

Abstract
Objectives
To assess clinical, laboratory and radiographic findings associated with outcomes and to clarify more practical ways to predict hospital mortality in patients with acute exacerbation (AE) of chronic fibrosing interstitial pneumonia (CFIP).
Design
Single-centre retrospective cohort study.
Setting
University Hospital in Japan.
Participants
We identified 51 consecutive patients with AE of idiopathic CFIP through multidisciplinary discussion. Patients who had connective tissue disease, drug-induced lung disease, pneumoconiosis, hypersensitivity pneumonitis, sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis and eosinophilic pneumonia were excluded.
Interventions
There were no interventions.
Main outcome measures
The main outcome was determination of in-hospital mortality predictors. Other outcomes included clinical, laboratory and radiographic differences between non-survivors and survivors in patients with AE of CFIP.
Results
The mean age of the patients with AE of CFIP was 71 years. Compared with survivors, non-survivors had a significantly shorter duration of symptoms before admission, lower prevalence of peripheral distribution of ground-glass opacity and centrilobular emphysema (CLE) on thin-section CT, lower peripheral lymphocyte count, higher brain natriuretic peptide titre, lower Pao2:Fio2 (P:F) ratio, higher prevalence of systemic inflammatory response syndrome (SIRS) and higher SIRS score on admission (p=0.0069, 0.0032, 0.015, 0.040, 0.0098, 0.012, 9.9×10−7 and 5.4×10−6, respectively). Multivariate analysis revealed SIRS (HR=6.2810, p=0.015), CLE (HR=0.0606, p=3.6×10−5) and serum procalcitonin level (HR=2.7110, p=0.022) to be independent predictors of in-hospital mortality. A Kaplan-Meier estimate on the basis of stratification according to the presence or absence of SIRS and CLE demonstrated a distinct survival curve for each subset of patients.
Conclusions
Distinct survival curves documented by stratification according to the presence or absence of SIRS and CLE may provide basic information for a rational management strategy for patients with AE of CFIP on admission.
Keywords: Idiopathic pulmonary fibrosis, Centrilobular emphysema, Systemic inflammatory response syndrome
Article summary.
Article focus
Several independent predictors of mortality in patients with acute exacerbation (AE) of idiopathic pulmonary fibrosis have been identified. However, more practical ways to predict hospital mortality, which may be of use in routine medical care, are required for AE of chronic fibrosing interstitial pneumonia (CFIP).
This study was undertaken to identify practical mortality predictors in patients with AE of CFIP.
Key messages
Systemic inflammatory response syndrome and centrilobular emphysema, which have not previously been evaluated as factors possibly affecting outcome, were the most significant predictors of in-hospital mortality in patients with AE of CFIP. Stratification according to the presence or absence of these two factors documented distinct prognoses for the subsets of patients and thus may be helpful for enabling more appropriate management strategies in the future.
Strengths and limitations of this study
This study's strength was the identification of the novel, non-invasive and easily applicable predictors of in-hospital mortality in patients with AE of CFIP. The major limitation of the study was the single-centre retrospective design.
Introduction
In patients with a pre-existing chronic process of pulmonary fibrosis, such as idiopathic pulmonary fibrosis (IPF), an acute exacerbation (AE) is the development of an acute lung injury superimposed on the underlying disease. Although AE was first described in patients with IPF,1 it has also been reported in underlying diseases other than IPF, such as interstitial pneumonia associated with connective tissue disease,2 3 fibrotic non-specific interstitial pneumonia (NSIP),2 3 hypersensitivity pneumonitis4 and asbestosis.5 This suggests that a variety of chronic fibrosing processes of the lung can present a potential risk for AE.
Because a specific treatment for AE has not yet been established, the mortality rate in patients with AE of IPF remains high.6 Furthermore, patients with secondary usual interstitial pneumonia (UIP) or a pathological pattern of NSIP, who generally have a more favourable prognosis than those with IPF, also show high mortality from AE.2 3
Recently, factors predicting the onset of AE have been described in patients with IPF. These include a high modified Medical Research Council score, a high body mass index, a decline in forced vital capacity (FVC) at 6 months from diagnosis7; a low FVC.8 Factors affecting survival in patients with AE of IPF have also been described9 and include high-resolution CT (HRCT) patterns (diffuse/multifocal/peripheral) of acute pulmonary infiltrates, a degree of CT involvement and a serum lactate dehydrogenase titre. Another report has found that the extents of ground-glass attenuation with traction bronchiectasis or bronchiolectasis and honeycombing on HRCT were the two independent prognostic factors in patients with AE of IPF.10 CT findings directly associated with disease severity may predict patient survival; however, the exploration of more objective and easily applicable predictors of mortality would be of value for developing rational management strategies, including novel therapeutic approaches.11
In the present study, we retrospectively analysed 51 consecutive patients with AE of idiopathic chronic fibrosing interstitial pneumonia (CFIP) in order to identify novel in-hospital mortality predictors that are present on admission.
Methods
Subjects
Consecutive patients with AE of idiopathic CFIP admitted to our department between January 2009 and May 2012 were retrospectively studied. During that interval, 56 patients were admitted for AE of CFIP. Of those, 19 patients were successfully managed to hospital discharge (survivors) and 37 patients died during hospitalisation (non-survivors). Of the latter, five patients who died from causes other than AE were excluded: two acute myocardial infarction (both of the patients had a medical history of old myocardial infarction), one rupture of aortic aneurysm after the graft replacement was performed 3 months ago and two advanced lung cancer (AE was improving or did not progressively worsen by administration of glucocorticoids). Eight patients with suspected AE who were diagnosed with lower respiratory tract infection by bacterial culture of specimens from the lower respiratory tract (4 patients), bacterial antigens in the urine (2 patients) and an elevated serum β-d-glucan (2 patients) had been excluded in advance.
