

Significance
Thymocytes express a diverse repertoire of T-cell antigen receptors. Stringent selection processes eliminate autoreactive cells and guide useful thymocytes to develop into CD4 or CD8 lineages. Development always generates more CD4 than CD8 T cells, but it is not understood why. Our study used mathematics to investigate the basis of this asymmetric lineage development. Although similar numbers of CD4 and CD8 precursors start selection, our analysis revealed unexpectedly high death rates in developing thymocytes. In particular, CD8 precursors were more susceptible to death than CD4 lineage cells, and this was a major contributor to the high CD4:CD8 ratio of development.
Keywords: CD4 T cells, CD8 T cells
Abstract
It has long been recognized that the T-cell compartment has more CD4 helper than CD8 cytotoxic T cells, and this is most evident looking at T-cell development in the thymus. However, it remains unknown how thymocyte development so favors CD4 lineage development. To identify the basis of this asymmetry, we analyzed development of synchronized cohorts of thymocytes in vivo and estimated rates of thymocyte death and differentiation throughout development, inferring lineage-specific efficiencies of selection. Our analysis suggested that roughly equal numbers of cells of each lineage enter selection and found that, overall, a remarkable ∼75% of cells that start selection fail to complete the process. Importantly it revealed that class I-restricted thymocytes are specifically susceptible to apoptosis at the earliest stage of selection. The importance of differential apoptosis was confirmed by placing thymocytes under apoptotic stress, resulting in preferential death of class I-restricted thymocytes. Thus, asymmetric death during selection is the key determinant of the CD4:CD8 ratio in which T cells are generated by thymopoiesis.
Development of CD4 and CD8 lineage cells from common thymic precursors is one of the most fundamental developmental processes in the adaptive immune system. The predominance of CD4 over CD8 T-cell populations in the periphery has been apparent since helper and cytotoxic T cells were first delineated more than 30 y ago (1), but the cause of this signature bias has remained obscure. A key contribution arises from the ratio in which CD4 and CD8 lineage T cells are generated by the thymus. Single-positive (SP) thymocytes exist in the thymus at ∼4:1, a ratio that is highly conserved across mouse strains and other species, suggesting that the developmental mechanisms involved are fundamental to the processes that give rise to mature T cells in the thymus. During thymocyte development, T-cell antigen receptor (TCR) genes undergo somatic rearrangements to generate a broad repertoire of TCR structures. Negative and positive selection of thymocytes results in the deletion of autoreactive thymocytes and ensures that class I and class II MHC reactivity is correlated with CD8 and CD4 lineage specification. The molecular mechanisms underpinning these processes are increasingly well understood. Although survival of thymocytes is regulated by expression of Bcl2 family members, up-regulation of the BH3-only family member Bim has been specifically implicated as a key event in negative selection of thymocytes (2). During positive selection, class II recognition is thought to induce strong persistent signaling that results in a cascade of transcriptional regulation by factors such as GATA3 and T-helper-inducing POZ/Krüppel-like factor, which results in fixation of cells to the CD4 lineage (3, 4). In contrast, weaker or transient signaling by class I MHC results in the cytokine-dependent induction of a Runx3-mediated transcriptional program that induces CD8 lineage commitment (5–8).
Despite this extensive characterization of molecular mechanisms, the fundamental question remains: What is the source of the bias toward the generation of CD4 lineage cells during thymic development? In principle, this bias can arise from two sources, which are not mutually exclusive: (i) There may be a substantial difference in the numbers of precursor CD4 CD8 double-positive (DP) thymocytes expressing functional TCR rearrangements that are able to recognize MHC class I vs. MHC class II, or (ii) selection of the two lineages may occur with different efficiencies as they progress toward CD4 SP or CD8 SP status, that is, the probabilities with which cells mature or die at each stage of development. However, because cells of each lineage cannot be enumerated separately until late in development, identifying where lineage-specific selection bottlenecks and/or asymmetries between lineages originate is difficult. To complicate the picture, our previous work has established that the two lineages develop with very different dynamics: CD4 lineage cells develop in 24–36 h from DP precursors, whereas CD8 lineage cells take in excess of 72 h to develop (9). Thus, at steady state in the thymus, each developmental stage comprises a mixture of lineages dynamically turning over, and maturing and dying, at potentially different rates. Disentangling and quantifying these processes to understand how the CD4 SP:CD8 SP ratio arises is therefore challenging.
