

Abstract
Despite advances in HIV therapy, viral resistance and side-effects with current drug regimens require targeting new components of the virus. Dual antagonist peptide triazoles (PT) are a novel class of HIV-1 inhibitors that specifically target the gp120 component of the viral spike and inhibit its interaction with both of its cell surface protein ligands, namely the initial receptor CD4 and the co-receptor (CCR5/CXCR4), thus preventing viral entry. Following an initial survey of 19 gp120 alanine mutants by ELISA, we screened 11 mutants for their importance in binding to, and inhibition by the PT KR21 using surface plasmon resonance. Key mutants were purified and tested for their effects on the peptide’s affinity and its ability to inhibit binding of CD4 and the co-receptor surrogate mAb 17b. Effects of the mutations on KR21 viral neutralization were measured by single-round cell infection assays. Two mutations, D474A and T257A, caused large-scale loss of KR21 binding, as well as losses in both CD4/17b and viral inhibition by KR21. A set of other Ala mutants revealed more moderate losses in direct binding affinity and inhibition sensitivity to KR21. The cluster of sensitive residues defines a PT functional epitope. This site is in a conserved region of gp120 that overlaps the CD4 binding site and is distant from the co-receptor/17b binding site, suggesting an allosteric mode of inhibition for the latter. The arrangement and sequence conservation of the residues in the functional epitope explain the breadth of antiviral activity, and improve the potential for rational inhibitor development.
Keywords: entry inhibitor, KR21, dual antagonism, binding site
INTRODUCTION
HIV-1 (hereafter referred to as HIV) is the causative agent of Acquired Immune Deficiency Syndrome (AIDS), a fatal disease that compromises the immune system. Thanks to highly active antiretroviral therapy (HAART), a drug cocktail targeting different components of the virus life cycle, patients can live symptom-free, as long as they have access to treatment. Hence the death rate has reduced since HAART was introduced in 1995 [1]. Nonetheless, HAART is not a cure, and the number of people living with HIV-1 worldwide remains at a very high level, 34 million in 2010 [1]. Moreover, occasional toxicity and resistance occur with HAART [2, 3]. Thus, in spite of the success of HAART, there is continuing need for inhibitors of new targets in fighting HIV. Even with protease inhibitors and 2 classes of reverse transcriptase inhibitors currently used in combination therapy, virologic failure occurs [4, 5] when patients’ virus develops resistance to the drug regimen [5].
Other drugs approved for HIV therapy include the entry inhibitors T-20 and maraviroc. Despite the advantage of entry inhibitors in stopping viral infection at the earliest steps of host cell exposure, both entry inhibitors in the market have drawbacks. Resistance to T-20 develops relatively easily. In addition, both it and its newer derivatives such as T-1249 suffer from injection site and other adverse reactions [5]. Maraviroc is an orally-bioavailable small molecule that blocks the cellular HIV co-receptor CCR5 and as such is not threatened by changes in its target; however, it is only effective against solely CCR5-utilizing virus, and not CXCR4-utilizing or dual-tropic virus strains which make up the virus in about half of the treatment resistant patient population [4]. Moreover, treatment with maraviroc can lead to emergence of resistant viruses with altered tropism in patients, and to an increased incidence of upper respiratory tract infections [4]. Raltegravir is another small molecule that inhibits the HIV integrase, and is preferable to T-20 for patients due to its oral route of delivery [6]; however, it was recently found to be vulnerable to significant resistance-conferring mutations that emerge during periods of low viral load [7]. All three additions to the anti-HIV armamentarium are only approved for treatment-experienced patients as an addition to the optimized background regimen (OBR), and not approved for treatment-naïve patients.
The peptide triazoles (PT) represent a novel, developing class of entry inhibitors. These agents target and antagonize the HIV Envelope (Env) component gp120. PTs were derived from a 12-residue scaffold that was discovered in a phage display library screen in 1999 [8] to bind the HIV viral envelope component gp120 with micromolar affinity. PTs are formed by Cu(I) catalyzed 3+2 cycloaddition (click chemistry) of alkynes on azidoproline introduced synthetically into the original peptide [9]. They bind gp120 and inhibit its interaction of gp120 with both the cell surface receptor CD4 and the co-receptor surrogate mAb 17b with low nanomolar KDs and IC50s [10], respectively. PTs were found to neutralize virus infection of cells; this inhibition was shown to be effective against a broad range of virus subtypes in both single round and well as fully infectious virus assays [11]. Some variants of peptide triazoles have been found to cause specific HIV-1 lytic inactivation [12].
Given the increasing evidence for strong potency, breadth of action and virus inactivation potential of PTs, we sought in the current study to determine the functional site for peptide triazole binding and receptor site antagonism on gp120. We tested residues on gp120 selected based on the following criteria: (1) residues implicated as important for inhibition by parent peptide 12p1 [13] and follow-up peptides HNG-105 and HNG-156 (Zentner, Madani, personal communication); (2) solvent-exposed and hydrophobic residues around the 12p1 site, including those within the CD4 F43 binding cavity based on the importance of hydrophobic elements in the PT sequence; (3) residues in depressions near the putative binding site. We made alanine mutations of the selected residues and characterized the effects of the mutations on binding to, and inhibition by, the PT KR21.
MATERIALS AND METHODS
Peptide Triazole Production and Validation
Peptide Synthesis
Peptides used for direct binding, inhibition and neutralization studies on gp120 and virus containing mutations in the putative peptide site were synthesized, conjugated and purified in-house.
Reagents were purchased from Sigma-Aldrich Co. LLC. unless otherwise stated. KR21 and HNG-156 were synthesized by solid phase peptide synthesis as done previously in our laboratory [10]. Briefly, 0.25 mMol capacity Rink amide resin was swelled by dichloromethane, washed with dimethylfluoride (DMF), and the Fmoc protection for groups on the amines of the resin were cleaved by half-hour incubation with 30% piperidine in DMF. Resin was washed with DMF and 1 mMol Fmoc-protected amino acid (C-terminus in sequence) in 1:1 mixture of 1 mMol N-Hydroxybenzotriazole (HOBt)/O-Benzotriazole-N, N, N′, N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) in N-methylpyrrolidone (NMP) was added, and kept shaking in the synthesis syringe (Torvig) for 2 hours (HOBt and HBTU were purchased from Chem-Impex International Inc). Following coupling, the reaction syringe was washed with DMF, and completion of coupling was monitored by Kaiser test (addition of 3 drops each of 5% (w/v) ninhydrin in ethanol, phenol in ethanol (40 g in 10ml) and 0.02mM KCN (0.5 ml of 2mM in water in 49 ml pyridine) to resin sample and 1 min incubation at 95°C, blue beads indicate free amines). Fmoc protection groups on the amino termini were removed by incubation in 30% piperidine in DMF for 10 minutes. The next (N-terminal) amino acid was added at 1 mM scale in HOBt/HBTU in NMP. The above steps were repeated for addition of each subsequent amino acid. Completion of coupling was monitored by Kaiser test except for addition of isoleucine after azidoproline which cannot be distinguished from a deprotected (azido)proline by Kaiser test and thus was simply repeated. Synthesized peptides were conjugated to ethynylferrocene (Sigma-Aldrich) by 3+2 cycloaddition (click chemistry) on the azidoproline as done previously in our lab [14] and cleaved from resin using a cocktail of (95:2:2:1) trifluoroaceticacid:ethanedithiol:water:thioanisole. Cleaved peptide slurry was cleaned by addition of ice-cold diethylether, reconstituted in water and purified by RP-HPLC using a C18 semiprep column (Phenomenex) run by a Beckman-Coulter System Gold 168 HPLC system. Isolated peptide peaks were freeze-dried using a lyophilizer (Labconco) and submitted to MALDI-TOF analysis (Wistar Institute, Philadelphia, PA). Azidoproline was produced in-house from the commercially available methyl ester of tert-butoxycarbonyl-4-hydroxyproline (Boc-Hyp-OMe) as done previously [9]. All other amino acids were purchased as Fmoc-protected species from Chemimpex Inc. (Wood Dale, IL).
Peptide verification by CD4 and 17b inhibition ELISA
Dual-antagonism and potency of the peptide triazoles were verified by CD4 and 17b inhibition ELISA.
For CD4 inhibition assays 100 ng soluble 4-domain CD4 (hereafter referred to as CD4) was adsorbed per well of a 96-well high-binding plate (Corning) in PBS. gp120 purified by F105 or nickel affinity was mixed with KR21 to yield final gp120 concentration of 10 nM and KR21 concentration between 1 nM and 10 μM and incubated with the adsorbed CD4 for 1 hr at room temperature. Bound gp120 was detected with anti-gp120 mAb D7324, rabbit anti-sheep HRP mAb (eBiosciences) and OPD reagent (Sigma) at 450 nm in a Microdevices microplate reader as per standard ELISA protocols. Data were plotted as percent binding of uninhibited (no KR21) and IC50s were calculated by fitting to a 4-parameter sigmoidal model in Origin 7.0 (OriginLab) as described under “Inhibition of CD4 binding to gp120 mutants by KR21”.
For 17b inhibition experiments 100 ng gp120 was adsorbed to the wells and 5 nM 17b IgG was added in solution with and without KR21. Detection was done with anti-human HRP mAb (Ebiosciences) and OPD reagent as described above. Data analysis was done similarly to that for CD4 inhibition.
Protein reagent production
Proteins used in inhibition experiments were expressed and purified in-house or prepared from commercially available IgG.
Hexahis-tagged 4-domain soluble CD4 (CD4) was produced by transient transfection into 293F cells using standard protocols (Gibco). The pcDNA3.1 vector carrying CD4 was a gift from Dr. Navid Madani. CD4 was separated from the expression medium by Nickel affinity purification on HiTrap columns (GE) using an Akta FPLC System (GE). CD4 was further purified by size-exclusion on a Superdex 200 column (GE). Protein size and functionality were verified by SDS-PAGE and anti-gp120 ELISA, respectively.
17b IgG was purchased from Strategic Diagnostics Inc (Newark, DE).