During the study period, 218 patients with idiopathic CFIP had been under the management of our department and of them, 116 patients had been diagnosed with IPF. Of the 51 patients studied, 26 had been outpatients of our department and the remaining 25 were referred ones. The mean interval between diagnosis of CFIP and admission was 42.3 months, although the data excluded three patients who had not been previously diagnosed with CFIP. Eleven of the 51 patients had been treated with immunosuppressants: glucocorticoids, 8 patients and glucocorticoids and cyclosporine, 3 patients.
Diagnostic criteria
The following features based on the previously used criteria12 were used to define an AE event: (1) aggravation of dyspnoea within 1 month, (2) decline of ≥10% in absolute FVC or a decline of ≥10 torr in Pao2 or a decline of ≥5% in Spo2, (3) new ground-glass opacities or consolidation on the chest radiograph or thin-section CT (TSCT), (4) negative respiratory culture and serological test results for respiratory pathogens and (5) no clinical evidence of pulmonary embolism, congestive heart failure or pneumothorax as a cause of acute decline.
Significant decline in oxygenation was confirmed by previous Pao2 or Spo2 in every patient. In 5 of 26 referred patients, previous chest radiographs had not been obtained; however, all of them evidently showed honeycombing on TSCT on admission. Extensive ground-glass opacity (GGO) was compatible with acute onset of respiratory distress. Cultures of the sputum or tracheobronchial aspirate for common bacteria, mycobacteria and fungi, urinary antigens for Streptococcus pneumoniae and Legionella pneumophila serotype 1, antigens for influenza A and B viruses by pharyngeal swab, antigenaemia for cytomegalovirus, serum antigen and antibody for Aspergillus, and a titre of serum β-d-glucan were examined in every patient studied and those disclosed negative results. Bronchoalveolar lavage was performed in 17 patients and all the bronchoalveolar lavage fluids were negative for routine microbiological culture. Of those, PCR detection for genomes of certain microorganisms (common bacteria, mycobacteria, Aspergillus and Pneumocystis jirovecii) was also performed in seven patients and gave negative results.
The diagnosis of idiopathic CFIP was first based on TSCT results: presence of a diffuse parenchymal lung disease with significant pulmonary fibrosis, defined as reticular opacities with peripheral and basal predominance, honeycombing, architectural distortion and/or traction bronchiectasis or bronchiolectasis.13 Patients found to have other distinct diseases on the basis of clinical and/or radiographic findings, associated with the development of pulmonary fibrosis, such as connective tissue disease, drug-induced lung disease, pneumoconiosis, hypersensitivity pneumonitis, sarcoidosis, pulmonary histiocytosis and lymphangioleiomyomatosis, were excluded. The diagnosis of IPF was made according to the 2011 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) statement on IPF.14 For this study, we diagnosed patients with IPF when they displayed features that fit the criteria for UIP as assessed by HRCT or met the criteria for UIP after a combination of HRCT and surgical lung biopsy findings.
CT imaging
TSCT was performed for all patients, usually on the day of admission, using a Somatom Sensation 64 (Siemens Medical Solutions, Forchheim, Germany). Images were obtained with 1.5 mm collimation and 1.5 mm slice intervals from the pulmonary apex to the lung base, reconstructed with a high-spatial-resolution algorithm and a small field of view. The images were analysed at a window level of −700 HU and a window width of 1400 HU.
The extent of the TSCT findings characteristic of AE and CFIP were determined; a thoracic radiologist and a pulmonary physician, both of whom were experts in interstitial lung diseases, examined the CT images without knowledge of any of the clinical, functional and radiographic findings. GGO was defined as an area of slightly increased attenuation in which the vessels remained visible. Honeycombing was defined as an accumulation of cystic spaces with thickened walls. Emphysema was defined as well-demarcated areas of decreased attenuation in comparison with contiguous normal lung and marginated by a very thin (<1 mm) or no wall with upper zone predominance. The extent of GGO and honeycombing was scored to the nearest 10% at the six lung zones: right upper and middle lobes, left upper segment and lingula and bilateral lower lobes. The scores at the six lung zones were then averaged out to obtain a mean score.15 The results by the two readers were well correlated (GGO: r=0.88, p=2.27×10−17 and honeycombing: r=0.92, p=2.15×10−20). Finally, GGOs at the time of AE were classified as peripheral, multifocal or diffuse parenchymal patterns, as described previously.16
Because concurrent emphysema may affect an accurate CT diagnosis of UIP and NSIP,17 CT images before admission were also reviewed, especially for distinguishing honeycombing from emphysema with GGO. We could access previous CT images for 40 of the 51 patients. In the remaining 11 patients, serial thoracic CT images were also reviewed in addition to the CT image obtained on admission not to misdiagnose emphysema with GGO as honeycombing.
Systemic inflammatory response syndrome
The diagnosis of systemic inflammatory response syndrome (SIRS) was determined at the time of admission, according to the previously defined criteria: temperature >38°C or <36°C, heart rate >90 bpm, respiratory rate >20 breaths/min or Paco2 <32 torr and white blood cell count >12 000/μL, <4000/μL or >10% band forms. Participants who met two or more of the criteria were diagnosed with SIRS.18
Data expression and the statistical analysis
Clinical data are expressed as mean±SD for continuous variables. Group comparisons were made using the Mann-Whitney U test for continuous variables. The χ2 and Fisher's exact tests were used for categorical variables. Logistic regression analysis was performed to determine the relationships between clinical parameters. A Kaplan-Meier model was generated to evaluate survival. Univariate and multivariate analyses using the Cox proportional hazards regression models were used to identify independent patient characteristics, laboratory data and CT predictors of hospital mortality. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing V.2.13.0). More precisely, it is a modified version of R Commander V.1.6-3 designed to include statistical functions frequently used in biostatistics.19 Statistical significance was defined as a p value of <0.05.