Here, we addressed this problem using an experimental system that allows us to follow cohorts of thymocytes through development, in combination with a quantitative analysis that naturally describes dynamic populations. Together, these tools allowed us to measure the efficiencies with which cells of all lineages combined progress through development and to identify the asymmetries in the rates of maturation and death between class I and class II MHC-restricted populations that underlie both the emergence of the CD4 SP:CD8 SP bias and the different developmental kinetics of the two populations. Our analysis suggests that the CD4 bias derives predominantly from differences in lineage-specific rates of death at a key DP branch point, and not from any substantial imbalance in the numbers of class I- and class II-restricted precursors entering selection. Analyzing thymocytes placed under apoptotic stress confirmed the key prediction that CD8 lineage thymocytes are specifically more susceptible to apoptosis during selection and that this underlies the marked CD4 lineage bias in thymocyte development.
Results
Thymocyte Development Can Be Described Quantitatively with a Dynamical Mathematical Model.
To understand quantitative aspects of thymopoiesis better, we developed a model of differentiation and death across key stages of development. The model focused on selection of DP thymocytes to either CD4 SP or CD8 SP thymocyte populations. Previous studies have identified three populations of DP thymocytes that are at distinct temporal stages of development (9). DP1 thymocytes are TCRloCD5lo and constitute preselection thymocytes; DP2 thymocytes are TCRintCD5hi and consist of class I- and class II-restricted thymocytes in the first 12–48 h of development; and DP3 thymocytes are TCRhiCD5int and consist entirely of CD8 lineage cells developing ∼60 h after onset of selection (Fig. 1). Using this temporal developmental scheme, we defined a five-compartment differential equation model representing the three DP and two SP populations (Fig. 1). DP1 thymocytes are generated from CD4 CD8 double-negative (DN) thymocyte precursors at a constant rate λ (total cells per day). Cells in each DP population either develop to the next stage(s) or die. These behaviors are assumed to obey first-order kinetics with independent rate constants (Methods). Selection efficiency within each compartment is the net probability of surviving to maturation, which is (maturation rate)/(maturation rate + death rate). Thus, differences in selection efficiency can arise from differences in maturation rate and/or susceptibility to death. Finally, SP thymocytes can be lost by either thymic egress or death, and the combination of these processes is captured by a one loss-rate parameter for each SP subset. The model also assumes that there is no cell division among mature DP1 thymocytes and subsequent product populations as suggested elsewhere (10). The validity of this assumption was confirmed by analysis of carboxyfluorescein succinimidyl ester-labeled DP1 thymocytes following transfer to WT thymus. No cell division was observed in any DP populations, and only a small population of SP thymocytes underwent a single division by ∼day 6 (Fig. S1).
Fig. 1.

Compartmental model of thymocyte development. Density plots are of CD4 vs. CD8 expression by total WT thymocytes and CD5 vs. TCR by DP-gated thymocytes. Gates indicate CD4 SP and CD8 SP among total thymocytes and DP1 (TCRloCD5lo), DP2 (TCRintCD5hi), and DP3 (TCRhiCD5int) subsets of DP thymocytes. The mathematical model assumes a strict precursor relationship between thymic subsets (as shown), and the population kinetics are described by first-order differential equations. Parameters: δ1, δ2, and δ3 are death rates from the corresponding DP population; λ is the input rate to DP1 from DN thymocytes; μ12, μ23, μ24, and μ38 are rates of maturation between the indicated populations; and v4 and v8 are combined death and egress rates from SP populations.
