

Abstract
Traditional culturing methods take a long time for identification of pathogenic isolates. A protocol has been developed for the detection of Fusarium from soil samples in the early stage of infection. Seventeen soil samples from different locations were collected before the onset of rains to find out the presence of Fusarium spp. population present in the soil of guava orchards and to correlate its presence with incidence of wilt. A PCR based method was developed for the molecular characterization of Fusarium using Fusarium spp. specific primer. DNA extracted by this method was free from protein and other contaminations and the yield was sufficient for PCR amplification. The primer developed in this study was amplifying ∼230 bp in all infected samples while not in healthy soil. The specificity and sensitivity of primer were tested on several Fusarium spp. and found that this primer was amplifying 10−6 dilution of the fungal DNA. The present study facilitates the rapid detection of Fusarium spp. from infected soil samples of guava collected from different agroclimatic regions in India. A rapid detection method for pathogens and a diagnostic assay for disease would facilitate an early detection of pathogen and lead to more effective control strategies.
Keywords: Culture independent PCR, Fusarium species, Guava wilt, ITS sequences
1. Introduction
Guava (Psidium guajava Linn.), is considered as nutrient rich sources for humans globally as it contains vitamin C, pectin, calcium, phosphorous and trace elements. It has been grown in all regions of India while good quality of guava is produced in Allahabad, Uttar Pradesh.
Due to the wide occurrence of microbial pathogens the production is now decreasing drastically as about 177 different pathogens including fungi, bacteria, algae, nematodes and epiphyte, causing various pre and post-harvest diseases, are reported on various parts of guava plant (Misra and Prakash, 1990). Fusarium spp., one of the most important pathogens which causes wilt disease of guava (P. guajava L.) is a major threat to guava cultivation (Misra and Pandey, 1996; Misra, 2006). Varied chemical and non-chemical control measures have been applied to control the Fusarium spp., which has resulted in heterogeneity among the isolates (Misra, 2006; Misra and Gupta, 2007). Fusarium is a cosmopolitan soil borne fungus that colonizes into the vascular system of the host plant and thereby blocks the movement of water to the upper part of the host plant, which in turn causes yellowing, wilting and finally death of the host plant. It is very difficult to manage this disease in the early infection of the pathogen because symptoms are not visualized during the early stage of infection (Pandey et al., 2010). But recently molecular tools have become valuable for specific detection of the pathogen in the early stage of infection and analyzing of microbial populations or communities (Kumar and Anandaraj, 2006; Louws et al., 1999). Specific taxonomic groups can be identified and detected by using nucleic acid probes without culturing microbes. Previously, studies on the development of microbial communities required their isolation from soil samples, followed by a series of morphological and biochemical tests to identify them. Furthermore, culture dependent community structure analysis produces spatial and heavily biased results (Fatima et al., 2011). Molecular techniques allow to access the metabolic potential of microorganisms via the isolation of DNA from environmental samples, i.e., without the application of microbial culture techniques such as PCR using pathogen specific probes or oligo primers to detect the pathogens (Louws et al., 1999).
Various procedures for extracting microbial DNA from soil have been reported and these techniques employ extensive purification steps to ensure that the DNA is suitable for PCR (Tsai and Olson, 1991; Holben, 1994; Zhou et al., 1996; Miller et al., 1999; Roose-Amsaleg et al., 2001). Thus, the selection of an appropriate DNA extraction and purification procedure remains a major problem in the application of molecular techniques for studying of soil and sediment microbial communities.
In the present investigation an attempt was made to isolate fungal DNA from soil and to detect guava wilt pathogen ‘Fusarium spp.’ in soil using specific primer. We described the development of PCR primer derived from ITS sequences for the specific detection of Fusarium spp. from soil. The specificity and sensitivity of the reaction were tested on a range of wild Fusarium species. The sensitivity of the PCR assay was determined, and the PCR protocols were tested for their ability to detect Fusarium in diseased soil samples and Fusarium isolates collected in the field.