17b Fab was produced in-house from 17b IgG by papain cleavage and protein A negative purification (Pierce Fab preparation kit, Thermo Scientific) and size exclusion on a Superdex 200 column (GE).
Production of wild type and mutant HIV-1YU2gp120
gp120 and its mutants used in direct binding and CD4 inhibition screens contained a V5 epitope tag to allow SPR on-chip capture and thus were used directly as expression supernatant. Select mutants were purified using 17b affinity and tested by direct immobilization on SPR surfaces to obtain absolute KDs and IC50s for KR21.
The wild-type (WT) gp120YU2 construct was created by insertion of a V5 (GKPIPNPLLGLDST) coding sequence N-terminal to the C-terminal hexahis tag in a pcDNA3.1 vector carrying the mammalian codon optimized coding sequence for a CM5 secretion peptide and gp120YU2 (a gift from Drs. Navid Madani and Joseph Sodroski). An Age1 cleavage site was introduced before the hexahis epitope tag coding sequence of gp120 with the forward primer 5′-cagagcgagaagaccggtcaccaccaccaccaccactg-3′ and its reverse complement. We utilized the HindIII recognition site already present in the vector 5′ to the hexahis tag to insert the V5-tag coding sequence which contained ends complementary with Age1 and HindIII cleavage sites and was ordered as sense and antisense oligonucleotides from Invitrogen. Sense: 5′-ccggtggcaagcccatccccaaccccctgctgggcctggacagcacca-3′, antisense: 5′-ccggtggtgctgtccaggcccagcagggggttggggatggggatgggcttgcca-3′. The vector was cut with the Age1 and HindIII restriction enzymes (New England Biolabs) and the V5 tag coding sequence was ligated in using standard procedures.
Mutants of gp120 were created using Quikchange site-directed mutagenesis reagents and methods (Stratagene). The primers used for mutagenesis were custom synthesized at Invitrogen or IDTDNA. The following forward primers and their reverse complements were used (in the 5′-3′ direction): M95A: gagaacttcaacgcctggaagaacaac, K97A: aacttcaacatgtgggcgaacaacatggtggag, N98A: caacatgtggaaggccaacatggtggag, N99A: catgtggaagaacgccatggtggagcag, V101A: gtgggctaacaacatggcggagcagatgcacg, E102A: agaacaacatggtggcgcagatgcacgagga, K421A: ctgccctgccggatcgctcagatcatcaacatgtggc, V430A: catgtggcaggaggccggcaaggccatg, D474A: ggcccggcggcggcgccatgcgggacaactg R476A: ggcggcgacatggccgacaactggcgg, R480A: cgggacaactgggccagcgagctgtac (Invitrogen), T257A: cccgtggtgagcgcccagctgctgctg, E370A: ggcgaccccgccatcgtgacccac, I371A: ggcgaccccgaggccgtgacccacagc, V372A: gaccccgagatcgccacccacagcttc, M426A: cagatcatcaacgcctggcaggaggtg, M475A: ggcggcggcgacgcccgggacaactgg, V120A; ctgaagccctgcgccaagctgaccccc, K121A: gaagccctgcgtggccctgacccccctg, L122A: ccctgcgtgaaggccacccccctgtgc, V255A: catccggcccgtggcaagcacccagctg (IDTDNA). Mutagenesis was verified by sequencing (Genewiz Inc.) using the in-house primers T7 (for 5′) or BGHR (for 3′), or the custom synthesized primer 5′-atccccatccactactgggccc-3′ (IDTDNA) for residues distant from the N and C-termini of the protein.. DNA for transient transfections was prepared large scale by Qiagen maxiprep kit or small scale by Zymo Research transfection grade miniprep kit. DNA was transfected into confluent 293F cells at 1,000,000 cells/ml as per protocol (Gibco) using 293Fectin. Cells were harvested 5 days after transfection and spun down. Supernatants were filtered and tested by anti-gp120 western blot using mAb D7324 (Aalto Bioreagents, Ireland) in non-reducing conditions for presence of gp120 of correct molecular weight (non-aggregated) and by anti-CD4 ELISA for functionality.
For experiments done on purified protein, mutant and WT gp120 were purified on a 17b affinity column created by direct coupling of 17b mAb to an NHS activated HiTrap HP column (GE). After supernatants were passed over the column, the column was washed with 1x PBS until baseline was reached (typically 10 column volumes) and captured gp120 was eluted into collection tubes carrying 1/10 the fraction volume 1 M Tris base pH 9.0 with elution buffer (0.1 M glycine, 0.15 M NaCl, pH 2.4). Eluted protein was immediately buffer exchanged into PBS using spin-columns (Amicon Ultra Ultracell-30K, Millipore). Protein was filtered through 0.45 μm syringe filters (Millex-LH, Millipore) and separated by size exclusion on a HiLoad 26/60 Superdex 200 HR prepacked gel filtration column (GE). The fraction of the peak corresponding to monomeric gp120 was determined by Western blot, and this fraction was collected, concentrated using spin columns, filtered with 0.2 μm syringe filters and concentration was measured by absorbance at 280 nm using a Shimadzu UV-spectrophotometer. Protein was aliquoted and frozen in dry ice and stored at −80°C until use. Purity of samples and monomeric state of gp120 were verified by SDS-PAGE – Coomassie staining and anti-gp120 western blot (Fig. S3).
Screening of gp120 mutants for binding and inhibition by the PT KR21
Screen approach
An initial ELISA screen was performed with 19 gp120 alanine mutants (see Fig. 9A for locations) that were selected based on several criteria. The first was the putative peptide binding site [13] (K97, E102 and R476, using standard numbering) which was developed with more recent data to include D474 and K421 (Zentner, Madani, unpublished results). The second was a set of residues identified around the putative binding site by visual inspection of the gp120 structures, that contained either (1) hydrophobic (V120, L122, T257, I371, V372, M426, M475) or (2) solvent exposed and hydrophilic (K121, E370) residues. The third criterion was a set of residues within surface depressions near the putative binding site as determined using the site analysis tool in MOE (Chemical Computing Group Inc.) (M95, N98, N99, V101, R480). 8 mutants that showed some resistance to PT inhibition, 2 additional mutants based on criteria 3 (V255A) and 1 (V430A) and 1 mutant that showed no effect were further screened for KR21 direct binding and CD4 inhibition by an SPR on-chip capture method previously employed to study binding of IL5 mutants without pre-purification of proteins from expression supernatant [15]. The screen approach is schematically outlined in Figure 1.
Figure 9. Env gp120 residues important for binding of, and inhibition by KR21 in the context of the mAb F105-bound gp120 crystal structure.
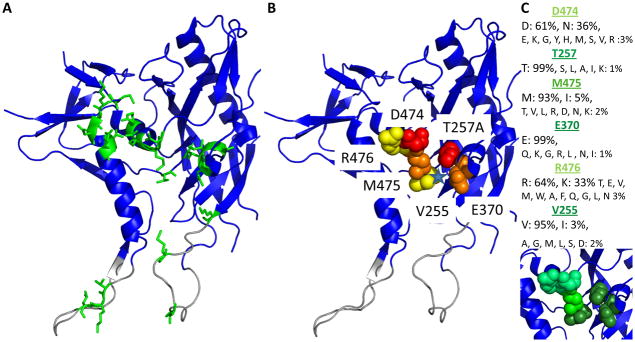
A. All gp120 residues (green sticks) that were tested in ELISA and/or SPR screens for inhibition by PTs are shown on the mAb F105 bound structure of core gp120YU2 (blue ribbon, PDB ID: 3hi1). Residues that form the bridging sheet in the CD-bound state (see Fig. S9) but are not structured in this conformation are shown in grey. B. Residues important for direct KR21 binding or CD4/17b competition/viral neutralization are shown as spheres. Tier 1: red; tier 2: orange; tier 3: yellow. Center of the components of the CD4 F43 binding pocket from the CD4-bound conformation is indicated with a blue star. C. Conservation of KR21-sensitive residues among the 3730 variants in the LANL database. The same structure as in (A) and (B) is used. Residue conservation is indicated as light green: <65%, medium green: <94% and dark green: >94%. Conservation values and changes observed are annotated above the structure.
Figure 1.
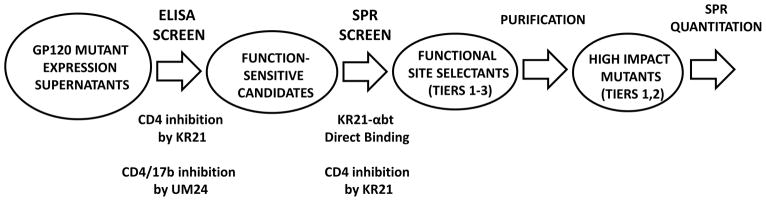
Scheme of the screen pathway for identifying peptide triazole functional site residues. The initial stage tested 19 gp120 alanine mutants for PT competition of CD4 and 17b binding by ELISA. Eliminating mutants with no effect on PT competition, expression supernatants for 11 mutants were tested for direct binding to KR21-αbt and for CD4 competition in an SPR screen. This screen identified 6 mutants with 3 tiers of impact on KR21 binding and inhibition (“Functional Site Selectants”). The mutants from the top two tiers (“High Impact Mutants”) were further analyzed as purified proteins in direct binding and inhibition assays.
ELISA screen for PT inhibition of CD4/17b binding
Serial dilutions of filtered expression supernatants were added to 96-well ELISA plates containing 100 ng CD4/17b Fab per well. Bound gp120 was detected with anti-gp120 antibody (D7324, Aalto Bioreagents) and anti-sheep HRP antibody and readout of OPD (o-phenylenediamine, Sigma) signal at 450 nm in a Microdevices microplate reader (not shown). The highest concentration of supernatant that was in a linear range of binding was chosen for each mutant to be used in subsequent inhibition experiments.
