungi with small effective population sizes. Mol Biol Evol. 2003;20:1545–1555. doi: 10.1093/molbev/msg167. [DOI] [PubMed] [Google Scholar]
- Woolfit M, Bromham L. Population size and molecular evolution on islands. Proc Biol Sci. 2005;272:2277–2282. doi: 10.1098/rspb.2005.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SD, Gillman LN, Ross HA, Keeling DJ. Slower tempo of microevolution in island birds: implications for conservation biology. Evolution. 2009;63:2275–2287. doi: 10.1111/j.1558-5646.2009.00717.x. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang J. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol. 2000;50:56–68. doi: 10.1007/s002399910007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.