Results
Patient profiles and the symptom duration
The flow diagram in figure 1 shows how the patients were identified. We finally identified 51 patients with AE of idiopathic CFIP (37 men and 14 women) between January 2009 and May 2012. The mean age, male-to-female ratio, smoking history and the mean smoking index were similar in non-survivors and survivors. The mean duration of symptoms (cough or dyspnoea) previous to admission was significantly shorter in non-survivors (p=0.0069, table 1). The mean durations of hospitalisation were 33 days in non-survivors and 72 days in survivors.
Figure 1.
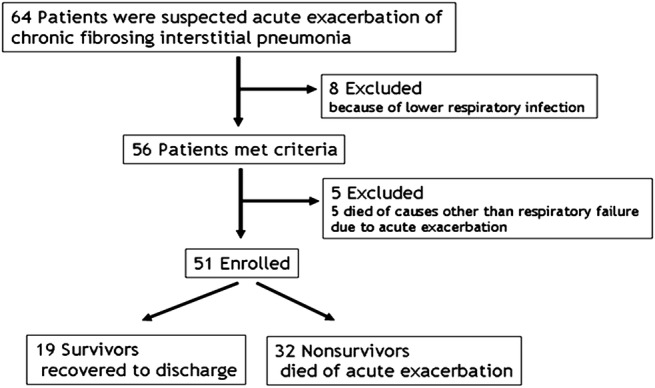
Flow diagram of patients with acute exacerbation of chronic fibrosing interstitial pneumonia.
Table 1.
Clinical characteristics of the patients studied
| Non-survivors | Survivors | p Value | |
|---|---|---|---|
| Age (years) | 72±10 | 69±5 | 0.22 |
| Male/female | 23/9 | 14/5 | 0.91 |
| Smoking history (±) | 23/9 | 15/4 | 0.48 |
| Smoking index (pack-years) | 28±25 | 45±37 | 0.071 |
| Duration of symptoms before admission (days) | 7±4 | 14±9 | 0.0069 |
Comparison between groups was performed by Mann-Whitney's U test (for continuous variables) or the χ2 test (for categorical variables).
Diagnosis of idiopathic CFIP and TSCT findings
In this cohort, 30 patients were diagnosed with IPF according to the 2011 criteria,14 and the remaining 21 patients were diagnosed with non-IPF. The latter group included one patient with upper lobe-dominant pulmonary fibrosis and two patients with familiar pulmonary fibrosis. The remaining 18 patients showed findings consistent with fibrotic NSIP on TSCT.20
The prevalence of IPF, the extent of GGO and the extent of honeycombing were not significantly different between non-survivors and survivors (table 2). The peripheral distributions of GGO and centrilobular emphysema (CLE) were significantly less prevalent in non-survivors (p=0.0032 and 0.015, respectively).
Table 2.
Classification of the CFIP and TSCT findings
| Non-survivors | Survivors | p Value | |
|---|---|---|---|
| IPF/non-IPF | 19/13 | 11/8 | 0.94 |
| Extent of honeycombing (%) | 17±13 | 11±10 | 0.12 |
| Extent of GGO (%) | 56±15 | 49±15 | 0.10 |
| Distribution of GGO: diffuse/multifocal/peripheral | 29/1/2 | 12/1/6 | 0.0032 |
| CLE (±) | 7/25 | 11/8 | 0.015 |
| PSE (±) | 15/17 | 12/7 | 0.39 |
Comparison between groups was performed by Mann-Whitney's U test (for continuous variables) or the χ2 test (for categorical variables).
CFIP, chronic fibrosing interstitial pneumonia; CLE, centrilobular emphysema; GGO, ground-glass opacity; IPF, idiopathic pulmonary fibrosis; PSE, paraseptal emphysema; TSCT, thin-section CT.
Laboratory data and SIRS
The mean peripheral blood lymphocyte count was significantly lower in non-survivors (p=0.040, table 3). The mean serum brain natriuretic peptide (BNP) titre was elevated in both groups and significantly higher in non-survivors (p=0.0098). The mean white blood cell counts, serum C reactive protein, serum Krebs von den Lungen-6, serum surfactant protein D, serum procalcitonin (PCT) and the mean D-dimer titres were elevated in both groups, but were not significantly different between groups. The mean Pao2:Fio2 (P:F) ratio on admission was significantly lower in non-survivors (p=0.012). On admission, 28 of 32 non-survivors and 4 of 19 survivors fulfilled the criteria for SIRS (p=9.9×10−7, table 3). The mean SIRS score was significantly higher in non-survivors (p=5.4×10−6). The SIRS score and the serum PCT level were positively correlated (p=0.0045, figure 2).
Table 3.
Laboratory data and SIRS on admission
| Non-survivors | Survivors | p Value | |
|---|---|---|---|
| WBC (/μL) | 12 041±6450 | 9712±3315 | 0.26 |
| Lymphocyte (/μL) | 1182±780 | 1447±570 | 0.040 |
| CRP (mg/dL) | 9.7±6.0 | 10.4±8.7 | 0.90 |
| KL-6 (U/mL) | 1513±687 | 1535±1013 | 0.63 |
| SP-D (ng/mL) | 427±321 | 360±262 | 0.54 |
| BNP (pg/mL) | 168±164 | 93±171 | 0.0098 |
| D-dimer (mg/mL) | 4.1±4.6 | 6.2±10.5 | 0.35 |
| PCT (ng/mL) | 0.33±0.60 | 0.13±0.12 | 0.17 |
| Pao2:Fio2 ratio | 157±53 | 213±81 | 0.012 |
| SIRS (±) | 28/4 | 4/15 | 9.9×10−7 |
| SIRS score | 2.5±0.9 | 1.1±1.0 | 5.4×10−6 |
Comparison between groups was by Mann-Whitney's U test (for continuous variables) or the χ2 test (for categorical variables).