2. Materials and methods
2.1. Sample collection and maintenance
Seventeen soil samples from different locations (Table 1) were collected before the onset of rains to find out the presence of Fusarium spp. population present in the soil of guava orchards and to correlate its presence with incidence of wilt. Fusarium spp. could be isolated from all the locations. All isolates were stored on potato dextrose agar (PDA) at 4 °C and maintained in collection at the Department of Molecular Plant Pathology, Central Institute for Subtropical Horticulture, Rehmankhera, Lucknow, UP, India.
Table 1.
Soil samples collected from different guava orchards.
| Sample ID | Location | Wilt (%) |
|---|---|---|
| W-1 | 1st Block, CISH, Lucknow (UP) | 70–80 |
| W-2 | 3rd Block, CISH, Lucknow (UP) | 50–70 |
| W-3 | 3rd Block, CISH, Lucknow (UP) | 100 |
| W-4 | RB road campus, CISH, Lucknow (UP) | 70–75 |
| W-5 | RB road campus, CISH, Lucknow (UP) | 100 |
| W-6 | RB road campus, CISH, Lucknow (UP) | 100 |
| W-7 | Puskar (Rajasthan) | 100 |
| W-8 | Puskar (Rajasthan) | 80–90 |
| W-9 | Puskar (Rajasthan) | 100 |
| W-10 | Muzaffarnagar (UP) | 100 |
| W-11 | Muzaffarnagar (UP) | 100 |
| W-12 | Bihar sample (Bihar) | 90 |
| W-13 | Bihar sample (Bihar) | 100 |
| W-14 | Allahabad sample (UP) | 60–70 |
| W-15 | Allahabad sample (UP) | 100 |
| W-16 | Meadow orchard CISH, Lucknow | 100 |
| W-17 | Meadow orchard CISH, Lucknow | 100 |
2.2. DNA extraction method
DNA was prepared by the modified method of Fatima et al. (2011). Approximately 500 mg of soil samples were suspended in 0.5 ml DNA extraction buffer containing 200 mM Tris–HCl (pH 8.0), 0.02 M Na2EDTA (pH 8.0), 5 M NaCl, 10% SDS, 10% CTAB, 10 μl of Proteinase K (10 mg/ml) and 1 M mannitol in centrifuge tubes and incubated at 65 °C in a water bath for 1 h with occasional stirring and homogenizing the slurry horizontally at 37 °C on a vortex mixture for 10 min. This was followed by centrifugation at 12,000 rpm for 15 min at 4 °C. The supernatant was extracted with an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1) followed by centrifugation at 12,000 rpm at 4 °C. Aqueous layer of PCI was precipitated with 1/10th volume of 3 M sodium acetate (pH 5.2) and 2 volumes of ethanol and the pellet was recovered by centrifugation at 12,000 rpm and dried, dissolved in 50 μl of sterile water or 1× TE buffer (10 mM Tris–HCl and 1 mM EDTA, pH 8.0), and used as a template for PCR amplification or stored at −20 °C until use.
2.3. Quantification of DNA
The concentration of the DNA was determined by following the absorbance at 260 nm (Sambrook et al., 1989). Sample dilution was adjusted to get the absorbance between 0.1 and 1.0. The ratio of the readings at 260 nm and 280 nm provides an estimate of the purity of DNA with respect to contaminants that absorb UV.
2.4. PCR amplification of 18S rDNA regions
The 18S rDNA regions were amplified using 18SF (5′-ATTGGAGGGCAAGTCTGGTG-3′) and 18SR (5′-CCGATCCCTAGTCGGCATAG-3′) primer pair (Einsele et al., 1997). Amplification was carried out in 25 μl reaction mixture containing 2.5 μl of 1× PCR buffer, 2.5 mM of MgCl2, 0.5 mM of each dNTPs, 0.5 μM of each primers (10 pmol), 1.25U of Taq polymerase (Fermentas), 5% (v/v) of DMSO (Sigma–Aldrich Inc. USA) and 1 μl (1:10 dilution) of community DNA. Amplification was performed with an Eppendorf Thermal Cycler in a program comprising of 34 cycles of denaturation at 94 °C for 60 s, annealing at 53 °C for 60 s, and extension at 72 °C for 1.5 min with an initial denaturation of 5 min at 94 °C before cycling and final extension of 5 min at 72 °C after cycling. The PCR products were analyzed by 1.2% agarose gel electrophoresis.