To estimate the rates of differentiation and death within each population, we required measurements of thymocyte development in vivo over time; in particular, we needed to follow the system as it approached equilibrium from its arrested state. The dynamics of this process are rich in information regarding death and maturation across the different developmental stages. To generate these data, we used Zap70−/− mice in which thymocyte development, blocked at the DP stage, can be reconstituted following induction of a tetracycline-inducible Zap70 transgene (TetZap70; Methods). Restoration of thymic development in Rag1−/− irradiation chimeras reconstituted with bone marrow from TetZap70 mice was measured over 10 d, allowing us to follow the dynamics of the different compartments as they reached equilibrium. In addition, we generated similar time course data using chimeric mice in which TetZap70 development occurred in the absence of either class I or class II MHC in Rag1−/−b2m−/− and Rag1−/−Mhc-II−/− chimeric hosts, respectively. In this way, both simultaneous and independent development of CD4 or CD8 T cells was assessed. We estimated the three associated sets of rate constants from these time courses (Methods). The predicted dynamics closely matched the observations in all three cases (Fig. 2), with the most striking agreement for class II- and class I-restricted development occurring in isolation (Fig. 2 B and C, respectively). Fig. 3 A and B shows the estimated rates of differentiation and death across each compartment for all three developmental scenarios, and Fig. 3C shows the partitioning of fates across each compartment, derived from the point estimates of the parameters. To minimize the complexity of the model and ensure identifiability of parameters, the model did not include populations such as natural killer T cell (NKT), regulatory T cell (Treg), or intraepithelial lymphocyte precursor populations. NKT and Treg are of low abundance in TetZap70 mice (9), whereas intraepithelial populations were similar between b2m−/− and MhcII−/− TetZap70 chimeras; thus, their omission was not expected to introduce significant bias to parameter estimates (Fig. S2).
Fig. 2.

Model of thymic development accurately describes kinetics of selection. Irradiation chimeras were generated using bone marrow from TetZap70 donors to reconstitute Rag1−/−, b2m−/−Rag1−/−, and Mhc-II−/−Rag1−/− hosts. Six weeks later, mice were fed doxycycline to induce Zap70 expression. Groups of mice were taken at days 0–10, and frequencies of DP1, DP2, DP3, CD4 SP, and CD8 SP thymocytes among induced huCD2+ thymocytes were determined by fluorescence-activated cell sorting (n > 8 per day). Graphs show the experimentally observed percentage of total thymus (black symbols) for the indicated subset with time ±SEM in Rag1−/− (A), b2m−/−Rag1−/− (B), and Mhc-II−/−Rag1−/− (C) hosts. Time course data from each host were used to identify best-fit parameter values for the model. Red lines show time courses of subset development as predicted by the best-fit model. Dashed lines indicate the frequency of the thymocyte subsets observed in control WT (A), b2m−/− (B), and Mhc-II−/− (C) hosts. Data are pooled from three or more independent experiments.
Fig. 3.
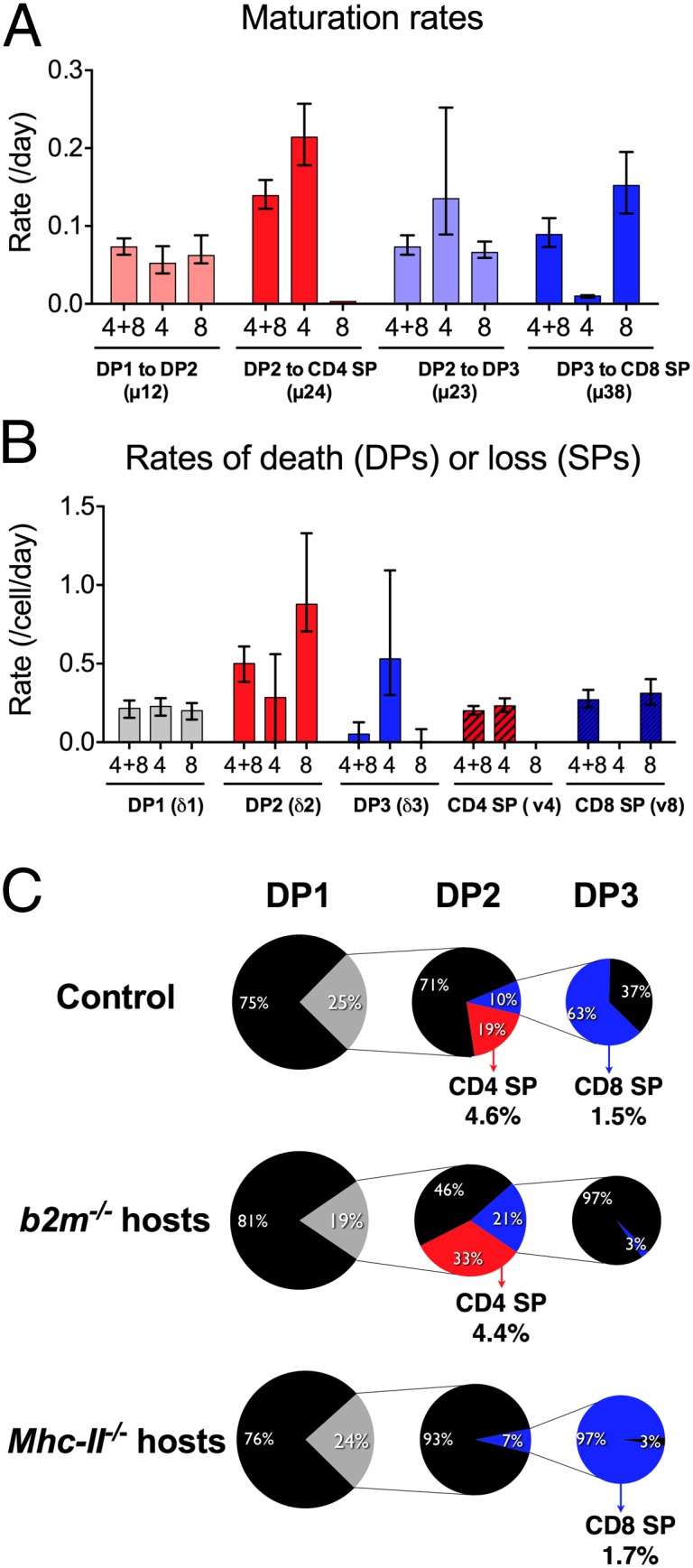