BNP, brain natriuretic peptide; CRP, C reactive protein; KL-6, Krebs von den Lungen-6; PCT, procalcitonin; SIRS, systemic inflammatory response syndrome; SP-D, surfactant protein D; WBC, white blood cell count.
Figure 2.
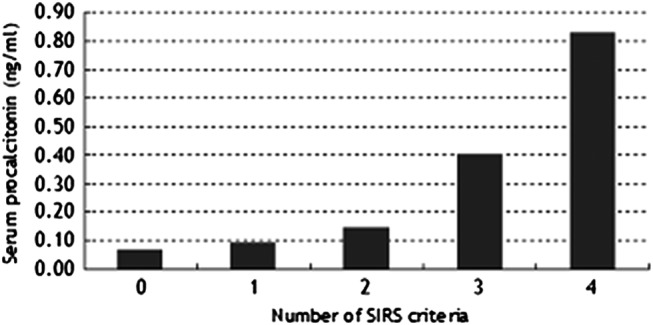
The number of systemic inflammatory response syndrome (SIRS) criteria was positively correlated with the serum procalcitonin concentration (p=0.045). The number of subjects was as follows: 7 (SIRS score 0), 12 (score 1), 11 (score 2), 13 (score 3) and 4 (score 4), respectively. Logistic regression analysis was performed for significance.
Treatment and the response
Glucocorticoid pulse therapy (methylprednisolone at 1000 mg/day for 3 consecutive days) was performed in 31 of 32 non-survivors and all of the 19 survivors (table 4). Immunosuppressants other than glucocorticoids and mechanical ventilation were used at similar rates in the two groups. The mean P:F ratio on the day after glucocorticoid pulse therapy was significantly lower in non-survivors (p=0.00018).
Table 4.
Treatment and the response
| Non-survivors | Survivors | p Value | |
|---|---|---|---|
| Glucocorticoid pulse therapy (±) | 31/1 | 19/0 | 1.0 |
| Immunosuppressants other than glucocorticoids (±) | 11/21 | 2/17 | 0.096 |
| Mechanical ventilation (±) | 12/20 | 3/16 | 0.12 |
| Pao2:Fio2 ratio on the day after glucocorticoid pulse therapy | 147±67 | 267±108 | 0.00014 |
Comparison between groups was performed by Mann-Whitney's U test (for continuous variables) or the χ2 test (for categorical variables).
Prognosis
In non-survivors, the median survival time was 33 days. In survivors, two patients died from a second AE event after discharge from hospital. The overall survival was 67% at 30 days, 43% at 60 days and 40% at 90 days after admission. The Kaplan-Meier estimate for overall survival revealed that the survival rate reached a plateau of approximately 35% at approximately 180 days from admission (figure 3). In this cohort, the TSCT classification (a definite UIP pattern, a possible UIP pattern and inconsistent with the UIP pattern) did not reveal a significant difference in survival by the Kaplan-Meier estimate (figure 4, p=0.51).
Figure 3.
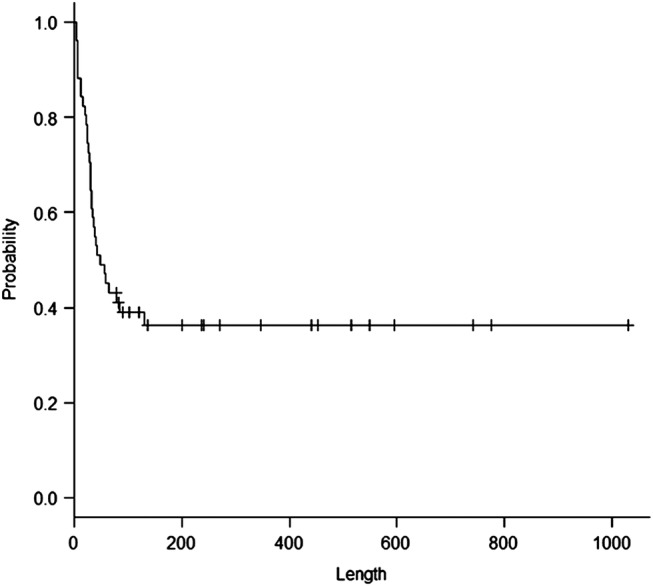
A Kaplan-Meier estimate for overall survival. The x axis indicates days after admission.
Figure 4.
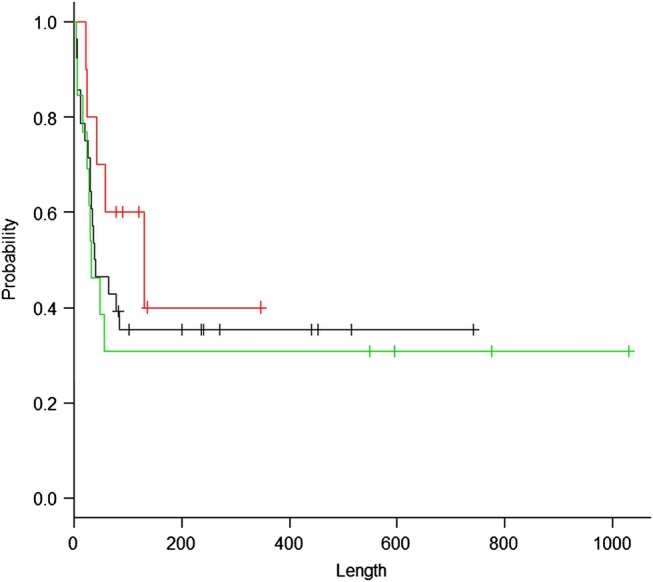
A Kaplan-Meier estimate for survival according to the thin-section CT classification: define usual interstitial pneumonia (UIP) pattern (black, 28 patients), possible UIP pattern (green, 13 patients) and inconsistent with UIP pattern (red, 10 patients). The x axis indicates days after admission.