2.5. Taxon specific primer designing
Prior to primer designing, the core parameters used in the primer design include the following: (1) the primer length is between 18 bp and 25 bp, (2) the percentage of GC is between 35% and 60%, (3) the Tm of the primers is over 40 °C, which was calculated using standard PCR conditions. Species-specific primers were designed using CLUSTALW based on the DNA sequence retrieve from the NCBI database. The specificity of the primers for Fusarium spp. was also validated through BLAST searching the primer sequences against the NCBI sequence database (http://www.ncbi.nlm.nih.gov/BLAST).
2.6. Specific detection of Fusarium species
PCR amplification for detection of Fusarium spp. in soil was performed using the DNA isolated from soil as template. Reaction volume 25 μl contains 2.5 μl of 1× PCR buffer, 2.5 mM of MgCl2, 0.5 mM of each dNTPs, 5% (v/v) of DMSO (Sigma–Aldrich Inc. USA), 0.5 μM of each primers ITS1F (5′-CCAGAGGACCCCCTAACTCT-3′) and ITS1R (5′-GCCTGAGGGTTGTAATGACG-3′), 1.25U of Taq polymerase (Fermentas) and 1 μl (1:10 dilution) of community DNA. Amplification was performed with an Eppendorf Thermal Cycler. PCR was performed by 34 cycles of denaturation at 94 °C for 60 s, annealing at 52 °C for 45 s, and extension at 72 °C for 60 s with an initial denaturation of 5 min at 94 °C before cycling and final extension of 5 min at 72 °C after cycling. The gel was stained with Ethidium bromide and photographed on an UV transilluminator.
3. Results and discussion
3.1. Survey of wilt disease
A total of 17 soil samples were collected from different agroecological regions in India. Out of 17 samples, six samples were selected due to some of samples from same regions thus, it can be considered as same soil types and agroclimatic conditions. The wilt diseases were periodically recorded and Fusarium oxysporum f. sp. psidii were identified from all the locations (Mishra et al., 2012). The percentage of wilt symptoms in guava were periodically recorded and given in Table 1 (Misra and Pandey, 2000).
3.2. DNA extraction and PCR amplification of 18S rDNA
The main objective of soil DNA extraction is that the soil DNA should be free from those PCR inhibitors or the concentration of those inhibitors must be low enough so that they do not interfere in the activity of DNA polymerase used in PCR. Soil DNA when isolated directly would accumulate impurities from soil that are potential inhibitors of restriction enzymes or polymerase enzyme (Tsai and Olson, 1992). DNA extraction from soil has three requirements: extraction of high molecular weight DNA; extraction of DNA free from inhibitors for subsequent molecular biological manipulations to be performed; and representative lysis of microorganisms within the sample (Yeates et al., 1998). The extracted DNA from six soil samples were used for PCR amplification and also obtained 500 bp bands in all samples using universal primers based on 18S rDNA sequences (Figs. 1 and 2). No variations were observed in PCR products of these samples. A series of dilutions that contain the 500 bp DNA template were made to evaluate the sensitivity of the universal primer. All the diluted templates gave the target band compared to the negative (blank) control.
Figure 1.
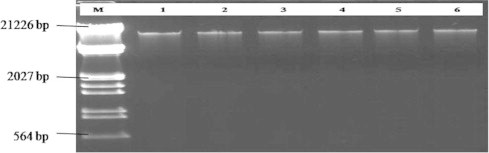
DNA extracted from six soil samples of wilted guava plant, Lane M: DNA size marker (DNA double digested with EcoRI and Hind III) (Fermentas). Lanes 1–6: Soil samples.
Figure 2.
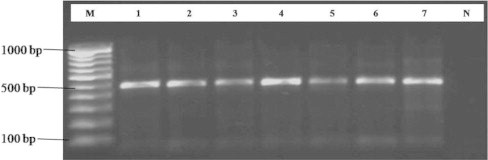
18S rDNA PCR amplification of uncultured fungi, Lane M: 100 bp DNA Ladder (Fermentas), Lanes 1–6 soil samples, Lane 7: Positive control, Lane N: Negative control.