Model parameters describe the cellular dynamical behavior and cell fates. The data depicted in Fig. 2 were used to estimate the best-fit parameter values for models fitted to TetZap70 chimeras of Rag1−/− hosts (n = 4 + 8), b2m−/−Rag1−/− hosts (n = 4), and Mhc-II−/−Rag1−/− hosts (n = 8). (A) Bar charts show best-fit parameter values for the indicated maturation rates in different hosts. Error bars indicate 95% CIs. (B) Bar charts show best-fit parameter values for the indicated death rates in different hosts. Error bars indicate 95% CIs. (C) Pie charts show the proportion of each subset undergoing onward differentiation (colored) or dying at the indicated stage (black). Numbers indicate percentage size of the associated pie slice. Numbers next to CD4 SP and CD8 SP labels indicate the frequency of DP1 thymocytes estimated to give rise to the indicated population.
Model of TetZap70 Thymic Development also Describes WT Development.
Although the best-fit parameter values gave an accurate description of thymic development in TetZap70 mice, we have previously reported that these mice have a specific defect in DP3-CD8 SP differentiation, and consequently generate fewer CD8 SPs than WT mice. This defect was evident in the time courses obtained from class II-deficient TetZap70 mice, in which CD8 SP generation was reduced compared with WT (Fig. 2). In addition, ongoing analysis of class I-deficient TetZap70 mice revealed that CD4 lineage development may be suboptimal in TetZap70 mice compared with WT because these mice develop a much larger DP3 population than is found in class I-deficient mice expressing WT Zap70 (Fig. 2B). This appears to be a developmental dead-end for class II-restricted cells in TetZap70 mice, because none of these cells give rise to mature CD8 SPs. The model estimates that ∼20% of class II-restricted thymocytes misdirect to the DP3 stage in TetZap70 mice (Fig. 3C). In light of these differences with WT, we were concerned as to how applicable the model parameters estimated from TetZap70 data would be to WT development. We have previously shown that purified WT DP1 thymocytes develop with dynamics similar to those observed in the TetZap70 mice following their transfer to WT thymus (9). Absolute cell recoveries following intrathymic injections are subject to too much biological variation to be used to identify parameter values directly. Instead, TetZap70 parameter estimates were used to predict the development of WT DP1 thymocytes following their intrathymic transfer. Up to day 4 after transfer, the model reproduced the kinetics of all the downstream populations. After day 4, DP2 and CD4 SP kinetics were described with less accuracy but still within the 95% confidence interval (CI) parameter envelope (Fig. 4). However, the model overestimated WT DP3 frequencies and underestimated WT CD8 SP frequencies. Because TetZap70 mice have a known block in CD8 SP development, the rate of differentiation from DP3 to CD8 SP (µ38 parameter) estimated from these mice is very likely to be lower than the true rate for WT DP3 thymocytes. Significantly, simply increasing this rate was sufficient to improve the model’s prediction of WT thymocyte behavior and, unsurprisingly, revealed an inverse relationship between the rate of maturation from DP3 into CD8 SP (µ38) and the 4:8 ratio (Fig. 4B). Increasing µ38 simultaneously reduced DP3 representation and increased the size of the CD8 SP population (Fig. 4C). This strongly suggests that the value of µ38 in WT mice is greater than the value estimated from TetZap70 time course data, and that selection within DP3 is thus more efficient. Finally, we compared key estimates of thymocyte behavior from the present model with those made in previous studies of mouse thymic development and found broad agreement (Table 1). Taken together, these results suggest that despite the defects evident in the TetZap70 thymocyte selection, the estimated parameters were applicable to WT thymocyte behavior, with the exception of the transition from DP3 to CD8 SP.