Univariate and multivariable analyses of survival
Univariate analysis (table 5) revealed that symptom duration prior to admission, the extent of honeycombing on CT, the presence of CLE, serum PCT level, P:F ratio on admission, the presence of SIRS and the SIRS score were significant predictors of mortality. The P:F ratio on the day after glucocorticoid pulse therapy was also significant in univariate analysis. Multivariable analysis revealed that the serum PCT (HR per 10% increase was 2.7110, 95% CI 1.1770 to 6.4890; p=0.022), the presence of CLE (HR 0.0606, 95% CI 0.0161 to 0.2290; p=3.6×10−5) and the presence of SIRS (HR 6.2810, 95% CI 1.4220 to 27.7500; p=0.015) remained significant predictors after adjusting for age, sex, P:F ratio on admission and extent of honeycombing (table 6). When five patients in whom the cause of death was other than AE were included in the multivariate analysis, PCT (HR 2.3980, 95% CI 1.1000 to 5.2240; p=0.028), CLE (HR 0.1752, 95% CI 0.06269 to 0.4894; p=0.00089) and SIRS (HR 5.2600, 95% CI 1.4950 to 18.5100; p=0.0097) continued to be significant and, in addition, age also showed significance (HR 1.0440, 95% CI 1.0020 to 1.0870; p=0.039).
Table 5.
Univariate analysis of survival
| HR | 95% CI | p Value | |
|---|---|---|---|
| Age (years) | 1.018 | 0.973 to 1.065 | 0.35 |
| Male sex | 1.083 | 0.50 to 2.345 | 0.84 |
| Duration of symptoms before admission (days) | 0.893 | 0.824 to 0.969 | 0.0066 |
| Smoking index (pack-years) | 0.9995 | 0.9989 to 1.0001 | 0.14 |
| IPF/non-IPF | 1.024 | 0.501 to 2.091 | 0.95 |
| Extent of ground-glass opacity (%) | 1.014 | 0.990 to 1.039 | 0.23 |
| Extent of honeycombing (%) | 1.031 | 1.004 to 1.06 | 0.023 |
| CLE (±) | 0.356 | 0.153 to 0.829 | 0.017 |
| PSE (±) | 0.785 | 0.392 to 1.574 | 0.50 |
| Lymphocyte (/μL) | 0.9996 | 0.9990 to 1.0002 | 0.22 |
| CRP (mg/dL) | 0.9929 | 0.947 to 1.041 | 0.77 |
| KL-6 (U/mL) | 1.000 | 0.9996 to 1.0004 | 0.88 |
| SP-D (ng/mL) | 1.0005 | 0.9992 to 1.0019 | 0.43 |
| BNP (pg/mL) | 1.001 | 0.9992 to 1.0028 | 0.27 |
| D-dimer (mg/mL) | 0.976 | 0.921 to 1.045 | 0.49 |
| PCT (ng/mL) | 1.889 | 1.063 to 3.545 | 0.030 |
| Pao2:Fio2 ratio on admission | 0.992 | 0.987 to 0.997 | 0.0038 |
| Pao2:Fio2 ratio on the day after glucocorticoid pulse therapy | 0.989 | 0.984 to 0.994 | 1.75×10−5 |
| SIRS (±) | 11.85 | 3.551 to 39.54 | 5.78×10−5 |
| Number of SIRS criteria | 1.98 | 1.448 to 2.707 | 1.87×10−5 |
Cox proportional hazards regression models were used for the statistical analysis.
BNP, brain natriuretic peptide; CLE, centrilobular emphysema; CRP, C reactive protein; IPF, idiopathic pulmonary fibrosis; KL-6, Krebs von den Lungen-6; PCT, procalcitonin; PSE, paraseptal emphysema; SIRS, systemic inflammatory response syndrome; SP-D, surfactant protein D.
Table 6.
Multivariate analysis of survival
| HR | 95% CI | p Value | |
|---|---|---|---|
| Age (years) | 1.0340 | 0.9847 to 1.0860 | 0.18 |
| Male sex | 2.4510 | 0.9260 to 6.4890 | 0.071 |
| PCT (ng/mL) | 2.7110 | 1.1770 to 6.2450 | 0.022 |
| Pao2:Fio2 ratio on admission | 0.9941 | 0.98232 to 1.0050 | 0.41 |
| Extent of honeycombing (%) | 1.0210 | 0.9881 to 1.0550 | 0.22 |
| CLE (±) | 0.0606 | 0.0161 to 0.2290 | 3.6×10−5 |
| SIRS (±) | 6.2810 | 1.4220 to 27.7500 | 0.015 |
Cox proportional hazards regression models were used for the statistical analysis.
CLE, centrilobular emphysema; PCT, procalcitonin; SIRS, systemic inflammatory response syndrome.
On the basis of the results of multivariate analysis, we divided 51 patients into four subgroups according to the presence or absence of CLE and SIRS (group 1: SIRS− and CLE+, group 2: SIRS− and CLE−, group 3: SIRS+ and CLE+, and group 4: SIRS+ and CLE−). A Kaplan-Meier estimate according to this subgrouping revealed a clear distinction in the prognoses between the four subgroups (figure 5, p=0.0025). All patients in group 1 recovered and survived to discharge; by contrast, all patients in group 4 died of AE.
Figure 5.