3.3. Quantification and digestion of soil DNA by TaqI and MspI
The protocol standardized in the present work yielded DNA with A260/280 ratio ranging from 1.53 to 2.14 and A260/230 ratio ranging from 1.27 to 1.97 (Table 2). A high 260/230 ratio (>2) is indicative of pure DNA, while a low ratio is indicative of humic acid contamination. The yield of soil DNA was ranging from 19.20 to 40.82 μg/g of soil samples by this method which proves an efficient extraction method. None of the crude extracts could be digested by EcoR I or Hind III. Furthermore, although a reduction in the brownish color of the crude DNA was observed after gel filtration, this DNA still could not be digested by EcoR I or Hind III (Tien et al., 1999). As we assume that there is enough purity in community DNA thus, it can be completely digested by EcoR I or Hind III. It may be the high concentration of humic acid in soils that may inhibit restriction enzymes. A total of six community DNAs were digested using two restrictions such as EcoR I and Hind III. It was completely digested, and indicated that these DNA samples were highly pure (Fig. 3a and b).
Table 2.
Amount and purity of DNA extracted from different soil samples.
| Sample ID | Purity |
Yield of DNA (μg/g) | |
|---|---|---|---|
| A260/A280 | A260/A230 | ||
| W-1 | 1.84 ± 0.07 | 1.27 ± 0.00 | 26.56 |
| W-2 | 1.86 ± 0.05 | 1.97 ± 0.02 | 40.82 |
| W-3 | 1.87 ± 0.05 | 1.91 ± 0.07 | 35.62 |
| W-4 | 1.85 ± 0.04 | 1.95 ± 0.00 | 38.64 |
| W-5 | 1.53 ± 0.01 | 1.78 ± 0.01 | 33.03 |
| W-6 | 2.13 ± 0.12 | 1.37 ± 0.02 | 19.20 |
Figure 3.
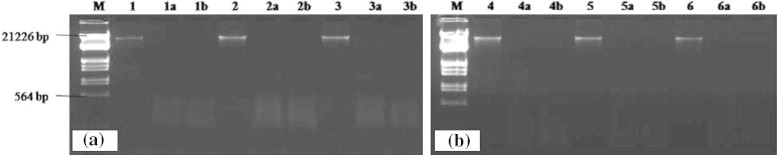
(a and b) Lane M: DNA size marker (DNA double digested with EcoRI and Hind III) (Fermentas), Lanes 1, 2, 3, 4, 5, 6 undigested soil DNA, Lanes la, 2a, 3a, 4a, 5a, 6a, TaqI digested soil DNA and Lanes 1b, 2b, 3b, 4b, 5b, 6b, soil DNA digested with MspI.
The yield of DNA ranging from 19.20 to 40.82 μg/g of soil samples by this method therefore proved an efficient extraction method like described by Roh et al. (2006). The humic materials in soil have similar size and charge characteristics to DNA resulting in their co-purification, evident by the extractions being brown in color (Holben, 1994). Humic contaminants also interfere in DNA quantification since this exhibit absorbance at both 230 nm and at 260 nm, the latter used to quantitate DNA (Liesack et al., 1997; Olson, 1992). Zhou et al. (1996) have reported 0.91 and 1.35 for A260/230 and A260/280, respectively. While More et al. (1994) reported soil DNA yield as high as 11.8 and 5.2 μg g−1 in the bead beating and freeze thawing methods, respectively. The bead beating direct lysis method described by Yeates et al. (1998) yielded DNA between 15 and 23.5 μg/g of soil. Extraction methods using small soil samples ranging from 5 to 100 mg of soil have extracted 9–25 μg/g (Porteous and Armstrong, 1991), 12 μg/g (Tsai and Olson, 1992), 1–100 μg/g (Porteous et al., 1994) and 2.5–26.9 μg/g of soil (Zhou et al., 1996).
3.4. Specific detection of Fusarium spp. in soil samples
PCR-based assays have already been applied to microbial ecology and environmental sciences for detection and monitoring of microorganisms in rhizosphere, soils and diagnose plant diseases. In the present study, culture independent PCR techniques were used for the specific detection of Fusarium species. Out of 17 soil samples, six were selected from different locations (Table 1) and used to evaluate the specificity and sensitivity of the newly developed primer.