Fig. 4.

TetZap70 model parameter values predict behavior of WT thymocytes. Purified WT DP1 thymocytes were injected intrathymically into WT CD45.1 hosts (n = 36). At different days after transfer, groups of mice were taken (n = 4–7) and the phenotype of the donor population was determined by fluorescence-activated cell sorting. (A) Line graphs show the representation of each indicated subpopulation with time in the donor cellular cohort ±SD. Using the frequency of the DP1 subset as input, model parameters estimated from time courses of TetZap70 development in control chimeras (Figs. 2 and 3) were used to estimate the changing representation of each subset. Red lines indicate this predicted time course. Error bars indicate 95% CIs of model predictions. (B) Line graph is of a 4:8 ratio vs. µ38 predicted by the model keeping all other parameters constant. The red diamond indicates the value of µ38 originally estimated from TetZap70 thymic development data. (C) Line graphs show the observed (gray symbols) and predicted (red lines) time courses of intrathymically injected WT DP1 thymocytes from A. Additional black lines are model predictions made using the indicated µ38 parameter values instead.
Table 1.
Comparison of model estimates with those from other studies
In Vivo Survival of Thymocyte Populations Reflects Model Estimates.
A key finding of the modeling was variation throughout thymocyte development in susceptibility to death, with the highest overall rate in DP2 thymocytes and the lowest in DP3 thymocytes. We had already established that the dynamics of TetZap70 thymocytes mirrored those of WT thymocytes. However, the possibility remained that rates of maturation and death in TetZap70 thymocytes could differ from those in WT but still yield similar overall dynamics. We therefore analyzed WT thymocyte survival to ask whether death rates that emerged from the modeling of TetZap70 development reflected the biology in WT thymocytes. Survival of purified WT thymocyte subsets was determined following their adoptive transfer into thymi of congenic mice. Purified thymocyte populations from CD45.1 donors were transferred by direct intrathymic injection, together with a control cohort of cell dye-labeled DP thymocytes from Zap70−/− mice. A measure of the overall survival of the donor population was made by comparing total recovery of donor cells with that of the control DP thymocytes from Zap70−/− mice. We first measured survival of DP1, DP2, DP3, CD4 SP, and CD8 SP WT populations. Human CD2+ (huCD2+) and huCD2− DP1 thymocytes from TetZap70 mice were also assessed as controls (Fig. 5A). Thymocytes from TetZap70 mice that have failed to induce the TetZap70 transgene following induction can be identified by the lack of huCD2 reporter expression and are Zap70-deficient (9). As anticipated, survival of huCD2− DP1 cells was virtually identical to that of the partner Zap70−/− DP thymocytes. Interestingly, survival of Zap70-sufficient huCD2+ DP1 thymocytes from TetZap70 mice was greater than that of Zap70−/− control DP thymocytes, and was indistinguishable from the survival of DP1 thymocytes from WT donors. From the model fits, death rates among DP2 thymocytes were estimated to be far higher than for DP1 cells, whereas the death rates of DP3 thymocytes were estimated to be much lower than for both DP1 and DP2 thymocytes (Fig. 3A). The recovery of WT DP2 and DP3 subsets mirrored the survival behavior estimated by the model. WT DP2 thymocyte survival was far poorer than WT DP1 survival. In contrast, WT DP3 thymocytes survived far better than the DP1 or DP2 subsets following transfer (Fig. 5A). Recoveries of SP thymocyte populations were highest of all the populations assayed.