For the inhibition experiments, 10 μM to 156 nM KR21 or 400 nM to 0.8 nM UM24 were mixed with the chosen dilution of supernatant and binding of gp120 to the adsorbed CD4/17b was measured as above. All mutants were tested with KR21, except for N98A and N99A which had been previously tested with UM24 and were not re-tested in the current assay. UM24 has the sequence Citrulline-N-N-I-X-W-S which is the minimal sequence with full potency [10]. We used UM24 to screen for effects of the alanine mutations around the putative PT binding site in order to identify those residues that were critical for binding and inhibition by PTs.
Preparation of peptide-antibody complex
The peptide triazole used in this study contained the sequence R-I-N-N-I-X-W-βAla-BtLys-G-NH2 where X is a ferrocenyltriazole conjugated proline [14], βAla is β-alanine (catalog no: 35737-10-1, Chem-Impex) and BtLys is a biotin-conjugated lysine (Ne-biotinyl-L-lysine, cat no: 04988, Chem-Impex) and was named KR21. We prepared non-covalent complexes of KR21 and anti-biotin antibody for use in studies of KR21 direct binding to gp120 mutants, as the added mass of the mAb yields measurable signals from binding the captured gp120, versus KR21 alone.
KR21 and mouse anti-biotin monoclonal antibody (Jackson Immunoresearch) were mixed at a 10:1 molar ratio in PBS. Concentration of KR21 was determined by absorbance at 280 nm (E= 6022/(cm*M)). Peptide-antibody mixture was incubated on a shaking platform at 4°C for 1 hour and applied to a PD-10 desalting column as per protocol (GE). Peptide-bound antibody (KR21-αbt) was eluted in 3.5 ml PBS, concentrated to 0.6 – 1.2 ml using an Amicon Ultra-15 centrifugal filter unit (Millipore), and concentration determined by absorbance at 280 nm (E = 252044/(cm*M)). Complex (KR21-αbt) concentrations were between 6 and 9 μM.
KR21-αbt binding to WT and mutant gp120
For binding experiments of KR21-αbt and gp120, concentrations of freshly made KR21-αbt were injected over gp120 and its mutants captured anew on the anti-V5 surface for each cycle of injection.
SPR reaction surfaces were created by covalent immobilization of α-V5 monoclonal antibody (catalog no: R960-25, Invitrogen) to 1000 – 2000 Response Units (RU) on a CM5 carboxydextran surface by EDC/NHS activation as per specifications using a Biacore 3000 SPR instrument (GE). All reagents were filtered through 0.2 μm syringe filters before injecting into the instrument. Antibody was mixed in acetate buffer pH 4.5 and injected over the activated surface until the required RU’s were immobilized. Unreacted groups were blocked by injection of 1M ethanolamine at 5 μl/min for 7 minutes. The first flow cell in each chip was derivatized with 1400 – 2400 RU 2B6R antibody to IL5 receptor α as a control surface for the α-V5+gp120 test surfaces (flowcells 2, 3 and 4).
30 – 40 μl supernatant containing gp120 was passed over each α-V5 surface at 5 μl/min to reach 400 –500 RU gp120 capture on the surface. Each surface was derivatized sequentially resulting in 2 minutes or more dissociation of each surface before the subsequent injection of buffer or KR21-αbt. Dissociation of captured gp120 from the surface was very slow, generally on the order of 0.1 RU/second (not shown).
Buffer or complex at increasing concentrations was injected over all four flow cells at 100 μl/min to a final volume of 250 μl. Dissociation was observed for 300 seconds. All surfaces were regenerated with 10 – 15 μl 10 mM glycine pH 1.5 at 100 μl/min until baseline was reached. At the next cycle, gp120 was again flowed over the surfaces and the next concentration of complex was injected.
The binding of KR21-αbt complex to captured gp120 was analyzed using Biaevaluation 4.0 software (GE). Sensorgrams were normalized by double reference subtraction in BiaEvaluation 4.0 as follows: Buffer injections were subtracted from the sample injection for the reference and α-V5+gp120 surfaces. Next, the buffer normalized reference surface sensorgram was subtracted from the buffer normalized α-V5+gp120 surface sensorgram to give the double reference subtracted sensorgram.
Analysis of binding sensorgrams
Kinetic parameters of the gp120-KR21-αbt interaction in each case were calculated through separate fits using dR/dt and ln(R1/Rn) plots for the on- and off-rates, respectively, as established previously [16, 17]. This was necessitated by the non-ideal fitting obtained when using global 1:1 Langmuir fitting in BiaEvaluation. The technique draws on the bimolecular interaction model in an SPR setting where one molecule (the ligand) is immobilized. Briefly, a bimolecular interaction is given by the equation
where A is the analyte (KR21-αbt in this case), B is the immobilized ligand (gp120) and AB is the complex formed between the two. Thus the rate of complex formation is
| (1) |
In SPR, the response (R) is directly related to the complex formed and free ligand (B) is equal to Rmax − R.
Thus replacing these two terms in (1):
| (2) |
and
| (3) |
Thus, plotting dR/dt vs R gives a slope which is equal to − (kon [A] + koff). This term is named −ks and is used to derive kon as
| (4) |
Thus, the slope of a ks vs [[A] (analyte concentration) plot gives kon.
In SPR, the analyte concentration is 0 during dissociation. Thus, plugging 0 in [[A] in equation 3 gives
| (5) |
Plugging in values for response at time 1 (beginning of dissociation) and n:
Thus, plotting ln(R1/Rn) vs time gives a slope of koff
8 – 130 seconds of association were used for the on-rate analyses, as this range overall covered the longest interval free of irregularities in sensorgrams due to injection start and stop. The 8 – 55 second interval was also used and gave similar results (not shown).
Dissociation rates approached zero with time, and this effect was more pronounced with the lower concentrations, most likely due to the lower signal/noise ratio at these concentrations. ln(R1/Rn) was plotted at different time intervals (first 20, 30, 40 and 50 seconds of dissociation) and the slope was observed to gradually decrease at each added interval, thus was only derived from the first 20 seconds of dissociation of the highest concentration of analyte injected for all experiments.
Inhibition of CD4 binding to gp120 mutants by KR21
We measured KR21 inhibition of the CD4-gp120 interaction to correlate these with the changes in affinity deduced from the above experiments. Anti-V5 capture of gp120 was used similarly as the above section, however only KR21 was used to inhibit CD4 binding, as only the CD4 signal, and not that from the KR21 was measured.
gp120YU2 and its mutants were captured on 2000 RU α-V5 surfaces similarly to the method of direct binding capture SPR explained above. A 2500 RU 2B6R surface was used as a control. CD4 was injected over WT, K421A and E370A gp120 at different concentrations and 250 nM concentration was chosen for further competition studies with KR21 since this concentration resulted in more than 50 RUs of analyte bound to surface after association for all mutants and would yield high signals for competition measurements (not shown).
For competition experiments, 250 nM CD4 alone and with increasing concentrations of KR21 (two-fold dilutions from 10 μM to 20 nM) were injected over the captured gp120 for 4 minutes at 10 μl/min flow rate. Dissociation was observed for 240 seconds. Free CD4 injections were done before and after the concentration range of KR21 to observe any change in the surface during the injections. There was minimal change (typically 10% or less) in the surface capacity throughout each concentration range, and in cases where there was a decrease, percent binding was calculated assuming a linear decrease in surface capacity with each injection. Injections of KR21 alone without CD4 were also done to subtract from the injections of CD4 plus KR21 in order to correct for signal arising from peptide alone, however, these values were in some cases higher than the injection of CD4 with the same concentration of peptide, so were not used in the final analysis of any mutant.
RUs used to calculate the amount of CD4 bound in the presence of KR21 were chosen from a time point that is both close to equilibrium and free from effects due to buffer change. Thus RUs at 225 seconds (15 seconds before the end of association) were chosen to plot binding vs. KR21 concentration for each wild-type and mutant gp120. Plots of Binding Response (RU) vs. KR21 concentration were fit to a 4-parameter sigmoidal model
in Origin 7.0 where A1 and A2 are the minimum and the maximum, respectively and x0 is the x coordinate that gives half-maximal binding (midpoint between A1 and A2). This value is reported as the concentration that results in 50% inhibition (IC50).
Follow-up Functional Verification for Major gp120 Escape Variants
KR21 direct binding to immobilized gp120
We wanted to determine KR21’s absolute affinity for the gp120 mutants that had 10-fold weaker affinity or inhibitory potency for gp120 compared to wild-type in the capture screen.
Purified WT, T257A, D474A, E370A and M475A gp120 were immobilized on a Biacore CM5 chip using amide coupling following activation with EDC/NHS (Biacore manual, GE). Briefly, carboxydextran surfaces were activated by injection of a 0.2M: 0.05M solution of 1-Ethyl-3- [3-dimethylaminopropyl]carbodiimide (EDC) and N-hydroxisuccinimide (NHS) (both reagents from Pierce), and gp120 diluted in acetate buffer pH 5.5 was injected to the desired coupling level using the manual inject command. Unreacted sites were blocked by ethanolamine and nonspecifically bound protein washed off by a pulse of 10 mM HCl. Approximately 2000 RU gp120 was immobilized on each surface. The first flow cell was derivatized with ~ 2000 RU mAb 2B6R to use as a control.
Increasing concentrations of KR21 were injected on the surfaces from 2 nM to 100 μM. 2 nM to 250 nM CD4 was injected in duplicate to control for overall functionality of the proteins. 0.8 to 400 nM or 3.1 to 400 nM 17b IgG was injected for a chip containing WT, E370A and M475A, and another chip containing WT, T257A and D474A gp120, respectively, in duplicate, to probe for the conformational effect of the mutations on gp120. All injections were done with the kinject command at 100 μl/min with a 250 μl injection volume. All bound peptide dissociated after flowing buffer (PBS + 0.005% Tween-20 + 0.1% NaN3) at 100 μl/min for 1000 seconds. 5 μl 10 mM HCl was injected at 100 μl/min flow rate after a 300 second dissociation following CD4 and 17b binding to regenerate the surfaces.
Following double reference subtraction with buffer and control surfaces, response units (RU) were plotted at the 145th second of association (Req) and fit to a steady state affinity model where steady state binding level is related to concentration (C) in M and the number of binding sites on the ligand (n, which was 1 in all cases) according to the applied Langmuir equation for KR21 binding using Biaevaluation 4.0 software (GE).