Among these samples one of them (W-1) was collected from the rhizosphere of partially wilted host plant that shows wilt symptoms and five of them (W-3, W-5, W-7, W-10 and W-13) were collected from the rhizosphere of completely wilted host plant. All six samples produced the target amplicon size of 230 bp (Fig. 4) which showed the presence of Fusarium spp. in the soil. Specificity and sensitivity of the primer are evaluated with several Fusarium isolates. The predicted amplicon size of 230 bp amplifies in all Fusarium isolates. No amplification was observed in control sample (Fig. 4). To determine the sensitivity of the newly designed primers based on ITS region they were tested with serial dilutions of genomic DNA from cultured Fusarium species. This PCR amplification was obtained up to 10−6 dilutions of DNA sample. ITS region was sequenced and found the homology with F. oxysporum (Mishra et al., 2012; Pandey et al., 2010). According to the results we can make a conclusion that this primer is sensitive enough to produce the target fragment even from one copy of the template DNA.
Figure 4.
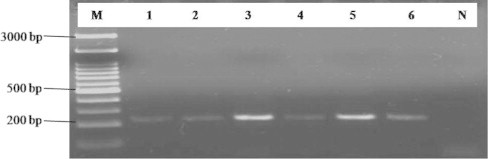
Specificity of PCR based assay for the detection of Fusarium spp. in soil samples. Lane M: 100 bp DNA Ladder (Biochem), Lane 1: W-1, Lane 2: W-3, Lane 3: W-5, Lane 4: W-7, Lane 5: W-10, Lane 6: W-13 soil samples, Lane N: control (DNA from healthy plant).
According to the specific detection results we can say that the primer we used was sensitive enough to detect the infection of Fusarium spp. in soil. Specificity and sensitivity of the primer pair were investigated using Fusarium isolates from these soil samples and we found 230 bp amplicon in all the Fusarium isolates tested. DNA detection tests from soil have been developed for several pathogens like Phytophthora species (Stammler and Seemuller, 1994; Grote et al., 2002; Hussain et al., 2005; Bilodeau et al., 2007), Ralstonia solanacearum (Kumar and Anandaraj, 2006), Phytophthora cinnamomi (O’Brien, 2008), Histoplasma capsulatum (Reid and Schafer, 1999) and Agrobacterium tumefaciens (Yang et al., 2011).
In present study, all these community DNAs were completely digested. Similarly, a number of reports are available on humic acids that pose a considerable problem and interfere in enzymatic manipulations of DNA and various protocols have been published in the past for the successful isolation of PCR amplifiable DNA from soil (Yeates and Gillings, 1998; Amorim et al., 2008). Due to the low level of contamination, DNA extracted by this method was easily digested by restriction enzymes TaqI and MspI and this DNA can be successfully employed for metagenomic studies of soil from wilted guava plant.
4. Conclusion
The PCR assay reported here is a sensitive, specific, efficient, very convenient and easy to be developed in actual diagnosis technique for detecting Fusarium spp. from the soil. Because this PCR assay also has significant practical applications in the detection of Fusarium infected soils as well as isolates, it constitutes a powerful tool for controlling the dispersion of Fusarium and developing disease control strategies. Detection of Fusarium spp. from soil is a time consuming process by traditional isolation methods, which can delay disease management decisions. The PCR detection method reported here can provide a definitive diagnosis of this pathogen in soils within hours, and can be used to more accurately survey the occurrence and distribution of the pathogen in soil. Moreover, the molecular markers developed in this study have potential to overcome the problems associated with existing methods and can even be used by non-expert biologists.
Acknowledgements
We are thankful to Director and Head, Division of Crop Protection, Central Institute for Subtropical Horticulture, Rehmankhera, Lucknow for their encouragement and support. We gratefully acknowledged the Indian Council of Agricultural Research for Financial support in the form of Senior Research Fellowship and Vice Chancellor, Integral University for necessary facilities.