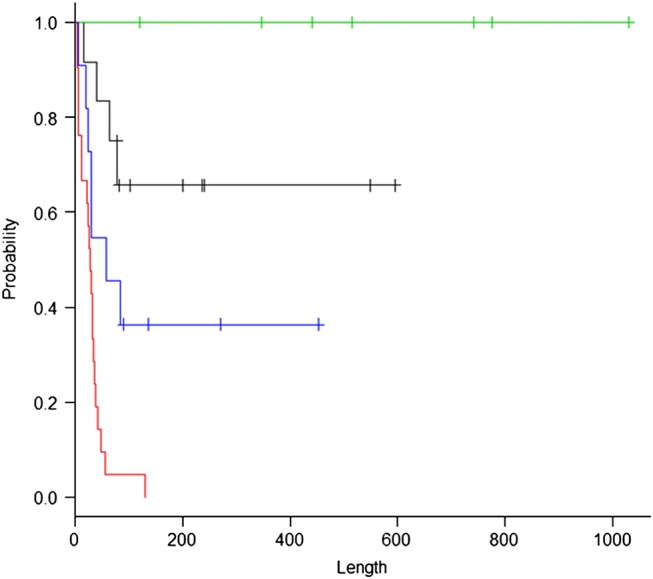
The stratification according to the presence or absence of centrilobular emphysema (CLE) and systemic inflammatory response syndrome (SIRS) revealed distinct survival curves of the four groups. Group 1 (green): SIRS− and CLE+ (n=7), group 2 (black): SIRS− and CLE− (n=12), group 3 (blue): SIRS+ and CLE+ (n=11) and group 4 (red): SIRS+ and CLE− (n=21). The x axis indicates days after admission.
Discussion
In the present study, the signs and symptoms of 18 of 21 patients diagnosed with non-IPF were suggestive of fibrotic NSIP on TSCT.20 There is the possibility that patients with IPF were included in the non-IPF group, and vice versa. However, whether a patient had IPF or not was not associated with the outcome in our cohort. Furthermore, the prognostic difference between patients with definite UIP, possible UIP and findings inconsistent with UIP on TSCT was not significant. Thus, we considered that there were convincing reasons to place all participants studied in the CFIP category. There have been a few reports on AE of non-IPF, including a study of patients with idiopathic NSIP2 3 in which the overall mortality rate was as high as 50%. This suggests that AE, a manifestation of diffuse alveolar damage, may commonly be fatal, irrespective of an individual pattern of CFIP.
Attention should be paid to the differences between survivors and non-survivors in clinical, laboratory and radiographic findings in this cohort. The relatively short symptom duration before admission in non-survivors might be dependent on the severity of the lung injury that has not yet been strictly objectified. Radiographically, factors for discriminating between non-survivors and survivors were the distribution pattern of acute pulmonary infiltrates and the presence of CLE, but not the presence of paraseptal emphysema. CT patterns have been shown to be associated with mortality in patients with AE of IPF;9 however, there may be a degree of subjectivity in the diagnosis of this condition, and interobserver differences must be considered. Among laboratory data, the serum BNP level and the P : F ratio on the day after glucocorticoid pulse therapy, as well as those on admission, were discriminative. We performed echocardiography in every patient with signs and symptoms clinically suggestive of cardiac dysfunction or those with increased serum BNP, and none showed left ventricular dysfunction. The higher P : F ratio after glucocorticoid pulse therapy suggests that this treatment might be effective for a certain subset of patients. The presence of SIRS and the number of SIRS criteria fulfilled were the factors that provided the most significant discrimination between survivors and non-survivors.
The univariate analysis revealed that the extent of honeycombing was associated with survival; this was not apparent in the group comparison. On the basis of the results of the univariate analysis, we performed a multivariate analysis of survival, and it revealed some notable results: the most significant factors for predicting in-hospital mortality were the presence of SIRS and the absence of CLE on TSCT, and the serum PCT level was also found to be associated with outcome.
The presence of SIRS, first defined in 1992, indicates a systemic inflammatory response to a variety of severe clinical insults, including non-infectious causes such as trauma, burns, pancreatitis, etc.18 The definition of SIRS is simple, and thus can be easily applied in routine medical care; however, the clinical significance of SIRS has been controversial. In infected participants, the number of SIRS criteria fulfilled did not influence patient outcome, despite the fact that the presence of organ dysfunction or shock showed prognostic significance.21 In our study, SIRS was significantly more prevalent in non-survivors, and was one of the most significant predictors of hospital mortality. The influence of SIRS on mortality in patients with AE of CFIP has not yet been examined, probably because the disease process has been recognised as a disorder compartmentalised to the lung. Compared with stable IPF patients, patients with AE of IPF has been reported to show increases in serum biomarkers associated with endothelial cell injury, such as thrombomodulin or plasminogen activator inhibitor-1 (PAI-1).22 In addition, increases in levels of interleukin 8 or intracellular adhesion molecule 1, known as molecules related to neutrophil recruitment to the lung, were reported to be independent mortality predictors in patients with acute lung injury/acute respiratory distress syndrome.23 In our cohorts, the mean ratio of bronchoalveolar neutrophils in 17 patients who underwent bronchoalveolar lavage was 20% (data not shown). These data suggest a possibility that acute lung injury mediates systemic inflammation via certain circulating molecules, as in the case of AE of CFIP. The significance of SIRS as a mortality predictor should be confirmed in future studies. Investigations to determine the mechanisms of SIRS development in patients with AE of CFIP may be essential to allow the development of more specific treatments.