CD4 and 17b competition on immobilized gp120
We wanted to determine KR21’s absolute inhibitory potency for the gp120 mutants that had 10-fold weaker affinity or inhibitory potency for gp120 compared to wild-type in the capture screen, using purified proteins which allowed us to lower CD4 concentration to 35 nM, which is a non-saturating concentration at or below the KD of CD4 to WT or mutant gp120’s (Table II).
Table II.
Comparison of KR21, CD4 and 17b affinities to purified gp120 mutants.
| gp120YU2 | KR21 KD (nM) | ± | Δ | ± | n | CD4 KD (nM) | ±(n=2) | Δ | ± | 17b IgG KD (nM) | ±(n=2) | Δ | ± | 17b Fab KD (nM) | ±(n=2) | Δ | ± |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 8 | 2 | 1 | 0.2 | 4 | 79 | 3 | 1 | 0.04 | 78 | 2 | 1 | 0.02 | 157 | 82 | 1 | 1 |
| T257A | 1090 | 320 | 134 | 39 | 4 | 72 | 2 | 1 | 0.02 | NA | NA | NA | NA | 408 | 140 | 3 | 1 |
| D474A | 1340 | 274 | 164 | 34 | 3 | 94 | 3 | 1 | 0.04 | 239 | 3 | 3 | 0.04 | 397 | 98 | 3 | 1 |
| WT | 23 | 8 | 1 | 0.4 | 9 | 22 | 1 | 1 | 0.1 | 93 | 0.1 | 1 | 0.001 | ||||
| E370A | 386 | 71 | 17 | 3 | 9 | 58 | 3 | 3 | 0.1 | 443 | 42 | 5 | 0.5 | ||||
| M475A | 196 | 34 | 8 | 1 | 9 | 29 | 1 | 1 | 0.04 | 316 | 30 | 3 | 0.3 |
indicates fold difference with WT for the value to the left.
indicates standard deviation.
NA (not available): no data could be obtained from the T257A surface in this experiment.
Ability of KR21 to inhibit binding of CD4 to surface immobilized gp120 was tested on the same chips that were used for measuring direct binding, where one flow cell contained a reference antibody (2B6R) and the other three contained the two mutant gp120’s and a wild-type gp120. If chip functionality decreased to a level that did not permit further experimentation, a new chip with identical surfaces was generated using EDC/NHS coupling and used for the next experiment. Peptide and CD4 were mixed such that concentration of CD4 was constant (35 nM) and that of KR21 varied from 6 nM to 100 μM (2.5 nM to 20 μM for E370A and M475A). These mixtures were injected over the immobilized gp120 at 50 μl/min (5 μl/min for E370A and M475A) flow rate for 5 minutes. Binding of 35 nM CD4 was measured before and after the injection of mixtures to determine levels of 100% (uninhibited) CD4 binding. Binding response of peptide alone at the same concentrations was subtracted from the signal to give binding signal of CD4 alone. RUs at 295th second of association (5 s. before end of injection) where equilibrium was reached were plotted to calculate IC50s by a sigmoidal-logistic fit in Origin 7.0 as done for CD4 competition using captured protein. Experiment was done in duplicate (triplicate for E370 and M475A) and average IC50s were reported.
The T257A and D474A inhibition assays were done with 1 μM 17b Fab as we had used this analyte to be free of avidity effects in our direct binding assays, and it gave a robust binding signal (>50 RU) at 1 μM concentration for the mutants tested. However the use of Fab versus IgG was not enough to yield measurable off-rates, and 1 μM 17b Fab and 400 nM 17b IgG were observed to have the same IC50’s for KR21 inhibition of binding to WT gp120 (within 10% of each other, not shown). Thus 400 nM 17b IgG was used in the later assay with E370A and M475A in order to increase the signal from binding. 17b was mixed with increasing concentrations of KR21 from 20 nM to 20 μM (T257A, D474A) or 2.5 nM to 40 μM (E370A, M475A) and injected over immobilized gp120 mutants for 5 min. at 5 μl/min with a 1 min. dissociation time (quickinject). Binding at 290 sec. was taken as the equilibrium (Req) value and plotted versus concentration after subtraction of signal that was obtained due to binding of the added concentration of KR21 alone. The resulting data points were fit to a 4-parameter sigmoidal logistic equation as described above.
Effects of Env changes on Single-round Infection of Cells
We tested the effects of the changes in gp120 that affected KR21 binding and potency in the context of viral infection.
Plasmid constructs expressing the full length envelope protein of HIV-1YU2 were mutated to contain the above-described mutations with the Quikchange XL kit (Stratagene) as explained in the methods above for “Protein Production”. The HIV-1 env/rev gene cassette in the pSVIIIenv vector, a vector carrying the HIV-1 Gag-Pol protein coding sequences (pCMVΔP1ΔenvpA) and a firefly luciferase-expressing vector (pHIV-1Luc) were co-transfected into 293T human embryonic kidney cells at a DNA ratio of 1:1:3 using the Effectene transfection reagent (Qiagen). Constructs carrying envelope glycoproteins of amphotropic murine leukemia virus (A-MLV) and a Rev expressing plasmid were substituted for the HIV-1 Env/Rev expressing plasmid in control viruses. Virus infectivity was tested without KR21 and virus infection of CD4, CCR5 and CXCR4 expressing Cf2Th cells was measured using a readout of luciferase activity as described previously [18]. Assays were done in triplicate and averaged. Assays were repeated using the original peptide HNG-156 [14] where the full C-terminus (SEAMM) replaces KR21’s βAla-BtLys-Gly.
Sequence alignment
Variation of hot-spot residues was obtained from pre-made alignment of the 3730 HIV-1 Env sequences from 2011, as made available at the Los Alamos National Laboratory (http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html). Filtered web alignment was run on the Env region for protein sequences, downloaded and visualized by Jalview (www.jalview.org). Sequence conservation and spatial relationship of the hot-spot residues were shown on the HIV-1 core gp120 x-ray structures with PDB IDs 3hi1 (F105 bound clade B strain YU2), 3tgq (unliganded, YU2), 3tgt (unliganded, clade A/E strain 93th057), 3tgr (unliganded clade C strain C1086) and 3tgs (C1086 bound to the CD4-mimic NBD-556) using PyMol 1.4 (Shrödinger).
RESULTS
KR21 synthesis and characterization
KR21 is a biotinylated dual-antagonist peptide triazole (PT [10]) that inhibits gp120 interactions with CD4 and 17b (not shown). The KR21 sequence shown in Figure 2A was derived from the ferrocenyl PT HNG-156 [14] with the deletion of non-critical residues from the C-terminus and addition of βAla-Lys(ε-Biotinyl)-Gly as an attachment point. This peptide was synthesized by solid phase peptide synthesis on Rink Amide resin and derivatized by click chemistry of ethynylferrocene on the azidoproline. Its mass was confirmed by MALDI-TOF mass spectrometry (Fig. S1). It inhibited binding of gp120YU2 to soluble CD4 (sCD4) and the co-receptor surrogate mAb 17b with IC50s similar to those of the full length peptide it is derived from (HNG-156 [14]) (Fig. S1).
Figure 2. SPR V5-capture assay setup.
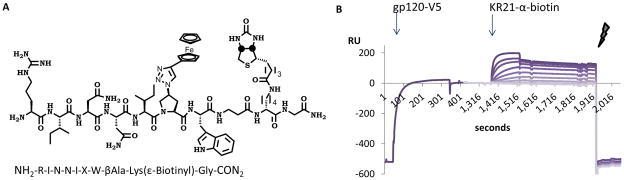
A. Chemical structure and amino acid sequence of the peptide KR21 used in direct binding and competition experiments.
B. Annotated sample sensorgram showing capture of V5-tagged gp120 on an α-V5 surface, followed by injection of the KR21-α-biotin complex and regeneration of surface. RU: Response Units.
ELISA screen of gp120 mutants for inhibition of CD4 and 17b binding by PTs
gp120 mutants’ binding to CD4 was measured in the presence of the PTs KR21 and UM24 [10] and 17b inhibition was further tested with UM24 in order to eliminate those mutants that had no effect on PT activity from further analysis. 50% inhibitory concentrations (IC50s) of the peptides were calculated from fitting the data to a 4-parameter sigmoidal equation in Origin 7.0 as explained in Materials and Methods under ‘Inhibition of CD4 binding to gp120 mutants by KR21’. The results are summarized in Fig. S2. Of these mutants, the following were chosen for a more detailed screen of KR21’s affinity and inhibitory potency on them: K97A, E102A and R476A since these mutations affected inhibition by the original peptide 12p1 [13] and showed differences with WT in the ELISA screen (see Fig. S2B); D474A and K421A since these mutants were shown to be important for peptide triazole function in later studies (not shown) and showed differences in the ELISA screen, E370A as this mutant was tested for the first time and showed a great difference with WT in the KR21 ELISA screen; T257A and M475A as these mutations had shown effects on antiviral potency (not shown) of the peptide triazole HNG-156 [14] and also affected CD4 inhibition by KR21 in the ELISA screen; M95A was a mutant selected as a control for no effect (less than 2-fold) on PT activity. N98A, N99A, K120A, V121A, L122A, V372A and M426A were inhibited by UM24 or KR21 as well as WT and were not carried over to the next study. Although I371A had a moderate effect (about 10-fold compared to WT) and V101A and R480A had smaller effects (less than 2-fold and 5-fold, respectively) on CD4 or 17b inhibition by the PTs, these effects were small and as there was no prior study to validate the importance of these mutations, these mutants were not carried over to the next phase of screening.