Footnotes
Peer review under responsibility of King Saud University.

References
- Amorim J.H., Macena T.N.S., Lacerda-Junior G.V., Rezende R.P., Dias J.C.T., Brendel M., Cascardo J.C.M. An improved extraction protocol for metagenomic DNA from a soil of the Brazilian Atlantic Rainforest. Genet. Mol. Res. 2008;7:1226–1232. doi: 10.4238/vol7-4gmr509. [DOI] [PubMed] [Google Scholar]
- Bilodeau G.J., L´evesque C.A., de Cock A.W.A.M., Duchaine C., Bri‘ere S., Uribe P., Martin F.N., Hamelin R.C. Molecular detection of Phytophthora ramorum by real-time polymerase chain reaction using TaqMan, SYBR Green, and molecular beacons. Phytopathol. 2007;97:632–642. doi: 10.1094/PHYTO-97-5-0632. [DOI] [PubMed] [Google Scholar]
- Einsele H., Hebart H., Roller G., Loffler J., Rothenhofer I., Muller C.A., Bowden R.A., van Burik J., Engelhard D., Schumacher U., Kanz L. J. Clin. Microbiol. 1997;35:1353. doi: 10.1128/jcm.35.6.1353-1360.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima F., Chaudhary I., Ali J., Rastogi S., Pathak N. Microbial DNA extraction from soil by different methods and its PCR amplification. Biochem. Cell. Arch. 2011;11:1. [Google Scholar]
- Grote D., Olmos A., Kofoet A., Tuset J., Bertolini E., Cambra M. Specific and sensitive detection of Phytophthora nicotianae by simple and nested-PCR. Eur. J. Plant Pathol. 2002;108:197–207. [Google Scholar]
- Holben W.E. Isolation and purification of bacterial DNA from soil. Soil Sci. 1994;5:727–751. [Google Scholar]
- Hussain S., Lees A.K., Duncan J.M., Cooke D.E.L. Development of a species-specific and sensitive detection assay for Phytophthora infestans and its application for monitoring of inoculums in tubers and soil. Plant Pathol. 2005;54:373–382. [Google Scholar]
- Kumar A., Anandaraj M. Method for isolation of soil DNA and PCR based detection of ginger wilt pathogen, Ralstonia solanacearum. Ind. Phytopathol. 2006;59:154–160. [Google Scholar]
- Liesack W.P., Janssen H., Rainey F.A., Ward-Rainey N.L., Stackebrandt E. Microbial diversity in soil: the need for a combined approach using molecular and cultivation techniques. Mod. soil Microbiol. 1997:375–439. [Google Scholar]
- Louws F.J.W., Rademaker J.L., de Bruijn F.J. The three D’s of PCR based genomic analysis of phytobacteria: diversity, detection, and disease diagnosis. Ann. Rev. Phytopathol. 1999;37:81–125. doi: 10.1146/annurev.phyto.37.1.81. [DOI] [PubMed] [Google Scholar]
- Miller D.N., Bryant J.E., Madsen E.L., Ghiorse W.C. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 1999;65:4715–4724. doi: 10.1128/aem.65.11.4715-4724.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A.K., Gupta V.K. Variability in Fusarium solani-a causal organism of wilt of guava. CISH Newsl. 2007;8:2. [Google Scholar]
- Misra A.K. Wilt of guava-a disease of national importance. Ind. Phytopathol. 2006;59:269–280. [Google Scholar]
- Misra A.K., Pandey B.K. Disease Scenario in Crop Plants. International Books and Periodical Supply Service; New Delhi: 1996. Present status of wilt disease of guava; pp. 61–70. [Google Scholar]
- Misra, A.K., Prakash, O.M., 1990. Guava diseases – (an annotated bibliography 1907–1990). Bishen Singh Mahendra Pal Singh, Dehradun, 132p.
- Misra A.K., Pandey B.K. Progressive natural wilting of guava plants during different months. Ind. Phytopathol. 2000;53:423–427. [Google Scholar]
- Mishra, R.K., Pandey, B.K., Pandey, A., Mohd. Zeeshan., Pathak N., 2012. Specific detection of Fusarium oxysporum f. sp. psidii (Fop) causing wilt disease of Psidium guajava L. in India. In: National Symposium on Microbes in Health and Agriculture, JNU, New Delhi, pp. 87.