PCT, a prohormone of calcitonin, is produced in the medullary C cells of the thyroid gland. It was described as an infection parameter in the early 1990s24 and is now considered to be a useful serum biomarker for early diagnosis of bacterial infections.25 On the other hand, increase of serum PCT levels caused by non-infectious processes have also been reported. In a retrospective analysis of patients who underwent cardiopulmonary bypass surgery but did not have postoperative infection, Hensel et al26 described a significant increase in PCT in patients with both SIRS and acute lung injury, compared with those with SIRS alone. In their cohort, PCT levels were also elevated in patients without SIRS as well as in patients with SIRS alone, and the levels were statistically similar; this indicates that acute lung injury might play a critical role in the facilitated generation of PCT. Rapid increases of PCT levels in patients with non-bacterial pneumonitis caused by inhalational burn have also been reported.27 In our cases of AE, serum PCT levels were of prognostic significance, and were also positively correlated with the number of SIRS criteria. The measurement of PCT has two key implications in our cohort. First, PCT was useful for excluding the possibility of respiratory infections, because most of the patients had PCT levels of <0.25 ng/mL (data not shown), which is below the level suggestive of severe pneumonia and/or bacteraemia.28 Second, in the absence of overt respiratory infection, a relatively slight but significant elevation of PCT should not be ignored. PCT was proved to be associated with hospital mortality in both the univariate and multivariate analyses. Although the source of PCT in patients with AE of CFIP has not been confirmed, pulmonary neuroendocrine cells are possible candidates, because an abundance of calcitonin per unit weight of lung tissue has been described.27 29 The serum PCT level was positively correlated with the number of SIRS criteria, suggesting that PCT could be a novel candidate biomarker for AE of CFIP.
One report has indicated that the absence of smoking history might positively influence the onset of AE in patients with IPF8; however, CLE has not been evaluated as a mortality predictor. There are a few possible reasons why the presence of CLE was associated with outcome. First, in some participants with both CFIP and CLE, diffuse alveolar damage, a histopathological hallmark of AE of CFIP, was not present. Second, the pathogenesis of CLE itself antagonises that of ongoing diffuse alveolar damage. Finally, glucocorticoids are more effective for participants who have background CLE. As mentioned above, we ruled out the possibility of obvious cardiac dysfunction by echocardiography in the patients studied. Six of 11 survivors with CLE underwent bronchoalveolar lavage soon after admission (data not shown), and none showed overt alveolar haemorrhage. Although AE in patients with combined pulmonary fibrosis and emphysema has recently been reported,30 its curability has not been described. In patients with chronic obstructive pulmonary disease (COPD), gene expression analysis and immunohistochemistry showed that urokinase plasminogen activator (PLAU) and urokinase plasminogen activator receptor (PLAUR) were found to be overexpressed in alveolar macrophages and the bronchial epithelium.31 In infant respiratory distress syndrome, a condition characterised by intra-alveolar fibrin deposition, the ratio of PAI-1 to PLAU in the tracheal aspirates was higher than that in control participants,32 suggesting a role for PLAU in intra-alveolar fibrinolysis. Lung-specific overexpression of PLAU in mice was shown to reduce the accumulation of collagen in the lung and reduced mortality after bleomycin-induced lung injury.33 Thus, constitutive activation of the PLAU–PLAUR system in participants with CLE, a histological hallmark of COPD, may antagonise the activation of PAI-1, a possible biomarker of AE of CFIP. Generally, COPD itself has been demonstrated to show glucocorticoid resistance.34 It remains to be determined whether there are any differences in the histological patterns of AE and the efficacy of glucocorticoids between patients with CFIP alone and those with both CFIP and CLE. The histological patterns could be unravelled by analysis of surgical lung biopsy specimens, and the efficacy of glucocorticoids could be clarified with the accumulation of data from future clinical cases.
The Kaplan-Meier estimate for survival according to the subgrouping by the presence or absence of SIRS and CLE clearly divided the patients into four subgroups. Every patient in group 1 (SIRS−CLE+) was successfully treated with glucocorticoids and discharged. Although an effective pharmaceutical therapy has not been established for patients with AE of IPF,12 the present study is the first to document a subgroup of CFIP patients who derive a possible benefit from glucocorticoid therapy during AE. Five of the seven cases in group 1 were of IPF. By contrast, all 22 patients in group 4 (SIRS+CLE−) died due to respiratory failure secondary to AE. In this subgroup, any treatment option such as polymyxin B-immobilised fibre column treatment11 should be considered. An indication for the use of glucocorticoids should also be discussed in the future.
The limitations of the present study are as follows. First, the analysis was retrospective. However, we consecutively enrolled every participant admitted with AE of CFIP from January 2009, and collected the hospitalisation medical records as completely as possible. Second, this study was undertaken in a single medical institution, and therefore the number of patients studied was limited. Third, the diagnosis of CFIP and the classifications were somewhat dependent on the TSCT findings. Because most of the patients were in moderate-to-severe respiratory failure (two-thirds of the patients studied had a P : F ratio of ≤200, data not shown), surgical lung biopsies on admission were difficult to perform. Half of the patients studied had been referred, and among those, none had undergone surgical lung biopsy. Consequently, five patients underwent surgical lung biopsy in this study. We tried, as much as possible, to rule out known entities associated with the development of pulmonary fibrosis. Finally, most of the referred participants had not undergone pulmonary function tests before admission. Therefore, we could not evaluate pulmonary function test results as mortality predictors.
Several mortality predictors have been identified in patients with AE of IPF. However, it is unclear how those factors could contribute to the management of this possibly fatal disorder in routine medical practice. We expect that the prognostic significance of CLE and SIRS, factors by which patients could be subgrouped on admission, could contribute to the rational management of patients with AE of CFIP. The utility of these prognostic factors should be prospectively investigated in future cohorts.