KR21-αbt binding to WT and mutant gp120
To validate the effects of the mutations first tested in the ELISA based inhibition assays and of two new mutants (V255A and V430A) that became available after the ELISA screen, we used an SPR based secondary screen to rank these mutants according to their binding affinity and inhibition by the biotinylated PT KR21 (Fig. 2B). We tested alanine mutation at the residues M95, K97, E102, V255, T257, E370, K421, V430, D474, M475 and R476. These mutants and WT gp120 carried V5 and His6 tags on the C-terminus so they could be captured on an SPR surface with immobilized mAb anti–V5 (α-V5). gp120 was captured on α-V5 antibody covalently immobilized to the carboxydextran surface. KR21-anti-biotin mAb complex (KR21-αbt) and CD4 were injected over captured gp120 in concentrations ranging from 8 nM to 4 μM and 4 nM to 1 μM, respectively. On- and off- rates were fit separately via dR/dt and ln(R1/Rn) plots, as shown for binding of KR21-αbt to WT gp120 in Figure 3 and explained in the Materials and Methods section under Analysis of Binding Sensorgrams. Sample sensorgrams are given for the binding of KR21-αbt to T257A, D474A, E370A and M475A gp120 in Figure 4.
Figure 3. A sample sensorgram set and analysis of KR21-αbt binding to WT gp120.

A. Overlaid, reference subtracted sensorgrams showing injection of 7.8 nM to 2 μM KR21-αbt in two-fold increments, shown from light to dark. RU: Response Units.
B. ks plot showing derivative of RU with time vs. RU of the first 2 minutes of each injection, keeping the same greyscale scheme as part A.
C. Plot of the ks (- slope from part B) vs. concentration for each injection of KR21-αbt, and the resulting linear regression whose slope gives the on-rate of KR21-αbt binding to WT gp120.
D. Natural logarithm plot of response during the first 20 seconds of dissociation vs. response at the first second of dissociation; over time. The slope of this line yields the off-rate of this interaction.
Figure 4. Representative, greyscale-coded overlaid sensorgrams of KR21-αbt binding to gp120 mutants.

Injections were done for 150 seconds, followed by 300 seconds of dissociation. 16 nM to 4 μM KR21-αbt were injected for T257A and D474A (64 nM to 4 μM are shown). 31.25 nM to 4 μM and 16 nM to 2 μM KR21-αbt were injected for E370A and M475A, respectively. Only the top 5 concentrations are shown for M475A. Higher concentrations are darker. y-axis: Response Units (RU), x-axis: seconds.
M95A, K97A, E102A and V430A gp120 were essentially identical to WT in their binding kinetics for KR21-αbt. (Table I, Fig. S5). K421A had a slightly faster off-rate that was balanced by a faster on-rate resulting in slightly lower (improved) dissociation constant (KD) than WT. M95A had a faster off-rate that was balanced by a faster on-rate resulting in no net change in KD.
Table I.
Kinetic parameters of KR21-αbt and CD4 binding to gp120 mutants.
| gp120YU2 | KR21-αbt binding KD (nM)a | ±b | Δc | ± | KR21-αbt kon (1/(Ms*1000)) | ± | KR21-αbt koff (M*1000) | ± | n | CD4 binding KD(nM) | ± | Δ | ± | CD4 kon (1/(Ms*10000)) | ± | CD4koff (1/(M*10000) | ± | n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 812 | 125 | 1 | 0.2 | 5 | 3 | 4 | 2 | 6 | 18 | 8 | 1 | 0.4 | 6 | 1 | 10 | 4 | 6 |
| M95A | 747 | ND | 1 | ND | 22 | ND | 17 | ND | 1 | 17 | ND | 1 | ND | 9 | ND | 15 | ND | 1 |
| K97A | 626 | ND | 1 | ND | 4 | ND | 3 | ND | 1 | 10 | ND | 1 | ND | 7 | ND | 7 | ND | 1 |
| E102A | 623 | ND | 1 | ND | 5 | ND | 3 | ND | 1 | 3 | ND | 0.2 | ND | 7 | ND | 2 | ND | 1 |
| V255A | 1709 | 1635 | 2 | 2 | 10 | 10 | 18 | 9 | 2 | 10 | 0.1 | 1 | 0.01 | 11 | 9 | 12 | 9 | 3 |
| T257A | >15000 | NA | >18 | NA | 9 | NA | 16 | NA | 2 | 5 | 4 | 0.3 | 0.2 | 13 | 4 | 6 | 3 | 2 |
| E370A | 3962 | 2448 | 5 | 3 | 6 | 2 | 25 | 5 | 4 | 25 | 13 | 1 | 0.7 | 7 | 5 | 18 | 20 | 4 |
| K421A | 502 | ND | 1 | ND | 19 | ND | 10 | ND | 1 | 23 | ND | 1 | ND | 3 | ND | 7 | ND | 1 |
| V430A | 923 | ND | 1 | ND | 12 | ND | 11 | ND | 1 | 51 | ND | 3 | ND | 6 | ND | 29 | ND | 1 |
| D474A | >15000 | NA | >18 | ND | NA | NA | NA | NA | 2 | 5 | 0.5 | 0.3 | 0.03 | 8 | 1 | 4 | NA | 3 |
| M475A | 3198 | 4243 | 4 | 5 | 5 | 3 | 15 | 3 | 2 | 28 | 23 | 2 | 1 | 12 | 0.1 | 32 | 27 | 2 |
| R476A | 2332 | ND | 3 | ND | 6 | ND | 15 | ND | 1 | 28 | 43 | 2 | 2 | 13 | 7 | 38 | 32 | 2 |
Average on and off rates and KDs were calculated from separate fits using ks and ln(R1/Rn) plots.
Standard deviation is shown if experiment was performed more than once (n>1).
Δ indicates fold difference with WT gp120 for the value to the left (standard deviation for fold differences are calculated by dividing the standard deviation by the value for WT).
NA (not available) indicates the value could not be obtained from the available data.
ND (not determined) indicates standard deviation could not be calculated as experiment was not repeated.
T257A and D474A mutants were specifically disrupted in binding to KR21-αbt (Fig. 4) and their affinities KR21-αbt could not be measured, despite retaining full affinity to CD4 (Fig. S4). The overall magnitude of KR21-αbt binding to these mutants was also greatly diminished, as they bound 23 and 47 RU, respectively, of CD4 at the end of association phase with the highest concentration of CD4 used (500 nM) vs. 6 and 11 RU of KR21-αbt, a molecule three times the molecular weight of CD4, at its highest concentration (4 μM). The same ratio is 92 vs. 80 RU for WT gp120 (see Fig. S5 for other mutants). Of the remaining mutants, E370A’s dissociation constant (KD) for KR21-αbt was 5-fold higher than that of WT, while V255A, M475A and R476A’s were 2, 4 and 3-fold higher, respectively (Table I). Experimental variation could be measured for V255A, E370A and M475A as repeat analysis was done with these mutants, and the experimental variation (standard deviation) was greater than the observed affinity difference with WT for V255A and M475A, but was less than the observed difference for E370A (affinity difference of 5 ± 3 fold, see table I). These changes were due to different off-rates (all more than 3-fold higher), as the on-rates for these mutants were relatively unchanged (Table I).
Effects of the mutations on CD4 binding were negligible or positive, indicating that these proteins were correctly folded. The only mutation that possibly had a detrimental effect on CD4 binding (V430A, ca. 3-fold) had no effect on KR21-αbt binding (Fig. S5, Table I).
Inhibition of CD4 binding to gp120 mutants by KR21
Aside from direct binding to gp120, inhibition of binding of gp120 to CD4 and the co-receptor surrogate mAb 17b are measures of the potency of dual antagonist peptide triazoles (PTs) like KR21. We wanted to confirm the significance of the direct binding results with a CD4 inhibition assay that measured the effect of the mutations on the functional outcome of KR21 binding to gp120. We screened mutants of gp120 for changes in their susceptibility to inhibition by KR21 using a capture SPR assay where the analyte is CD4 with or without KR21. We used 250 nM CD4 in the inhibition assays as this was the minimum concentration that yielded 50 RU or more of binding for the mutants tested and was not saturating (see Fig. S4, S7). Injection of 250 nM CD4 before and after the addition of different peptide concentrations gave a measure of the change in level of 100% i.e. uninhibited binding throughout the assay. Overall, the surface capacity changed very slightly, as visualized in Figure S6. Most of the signal in this assay was due to CD4, as the KR21 component of 1644 Da gave a negligible signal compared to CD4 of 45 kDa. It was not possible to calculate the contribution due to the KR21 component, as KR21 passed alone over captured gp120 at the highest concentrations tested gave a higher signal than the mixture of 250 nM CD4 and KR21 at the same concentrations, and thus could not be directly subtracted from the sensorgrams of the mixtures. However, the fact that the sensorgrams were reaching a stable baseline at the highest concentrations of added KR21 was used to fit plots of equilibrium response levels (Req) to a 4-parameter sigmoidal equation that assumes stable maxima and minima. KR21 was mixed with CD4 to yield final CD4 concentration of 250 nM, which yielded large enough RU’s for all mutants for reliable data analysis, and KR21 concentration in the range 10 μM – 19.5 nM (two-fold dilutions). The mixtures were incubated for varying times at 4°C as they awaited injection in the instrument, with the minimum incubation being 1.5 hr, and the maximum 7 hr. Injections were done from the highest to the lowest concentration of inhibitor to bias against the gradual decline in surface functionality seen with regenerated surfaces. Inhibition was concentration dependent for all mutants tested. Req were taken at 225 seconds (15 seconds before the end of association phase) and plotted as fraction of the average of the CD4 injections before and after the start of peptide concentrations. These data were fit to the sigmoidal model
to yield x0, the half-maximal inhibitory concentration (IC50).
Averages of 2 runs each for the mutants and 6 runs for the WT gp120 are shown in Figure 5. The most drastic change in KR21’s ability to inhibit CD4 binding was seen with the weak binders T257A and D474A, which had 3088 and 4049 nM IC50s that are 33 and 43 fold higher than the 94 nM IC50 of WT gp120, respectively. E370A and M475A were also refractory to inhibition by CD4, with 10 and 12 times higher IC50s than WT, respectively. V255A, T257A, K421A and R476A had 3 to 6 fold greater IC50s (Fig. 5). The differences seen with V255A, T257A, E370A, D474A, M475A and R476A were statistically significant (Fig. 5), and of these, 4 mutants (T257A, E370A, D474A and M475A) that had a difference of 10-fold or greater (before statistical analysis) were selected for further analysis.