- More M.I., Herrick J.B., Silva M.C., Ghiorse W.C., Madsen E.L. Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl. Environ. Microbiol. 1994:1572–1580. doi: 10.1128/aem.60.5.1572-1580.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien P.A. PCR primers for specific detection of Phytophthora cinnamomi. Aust. Plant Pathol. 2008;37:69–71. [Google Scholar]
- Olson B.H. Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl. Environ. Microbiol. 1992;58:2292–2295. doi: 10.1128/aem.58.7.2292-2295.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey B.K., Mishra R.K., Pandey A., Kamle M., Sareen P., Muthukumar M. National Symposium on Molecular Approaches for Fungal Diseases of Crop Plants. IIHR; Bangalore, Karnataka: 2010. Designing and validation of primers aided through bioinformatics tools for molecular genetic diversity assessment of Guava wilt Pathogen; p. 159. [Google Scholar]
- Porteous L.A., Armstrong J.L. Recovery of bulk DNA from soil by a rapid, small-scale extraction method. Curr. Microbiol. 1991;22:345–348. [Google Scholar]
- Porteous L.A., Armstrong J.L., Seidler R.J., Watrud L.S. An effective method to extract DNA from environmental samples for polymerase chain reaction amplification and DNA fingerprint analysis. Curr. Microbiol. 1994;29:301–307. doi: 10.1007/BF01577445. [DOI] [PubMed] [Google Scholar]
- Reid T.M., Schafer M.P. Direct detection of Histoplasma capsulatum in soil suspensions by two-stage PCR. Mol. Cell Probes. 1999;13:269–273. doi: 10.1006/mcpr.1999.0247. [DOI] [PubMed] [Google Scholar]
- Roh C., Villatte F., Kim B.G., Schmid R.D. Comparative study of methods for extraction and purification of environmental DNA from soil and sludge samples. Appl. Biochem. Biotechnol. 2006;134:97–112. doi: 10.1385/abab:134:2:97. [DOI] [PubMed] [Google Scholar]
- Roose-Amsaleg C.L., Garnier-Sillam E., Harry M. Extraction and purification of microbial DNA from soil and sediment samples. Appl. Soil Ecol. 2001;18:47–60. [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. 2nd ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. Molecular cloning: a laboratory manual. [Google Scholar]
- Stammler, G., Seemuller, E., 1994. Detection of Phytophthora fragariae var. rubi in infected raspberry roots by PCR. In: Modern Assays for Plant Pathogenic Fungi, pp. 135–139.
- Tien C.C., Chao C.C., Chao W.L. Methods for DNA extraction from various soils: a comparison. J. Appl. Microbiol. 1999;86:937–943. [Google Scholar]
- Tsai Y.L., Olson B.H. Rapid method for direct extraction of DNA from soil and sediments. Appl. Environ. Microbiol. 1991;57:1070–1074. doi: 10.1128/aem.57.4.1070-1074.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y.L., Olson B.H. Detection of low numbers of bacterial numbers in soils and sediments by polymerase chain reaction. Appl. Environ. Microbiol. 1992;58:754–757. doi: 10.1128/aem.58.2.754-757.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Wei, Lei Ji, Li-Rong Tan, Shi-Mo Li, Yan Wang, Hong-Xia Liu, Yu-Ming Luo. Sensitive and specific detection of Agrobacterium tumefaciens in soil using a rapid polymerase chain reaction (PCR) Afr. J. Microbiol. Res. 2011;5:708–713. [Google Scholar]
- Yeates C., Gillings M.R. Rapid purification of DNA from soil for molecular biodiversity analysis. Lett. Appl. Microbiol. 1998;27:49–53. [Google Scholar]
- Yeates C., Gillings M.R., Davison A.D., Altavilla N., Veal D.A. Methods for microbial DNA extraction from soil for PCR amplification. Biol. Proced. Online. 1998;1:40–47. doi: 10.1251/bpo6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Bruns M.A., Tiedje J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996;62:316–322. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]