Supplementary Material
Footnotes
Contributors: YU conceived and designed the study. YU, AK, FS, AS, KIK, KH and MK were involved in the acquisition, analysis, interpretation of data and in writing the manuscript before submission. All authors have read and approved the final version of the manuscript.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None.
Ethics approval: Saitama Medical University.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Kondoh Y, Taniguchi H, Kawabata Y, et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest 1993;103:1808–12 [DOI] [PubMed] [Google Scholar]
- 2.Park IN, Kim DS, Shim TS, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest 2007;132:214–20 [DOI] [PubMed] [Google Scholar]
- 3.Silva CI, Müller NL, Fujimoto K, et al. Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging 2007;22:221–9 [DOI] [PubMed] [Google Scholar]
- 4.Olson AL, Huie TJ, Groshong SD, et al. Acute exacerbations of fibrotic hypersensitivity pneumonitis: a case series. Chest 2008;134:844–50 [DOI] [PubMed] [Google Scholar]
- 5.Richard L, Kradin RL, Subba R, et al. Case records of the Massachusetts General Hospital. Case 12–2010: an 89-year-old man with progressive dyspnoea. N Engl J Med 2010;362:1522–31 [DOI] [PubMed] [Google Scholar]
- 6.Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40 [DOI] [PubMed] [Google Scholar]
- 7.Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010;27:103e10. [PubMed] [Google Scholar]
- 8.Song JW, Hong SB, Lim CM, et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011;37:356–63 [DOI] [PubMed] [Google Scholar]
- 9.Akira M, Kozuka T, Yamamoto S, et al. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008;178:372–8 [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto K, Taniguchi H, Johkoh T, et al. Acute exacerbation of idiopathic pulmonary fibrosis: high-resolution CT scores predict mortality. Eur Radiol 2012;22:83–92 [DOI] [PubMed] [Google Scholar]
- 11.Abe S, Azuma A, Mukae H, et al. Polymyxin B-immobilized fiber column (PMX) treatment for idiopathic pulmonary fibrosis with acute exacerbation: a multicenter retrospective analysis. Intern Med 2012;51:1487–91 [DOI] [PubMed] [Google Scholar]
- 12.Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007;176:636–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005;26:586–93 [DOI] [PubMed] [Google Scholar]
- 14.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichikado K, Suga M, Müller NL, et al. Acute interstitial pneumonia: comparison of high-resolution computed tomography findings between survivors and nonsurvivors. Am J Respir Crit Care Med 2002;165:1551–6 [DOI] [PubMed] [Google Scholar]
- 16.Akira M, Hamada H, Sakatani M, et al. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol 1997;168:79–83 [DOI] [PubMed] [Google Scholar]
- 17.Akira M, Inoue Y, Kitaichi M, et al. Usual interstitial pneumonia and nonspecific interstitial pneumonia with and without concurrent emphysema: thin-section CT findings. Radiology 2009; 251:271–9 [DOI] [PubMed] [Google Scholar]
- 18.Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992;101:1644–55 [DOI] [PubMed] [Google Scholar]
- 19.Kanda Y. doi: 10.1038/bmt.2012.244. Investigation of the freely-available easy-to-use software “EZR” (Easy R) for medical statistics. Bone Marrow Transplant 2013;48:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.American Thoracic Society/European Respiratory Society International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002;165:277–304 [DOI] [PubMed] [Google Scholar]
- 21.Alberti C, Brun-Buisson C, Goodman SV, et al. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med 2003;168:77–84 [DOI] [PubMed] [Google Scholar]
- 22.Collard HR, Calfee CS, Wolters PJ, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2010;299:L3–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClintock D, Zhuo H, Wickersham N, et al. Biomarkers of inflammation, coagulation and fibrinolysis predict mortality in acute lung injury. Crit Care 2008;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assicot M, Gendrel D, Carsin H, et al. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993;341:515–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert DN. Procalcitonin as a biomarker in respiratory tract infection. Clin Infect Dis 2011;52:S346–50 [DOI] [PubMed] [Google Scholar]
- 26.Hensel M, Volk T, Döcke WD, et al. Hyperprocalcitonemia in patients with noninfectious SIRS and pulmonary dysfunction associated with cardiopulmonary bypass. Anesthesiology 1998;89:93–104 [DOI] [PubMed] [Google Scholar]
- 27.Nylen ES, Jeng J, Jordan MH, et al. Pneumonitis-associated hyperprocalcitonemia. Am J Med Sci 1996;312:12–18 [DOI] [PubMed] [Google Scholar]
- 28.Berg P, Lindhardt BØ. The role of procalcitonin in adult patients with community-acquired pneumonia—a systematic review. Dan Med J 2012;59:A4357. [PubMed] [Google Scholar]
- 29.Becker KI, Silva OI, Snider RH. The pathophysiology of pulmonary procalcitonin.The endocrine lung in health and disease. Becker KI, Gazdar AF, eds. Philadelphia, WB Saunders 1984:277–99 [Google Scholar]
- 30.Kishaba T, Shimaoka Y, Fukuyama H, et al. A cohort study of mortality predictors and characteristics of patients with combined pulmonary fibrosis and emphysema. BMJ Open 2012;2:e000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang IM, Stepaniants S, Boie Y, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med 2008;177:402–11 [DOI] [PubMed] [Google Scholar]
- 32.Cederqvist K, Sirén V, Petäjä J, et al. High concentrations of plasminogen activator inhibitor-1 in lungs of preterm infants with respiratory distress syndrome. Pediatrics 2006;117:1226–34 [DOI] [PubMed] [Google Scholar]
- 33.Sisson TH, Hanson KE, Subbotina N, et al. Inducible lung-specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2002;283:L1023–32 [DOI] [PubMed] [Google Scholar]
- 34.Barnes PJ. Corticosteroid effects on signaling. Eur Respir J 2006;27:413–26 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.