Figure 5. Inhibition of CD4 binding by KR21.

A. CD4 binding to each mutant at varying concentrations of KR21. Amount of bound CD4 was recorded at 225 seconds into start of injection and plotted for each concentration of added KR21. Error bars indicate standard deviation of 5 experiments for WT and 2 experiments for mutants. ± indicates standard deviation for the value to the left of the symbol. A fit to a 4-parameter sigmoidal equation was overlaid on the data points for each protein. B. Calculated IC50s and fold differences with WT IC50. Error in IC50 is calculated by comparing results from different experiments.
Test of hot-spot residues with purified protein
Direct binding
The V5-capture screen presented three tiers of importance for PT binding and inhibition. We quantitated these differences by using purified mutants directly amine-coupled to an SPR surface. This configuration allowed greater signals to be observed with KR21 alone as analyte. WT, T257A, E370A, D474A and M475A gp120YU2 were purified by 17b affinity and size exclusion as described in the Materials and Methods section under Production of wild type and mutant HIV-1YU2gp120. These proteins were pure gp120 of the right size and monomeric, as shown by the Coomassie-stain and anti-gp120 western blots in Fig. S3. We measured the direct affinities of KR21, CD4 and 17b to these mutants by passing these molecules at increasing concentrations directly over the immobilized gp120 surfaces. CD4 and 17b binding was used to verify the functionality and deduce the impact of each mutant on overall conformation elsewhere in the protein. Dissociation constants were obtained by plotting steady state binding response immediately before the end of association (140 seconds) versus concentration of analyte and fitting the data points to a 1:1 Langmuir binding equation in BiaEvaluation 4.0 software (GE) (Fig. 6). (Color figures are available in the online version of this article).
Figure 6. Steady state affinity fits of KR21, CD4 and 17b binding to purified and immobilized WT and mutant gp120.

Percent binding of Rmax (maximum binding theoretically achievable as deduced from fits of steady state equilibrium binding values (Req) at 140 seconds to a 1:1 Langmuir binding equation in Biaevaluation 4.0) plotted versus concentration of analyte for 2 nM to 20 μM KR21 binding to (A) WT, T257A and D474A, and (B) WT, E370A and M475A gp120. C. Fits of 2 nM to 250 nM CD4 binding overlaid on data points. D. Data and fits for 1 nM to 500 nM CD4 binding to the indicated proteins. E. 3.1 nM to 400 nM 17b IgG binding to the indicated proteins. F. 0.8 to 400 nM 17b IgG binding to the indicated proteins. G. 16 nM to 2 μM 17b Fab binding to WT, T257A and D474A gp120YU2. Experiments were done in duplicate except for A and B (KR21 binding) which were done a total of 3 to 9 times. All results are those of averaged data from multiple repeats.
The T257A and D474A mutants, which were classified as tier 1 in the capture assay, proved to have drastically lowered affinities to KR21. T257A and D474A had 1.1 ± 0.3 and 1.3 ± 0.3 μM KDs to KR21 compared to 8 ± 2 nM for WT gp120 (Fig. 6A, Table II). These correspond to 130 ± 40 and 160 ± 30 fold greater KDs compared to WT. The KD values for CD4 binding were 80 ± 3, 70 ± 2 and 90 ± 3 nM for WT, T257A and D474A, respectively (Fig. 6C, Table II), showing that the dramatic decrease in KR21 affinity takes place without a decrease in CD4 affinity. Although the T257A mutation was previously observed to reduce CD4 binding roughly 30% in a pull-down assay using cell-lysates containing mutant full-length envelope protein [19], we observed much smaller differences in assays with our purified proteins, confirming that the overall CD4 binding site was not perturbed by these mutations.
E370A and M475A mutants had KR21 affinities of 386 ± 71 and 196 ± 34 nM, respectively, compared to 23 ± 8 for their WT control (Fig. 6B, Table II). The corresponding fold differences versus the WT KD are 17 ± 3 and 8 ± 2 for the two mutants. These values are statistically greater than the fold differences found with the mutants versus WT for CD4 binding, for which E370A had a 3 ± 0.1 fold lower affinity, while that of M475A was the same as WT. (Fig. 6D, Table II). The decreased affinities of V430A and E370A to CD4 seen in the SPR screen (Table I), and here, respectively, may be caused by losses of direct contacts, as these residues make critical contacts with R59 and F43 of CD4, respectively [20, 21]. While the KR21 affinity difference observed with purified E370A (17 ± 3) was barely statistically different than the differences observed with the V5-tag capture method and KR21-αbt as the analyte (5 ± 3 fold), the assays did not give statistically different results for M475A (4 ± 5 fold vs. 8 ± 3 fold, see Tables 1 and 2). Moreover, the ranking of affinity decreases is conserved between the capture and direct immobilization methods, supporting the initial classification of important mutants into three tiers.
The presence of the biotin tag on the C terminus of the peptide was found not to affect its affinity to gp120. An N-terminal acetylated version of KR21 that lacked the biotin tag and instead contained a free lysine had a KD of 13 nM to purified WT gp120 (not shown) compared to 8 nM for the biotin-containing KR21. Similarly, the V5 and His6 tags on the C-terminus of gp120 did not affect its functionality. The tagged WT gp120 had a CD4 affinity of 80 nM, compared to 36 nM when the same gp120YU2 lacking any tags was expressed in insect cells [22, 23]. This protein also bound PTs with KDs similar to tagged WT protein [24].
Mutation of D474 to N as seen naturally in HIV was recently seen to make virus about 100–fold more resistant to inhibition by the mAb 17b [25]. We tested the effect of the D474A, E370A and M475A mutations on 17b IgG binding (Fig. 6(E and F). At the time this experiment was done, the T257A surface had lost functionality and did not give measurable signals for 17b IgG binding. It was, however, previously tested with 17b Fab, and was shown to be only 3 ± 1 fold weaker than WT gp120 in its affinity to this molecule (Fig. 6G, Table II, Fig. S7E). As 17b’s off-rate was too small to be measured within the time-frame of the experiments (up to 10 minute dissociation time, see Fig. S7C and D), we plotted the steady state equilibrium binding values at each concentration versus the concentration and fit these data to a 1:1 Langmuir binding equation using the BiaEvaluation software. The binding sensorgrams (average of duplicate injections) for the chip containing WT and D474A gp120 (Fig. S7C) and that containing WT, E370A and M475A gp120 (Fig. S7D) show somewhat slower on-rates for these mutants. Plotting the equilibrium binding level of these proteins versus concentration (Fig. 6E and F) and fitting a 1:1 Langmuir binding equation to these data points, we obtained an affinity of 239 ± 3 nM for D474A compared to 78 ± 2 for its WT control, which amounts to a 3-fold decrease in affinity for 17b IgG. D474A also had a 3 ± 1 fold lower affinity for 17b Fab (Fig. 6G), indicating that the affinity difference observed with 17b Fab for T257A is comparable to the affinity difference that would have been observed if IgG were used. E370A and M475A had 17b IgG affinities of 443 ± 42 and 316 ± 30 nM, respectively, compared to 93 ± 0.1 for their WT control, which amounted to affinity differences of 5 ± 0.5 and 3 ± 0.3 fold (Fig. 6F). These differences may be due to conformational changes in the protein, but do not explain the 130, 160 and 17-fold affinity differences observed for KR21 binding to T257A, D474A and E370A. M475A, on the other hand, binds to KR21 only 8 ± 1.5 fold weaker than WT (Fig. 6B, Table II). Although this difference is significantly different than the increase in its KD for 17b binding (3 ± 0.3 fold compared to WT), we cannot rule out that a significant contribution to the loss in peptide affinity comes from a global conformational change induced by this mutation. Overall, the CD4 and 17b affinities of tier 1 and 2 mutants did not change or changed much less than the KR21 affinities for these mutants.
The CD4 and 17b affinities were higher than previously published [26]. This was especially apparent for 17b, which is sensitive to the orientation of the assay and tends to bind more readily when it is immobilized on the chip, and gp120, as the analyte (in solution), has more freedom to fold. This was the orientation used in the previous study. Also, analyzing sensorgrams with global fits, as was done in the previous study, often results in artificially low KDs when analytes with very low off-rates are used, which is the case with CD4, and even more so with 17b, as these rates cannot accurately be measured at the sensitivity of the instrument with the dissociation times generally used in the laboratory. These factors may have contributed to the differences in KDs in the two studies.
Inhibition by KR21
We investigated whether the tier 1 and 2 alanine mutations established so far to reduce KR21 affinity similarly reduced PT dual-antagonism of gp120. First, we quantitated the CD4 IC50s of mutants using purified proteins (Fig. 7A, C). Having a highly functional gp120 surface allowed us to use lower concentrations of CD4 (35 nM) compared to the experiments done using captured gp120 (250 nM), and thus increased the sensitivity of our assay by decreasing the amount of competition of KR21 by the CD4. 35 nM is close to the KD of CD4 for all WT and mutant gp120’s that we purified (20 – 90 nM, Fig. 6(C, D), Table II, Fig. S7A, B). For tier 1 mutants, IC50s calculated were 85 ± 6 nM for WT, 3.3 ± 0.1 μM for T257A and 4.3 ± 0.2 μM for D474A gp120. The fold differences in IC50 for T257A and D474A compared to WT (39 ± 1 and 51 ± 2, respectively) are comparable to those obtained using captured protein (33 ± 27 and 43 ± 1). As with direct binding affinity, the tier 2 mutants E370A and M475A had a similar effect on CD4 inhibition in both the capture and direct immobilization assays. E370A had a CD4 IC50 for KR21 of 998 ± 68 nM and M475’s was 686 ± 16 nM, which were 12 ± 1 and 8 ± 0.2 fold greater than that for WT, 70 ± 1 nM (Fig. 7A, C). These differences are not significantly different from the fold differences obtained in the V5 capture screen, which were 10 ± 4 and 11 ± 5, respectively.
Figure 7. KR21 inhibition of binding of CD4 and 17b to purified gp120 mutants.

KR21 inhibition of binding of CD4 (A) and 17b Fab (B) to WT, T257A and D474A gp120 and inhibition of CD4 (C) and 17b IgG (D) binding to WT, E370A and M475A gp120. Varying amounts of KR21 were mixed with a constant concentration of CD4 or 17b and injected over the covalently immobilized gp120. For CD4 binding, normalized binding responses (RUs) were measured at 5 seconds before the end of association and plotted. For 17b binding, signals were read 10 s. before end of association (290 s.), binding signal obtained from binding of KR21 alone was subtracted, and resulting signals plotted vs. peptide concentration. Fits obtained from applying a 4-parameter sigmoidal logistic equation to the data points are overlaid. Results are an average of two data sets. E. 50% inhibitory concentrations for KR21 inhibition of CD4, 17b Fab (WT, T257A, D474A) and 17b IgG (WT, E370A, M475). ± indicates standard deviation, and Δ indicates fold difference with WT gp120 for the value to the left.
To investigate the effects of the mutations on the second facet of PT dual-antagonism, namely 17b/co-receptor inhibition, we determined IC50s for 17b inhibition by KR21 using the purified proteins (Fig. 7B). The T257A and D474A inhibition assays were done with 1 μM 17b Fab instead of IgG to be free of avidity effects in our direct binding assays, however this was not enough to yield measurable off-rates. As 1 μM 17b Fab and 400 nM 17b IgG were observed to have the same IC50’s for KR21 inhibition of binding to WT gp120 (within 10% of each other, not shown), thus 400 nM 17b IgG was used in the later assay with E370A and M475A in order to increase the signal from binding. These concentrations are about 5 and 6-fold greater than the measured KD’s to WT gp120, but are at or less than 3-fold above the KD for the mutants (Table II). These high KDs explain why such high concentrations had to be used to get measurable signals, and suggest that the amount of competition and displacement of KR21 from gp120 by the free 17b was minimal, and that the measured KR21 IC50s, at least for the mutants, were valid. The effects of the mutations on 17b inhibition (Fig. 7B) did not differ drastically from their effects on CD4 inhibition (Fig. 7A). KR21 IC50s for T257A and D474A binding to 17b Fab were 2 ± 0.1 and 6.2 ± 1.7 μM, compared to 110 ± 15 nM for WT, with a difference of 18 ± 1 and 56 ± 15 fold. In comparison, those for E370A and M475A were 1.2 ± 0.2 μM and 422 ± 32 nM, compared to 87 ± 2 for WT, with differences of 10 ± 2 and 4 ± 0.4 fold, respectively (Fig. 7C). Thus, the effects of the mutations do not seem to differ for direct binding versus CD4/17b inhibition, except for a relatively larger increase in KDs compared to IC50s for T257A and D474A. This larger increase could be due to the lower sensitivity of the direct binding assay at low signal levels and non-ideal sensorgram conditions. These sensorgrams could not be fitted with a global 1:1 Langmuir binding equation and were instead fitted using the steady state binding values. Comparing IC50s of KR21 for CD4 versus 17b, T257A and M475A mutations seem to affect 17b binding only half as much as they affect CD4 binding (fold difference with WT: 38 vs. 18 for T257A and 8 vs. 4 for M475A). However, the concentrations of 17b used were high (1 μM Fab for T257A and D474A and 400 nM IgG for E370A and M475A compared to 35 nM for CD4). These concentrations of 17b were around or less than 3-times their KDs to the mutant gp120s, however 4–6 times their KDs to WT gp120. Greater binding of 17b possibly drove up the concentration of KR21 required to inhibit WT gp120 as less gp120 was free to bind to KR21 and some displacement of KR21 from gp120 by 17b possibly occurred. Alternatively, M475A’s resistance to inhibition of 17b binding by KR21 (only 4-fold higher IC50 compared to WT) may be caused by a global conformational change induced by this mutation, as its KD to 17b was also 3-fold higher than that of WT (Fig. 6F, Table II), albeit this difference (3 ± 0.3) being statistically different than that for the IC50s (4 ± 0.3).
KR21 inhibition of HIV-1 pseudotyped with Env mutants
HNG-156, the parent peptide triazole from which KR21 was derived by deletion of noncritical C-terminal residues and addition of βAla-Lys(ε-Biotinyl)-Gly as an attachment site, was previously shown to inhibit both CD4 and 17b binding to gp120 and infection of target cells by HIV-1BaL pseudotyped virus [14]. As peptide binding and dual receptor site antagonism are thought to be essential to viral inhibition, we tested whether the effects seen with the mutants carried over to viral inhibition. Viruses were made by co-transfection of producer cells with plasmids expressing the wild-type and mutant HIV-1YU2 Env glycoproteins, the HIV-1 Gag-Pol packaging construct and firefly luciferase. Infection of target cells expressing CD4 and CCR5/CXCR4 yielded greater than 1 × 106 RLU (relative light unit) for all mutant and WT gp160 pseudotyped viruses (not shown). Single cycle infection was done in the presence of KR21 using the same concentration of virus. Results agreed with those from CD4 and 17 inhibition in that the T257A and D474A mutations conferred the highest resistance to KR21, resistance of E370A and M475A was moderate, and R476A conferred low resistance (Fig. 8). V430A did not differ from wild-type, as expected. V255A was not tested for viral inhibition. No inhibition was observed with virus carrying the A-MLV envelope, demonstrating the specificity of KR21 to HIV-1 Env. Similar results were obtained with the full length peptide HNG-156 [14] where the C-terminus S-E-A-M-M replaces KR21’s βAla-BtLys-Gly (Fig. S8), indicating that the residue dependence of KR21 is not specific to the unique amino acids on this peptide, but applies to the general class of PTs.
Figure 8. Effect of mutations on neutralization of single-round cell infection by KR21.

Infection by virus with WT and mutant gp120 mutations in the presence of KR21.
Mixture of 100 to 1 μM (A) or 300 to 3 μM (B) KR21 and single-cycle infective virus carrying WT or mutant gp120YU2 (Env) and the luciferase gene was added to target cells expressing CD4 and CCR5/CXCR4, incubated for 2–4 hr., and the resulting luciferase signal after 48 hr. was plotted as percent of that with no peptide added. Infectivity of virus carrying the A-MLV envelope was used as the negative control for neutralization.
Conservation of hot-spot residues
The extent of conservation was evaluated for residues in the mutationally-derived hot-spot for peptide triazole function. Residues belonging to the top two tiers of importance for KR21 binding and inhibition established in this study (T257, D474, E370, M475) and those from a third tier suggested to be important by the V5-tag capture assay (V255 and R476) were found to be highly conserved among all HIV-1 variants (Fig. 9C). Moreover, the spatial relationship of the amino acids in the functional epitope to each other and the rest of the protein were found to be conserved between the CD4-bound [27], mAb F105 bound [28] and the unliganded [29] gp120 structures (Fig. S9). Compared to the spread of all residues that were screened (Fig. 9A) these residues form a cluster (Fig. 9B) that is not more than 19 Å across, and can be spanned by the shortest full-potency PT identified to date, namely UM24 [10]: 7 residues, ca. 19 Å.
DISCUSSION
The aim of this study was to elucidate determinants of gp120 binding and inhibition by peptide triazoles, a unique class of HIV inhibitors that have dual antagonistic character, i.e. they inhibit binding of gp120 at both the CD4 and chemokine receptor sites. We screened gp120 alanine mutants, using an initial ELISA screen for CD4 and 17b inhibition of mutant gp120s by PTs and secondary V5-capture assays for binding by the biotin-tagged PT KR21 and its inhibition of CD4 binding to chosen mutants. Mutants that showed the greatest effects in the latter screens were purified, and their effects on PT binding and inhibition were quantitated following direct coupling of these proteins to SPR chips. The screen identified residues categorized into three tiers by level of importance for peptide binding and inhibition potency. The experiments with purified proteins focused on the most important residues (tiers 1 and 2). Moreover, these alanine mutants were tested for their effects on the infection of a pseudotyped virus. Through this analysis, we elucidated a functional epitope comprising 4 residues of most major and 2 other residues with more fractional importance. The 6 residues form a cluster that overlaps with the CD4 F43 binding cavity. The effects of these mutations on CD4 and 17b binding were minimal, demonstrating their selective impact on PT binding (Tables 1 and 2).
Residues in gp120 identified in this study as important for PT action are all well conserved, and some (E370, D474 and M475) are also CD4 contacts. Moreover, naturally found variations at these residue positions are known to be tolerated by PTs [11]. These findings explain the breadth of potency of PTs that was previously reported [11, 30]. The peptide triazoles are unique as inhibitors in targeting a conserved site in gp120 with nanomolar affinity, as opposed to co-receptor binding site inhibitors that bind with μM affinity or need CD4 for higher affinity [31]. They are also unique in being non-activating molecules i.e. dual antagonists, as opposed to CD4 mimics that activate the co-receptor site and also have close to micromolar affinity [18, 32], and as opposed to the BMS class molecules that act in a different manner than inhibition of the CD4 or coreceptor sites [33].
The KR21 hot spot overlaps with the CD4 binding site but is distinct from the co-receptor binding site which is 5 Å away from E370 and 12 Å from T257. Combined with calorimetric data [10] from other peptide triazoles demonstrating stabilization of gp120 upon peptide binding, this suggests that inhibition of the co-receptor site is allosteric, and occurs through linkage between the PT binding site and disruption of the co-receptor site due to conformational entrapment of gp120. The PT HNG-156 can bind (manuscript in preparation) to constrained mutants of gp120 with β3–beta;5 loop deletions that mimic the unliganded state of gp120 [34], to which the binding of the coreceptor surrogate mAb 17b is undetectable [35]. Moreover, the peptide triazole UM24 has no effect on the binding of an antibody (38.1a) recognizing a linear epitope [36] in the β21 strand of the bridging sheet (Figs. 9A, S9) that is shared with 17b and co-receptor, further supporting the view that inhibition of 17b/co-receptor by PT is allosteric rather than steric. PTs inhibit binding of antibodies to multiple epitopes that prefer the CD4-bound conformation, further supporting stabilization of an unactivated conformation by the peptide triazoles (manuscript in preparation).
PTs were found not to bind CD4 or 17b (not shown). Thus, we believe that they affect gp120 directly. PTs have a single binding site on gp120, as shown in previous work with full length (HNG-156) and sequence minimized (UM24) peptide triazoles using isothermal titration calorimetry, where 1:1 binding was observed between PTs and gp120 [10]. This result further argues for allosteric inhibition of the co-receptor site.
While the above map of a site critical for peptide triazole function was devised with protein alanine mutants, single-round pseudoviral inhibition results demonstrated that mutational effects on function carried over directly to the virus. This finding supports the potential of peptide triazoles as leads for virus intervention and prevention, as the sites they target are conserved, minimizing the possibility of viral resistance due to mutations at these sites that may affect peptide binding directly or allosterically. The PTs HNG-156 and UM24 were found to neutralize 100 and 80%, respectively, of viruses tested from clades A, B, C and D, showing that they are broadly active against diverse HIV-1 subtypes [11]. Sequence comparison of the gp120 components from these viruses showed that naturally found variations at the hot-spot residues did not by themselves confer virus resistance to PTs, arguing for general applicability of therapeutics derived from PTs (not shown).
Our work demonstrates an epitope that is equally important for PT binding and inhibition. How this site forms specific contacts with PTs cannot be explicitly concluded from the current study. Among other things, mutational effects can be both conformational and steric. However, the minimal effects on CD4 and 17b binding upon mutations in the residues identified in this site rule out a global effect on protein conformation. Looking ahead, biophysical and specificity analysis of mutants already obtained will help define the functional epitope in greater detail. In addition, double cycle and complementary mutational analysis can show the interrelationships between residues on the protein and the peptide, respectively. Ultimately, co-crystallization and X-ray diffraction analysis would greatly advance our understanding of the high-resolution contact map between PTs and gp120.
In summary, PT binding is greatly affected by mutation of two residues, D474 and T257 to alanine, whereas mutation of other residues have intermediate effects (E370, M475), with a possible, though yet unproven contribution by V255 and R476. Mutagenic effects on binding translate directly to reduction in both dual-antagonism i.e. ability of the peptide to inhibit gp120 binding to its two different site ligands, CD4, and the co-receptor surrogate mAb 17b, and to the ability of the peptide to inhibit pseudoviral infection. All of the mutation-sensitive residues cluster together, defining a localized site for PT action. The functional epitope is well conserved, and overlaps the conserved CD4 binding site. As such, these findings help identify a structural target against which further inhibitor design can be directed for translation of the dual-antagonist character of the PTs into orally bioavailable and bio-stable small molecule scaffolds.
Supplementary Material
Acknowledgments
Research was sponsored by the NIH grant P01 GM 56550.
We thank Caitlin Duffy for performing part of the transfections. J.S. was supported by the Ragon Institute of MGH, MIT and Harvard, the International AIDS Vaccine Initiative, the Center for HIV/AIDS Vaccine Immunology, and the late William F. McCarty-Cooper.
Abbreviations
- HPLC
high performance liquid chromatography
- MALDI-TOF
matrix assisted laser desorption/ionization - time of flight
- RU
response unit
- KD
equilibrium dissociation constant
- IC50
50% inhibitory concentration
- HIV
human immunodeficiency virus
- SD
standard deviation
- PBS
phosphate buffered saline
- ELISA
enzyme linked immunosorbent assay
- SPR
surface plasmon resonance
- mAb
monoclonal antibody
- RLU
relative light unit
References
- 1.UNAIDS. UNAIDS World AIDS Day Report. 2011 [Google Scholar]
- 2.Lucas GM, Chaisson RE, Moore RD. Highly active antiretroviral therapy in a large urban clinic: risk factors for virologic failure and adverse drug reactions. Ann Intern Med. 1999;131(2):81–7. doi: 10.7326/0003-4819-131-2-199907200-00002. [DOI] [PubMed] [Google Scholar]
- 3.Yerly S, et al. Transmission of antiretroviral-drug-resistant HIV-1 variants. Lancet. 1999;354(9180):729–33. doi: 10.1016/S0140-6736(98)12262-6. [DOI] [PubMed] [Google Scholar]
- 4.Yost R, Pasquale TR, Sahloff EG. Maraviroc: a coreceptor CCR5 antagonist for management of HIV infection. Am J Health Syst Pharm. 2009;66(8):715–26. doi: 10.2146/ajhp080206. [DOI] [PubMed] [Google Scholar]
- 5.Lalezari JP, et al. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J Infect Dis. 2005;191(7):1155–63. doi: 10.1086/427993. [DOI] [PubMed] [Google Scholar]
- 6.HIV and Hepatitis.com. Available from: www.hivandhepatitis.com.
- 7.Gallien S, et al. Emerging integrase inhibitor resistance mutations in raltegravir-treated HIV-1-infected patients with low-level viremia. AIDS. 2011;25(5):665–9. doi: 10.1097/QAD.0b013e3283445834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrer M, Harrison SC. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J Virol. 1999;73(7):5795–802. doi: 10.1128/jvi.73.7.5795-5802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopi HN, et al. Click chemistry on azidoproline: high-affinity dual antagonist for HIV-1 envelope glycoprotein gp120. Chem Med Chem. 2006;1(1):54–7. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]
- 10.Umashankara M, et al. The active core in a triazole peptide dual-site antagonist of HIV-1 gp120. Chem Med Chem. 2010;5(11):1871–9. doi: 10.1002/cmdc.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McFadden K, et al. Antimicrob Agents Chemother. 2011. Antiviral Breadth and Combination Potential of Peptide Triazole HIV-1 Entry Inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bastian AR, et al. Cell-free HIV-1 virucidal action by modified peptide triazole inhibitors of Env gp120. Chem Med Chem. 2011;6(8):1335–9. 1318. doi: 10.1002/cmdc.201100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biorn AC, et al. Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry. 2004;43(7):1928–38. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- 14.Gopi H, et al. Introducing metallocene into a triazole peptide conjugate reduces its off-rate and enhances its affinity and antiviral potency for HIV-1 gp120. J Mol Recognit. 2009;22(2):169–74. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishino T, et al. Slow-dissociation effect of common signaling subunit beta c on IL5 and GM-CSF receptor assembly. Cytokine. 2008;42(2):179–90. doi: 10.1016/j.cyto.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlsson R, Michaelsson A, Mattsson L. Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J Immunol Methods. 1991;145(1–2):229–40. doi: 10.1016/0022-1759(91)90331-9. [DOI] [PubMed] [Google Scholar]
- 17.Morton TA, et al. Analysis of the interaction between human interleukin-5 and the soluble domain of its receptor using a surface plasmon resonance biosensor. J Mol Recognit. 1994;7(1):47–55. doi: 10.1002/jmr.300070107. [DOI] [PubMed] [Google Scholar]
- 18.Lalonde JM, et al. Structure-Based Design, Synthesis, and Characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J Med Chem. 2012;55(9):4382–96. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang SH, et al. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J Virol. 2002;76(19):9888–99. doi: 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393(6686):648–59. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potapov V. CMA: Contact Map Analysis. 2012 PDB ID 1RZK]. Available from: http://ligin.weizmann.ac.il/cma/
- 22.Culp JS, et al. Regulated expression allows high level production and secretion of HIV-1 gp120 envelope glycoprotein in Drosophila Schneider cells. Biotechnology (N Y) 1991;9(2):173–7. doi: 10.1038/nbt0291-173. [DOI] [PubMed] [Google Scholar]
- 23.Pancera M, et al. Soluble mimetics of human immunodeficiency virus type 1 viral spikes produced by replacement of the native trimerization domain with a heterologous trimerization motif: characterization and ligand binding analysis. J Virol. 2005;79(15):9954–69. doi: 10.1128/JVI.79.15.9954-9969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopi H, et al. Structural determinants for affinity enhancement of a dual antagonist peptide entry inhibitor of human immunodeficiency virus type-1. J Med Chem. 2008;51(9):2638–47. doi: 10.1021/jm070814r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grupping K, et al. MiniCD4 protein resistance mutations affect binding to the HIV-1 gp120 CD4 binding site and decrease entry efficiency. Retrovirology. 2012;9(1):36. doi: 10.1186/1742-4690-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gift SK, et al. Conformational and structural features of HIV-1 gp120 underlying the dual receptor antagonism by cross-reactive neutralizing antibody m18. Biochemistry. 2011;50(14):2756–68. doi: 10.1021/bi101160r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang CC, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317(5846):1930–4. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, et al. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326(5956):1123–7. doi: 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon YD, et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci U S A. 2012;109(15):5663–8. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cocklin S, et al. Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J Virol. 2007;81(7):3645–8. doi: 10.1128/JVI.01778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acharya P, et al. Structure-based identification and neutralization mechanism of tyrosine sulfate mimetics that inhibit HIV-1 entry. ACS Chem Biol. 2011;6(10):1069–77. doi: 10.1021/cb200068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lalonde JM, et al. Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg Med Chem. 2011;19(1):91–101. doi: 10.1016/j.bmc.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si Z, et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci U S A. 2004;101(14):5036–41. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rits-Volloch S, et al. Restraining the conformation of HIV-1 gp120 by removing a flexible loop. EMBO J. 2006;25(20):5026–35. doi: 10.1038/sj.emboj.7601358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gift SK, et al. Conformational and Structural Features of HIV-1 gp120 Underlying the Dual Receptor Antagonism by Cross-Reactive Neutralizing Antibody m18. Biochemistry. 2011;50(14):2756–2768. doi: 10.1021/bi101160r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKeating JA, et al. Monoclonal antibodies to the C4 region of human immunodeficiency virus type 1 gp120: use in topological analysis of a CD4 binding site. AIDS Res Hum Retroviruses. 1992;8(4):451–9. doi: 10.1089/aid.1992.8.451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.