Fig. 5.
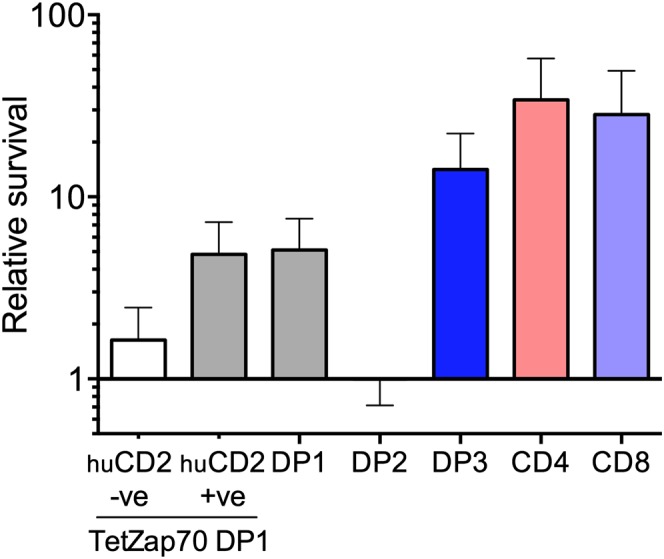
Measuring thymocyte subset survival in vivo confirms death rates estimated by the model. Survival of the indicated thymocyte populations was measured following intrathymic transfer to CD45.1 WT hosts. Thymic subpopulations were purified by cell sorting and, together with cell dye-labeled thymocytes from Zap70−/− donors as an internal control, were transferred to CD45.1 WT hosts by intrathymic injection. Two days later, cell recovery relative to the Zap70−/− partnering population was determined by flow cytometry. The bar chart shows survival relative to Zap70−/− thymocytes of the following populations: huCD2−DP1 thymocytes from doxycycline-fed donors (n = 6), huCD2+ DP1 thymocytes from doxycycline-fed donors (n = 9), WT DP1 and WT DP2 (n = 12), WT DP3 (n = 9), WT CD4 SP (n = 6), and WT CD8 SP (n = 8) thymocyte populations. +ve, positive; −ve, negative.
Low Death Rate Among SP Thymocytes.
The model of development did not explicitly estimate death among SP thymocytes but, rather, “loss,” which is the combination of both death and export of SPs. Given the unexpectedly high death rate of DP2 thymocytes, we sought to estimate the death rate among SP thymocytes, thought to be a key point at which negative selection can occur, so as to determine the thymic stage at which most death occurs. To achieve this, we took advantage of the S1P receptor inhibitor fingolimod (FTY720) to block egress of thymocytes from the thymus, which is induced by S1P. In the absence of egress, the loss of thymocytes from SPs calculated by the model represents an estimate of death among SP thymocytes. Treatment of WT mice with FTY720 resulted in a substantial accumulation of both CD4 SP and CD8 SP thymocytes, as previously reported (11) (Fig. 6A). We next generated time course data of thymic development in TetZap70 chimeras treated with FTY720. CD4 SP and CD8 SP populations grew in size in an identical manner to controls up to day 5 (Fig. 6B). From day 6 onward, SP populations in FTY720-treated mice continued to expand beyond the size observed in control groups. This coincided closely with the time when peripheral T cells are first detected in TetZap70 mice, previously reported as being ∼day 6 onward (9). Using these newly generated data, we estimated the loss parameters for the SP compartments, which would now be expected to reflect death alone. Model predictions using the new parameter estimates closely described the expansion of the SP compartments in FTY720-treated mice (Fig. 6B). Using the death rates calculated in this way, it was possible to calculate the export rate (Fig. 6C). For both CD4 SP and CD8 SP compartments, most of the loss due to thymocyte egress and death rates was estimated to be low compared with either DP1 or DP2 thymocytes.
Fig. 6.
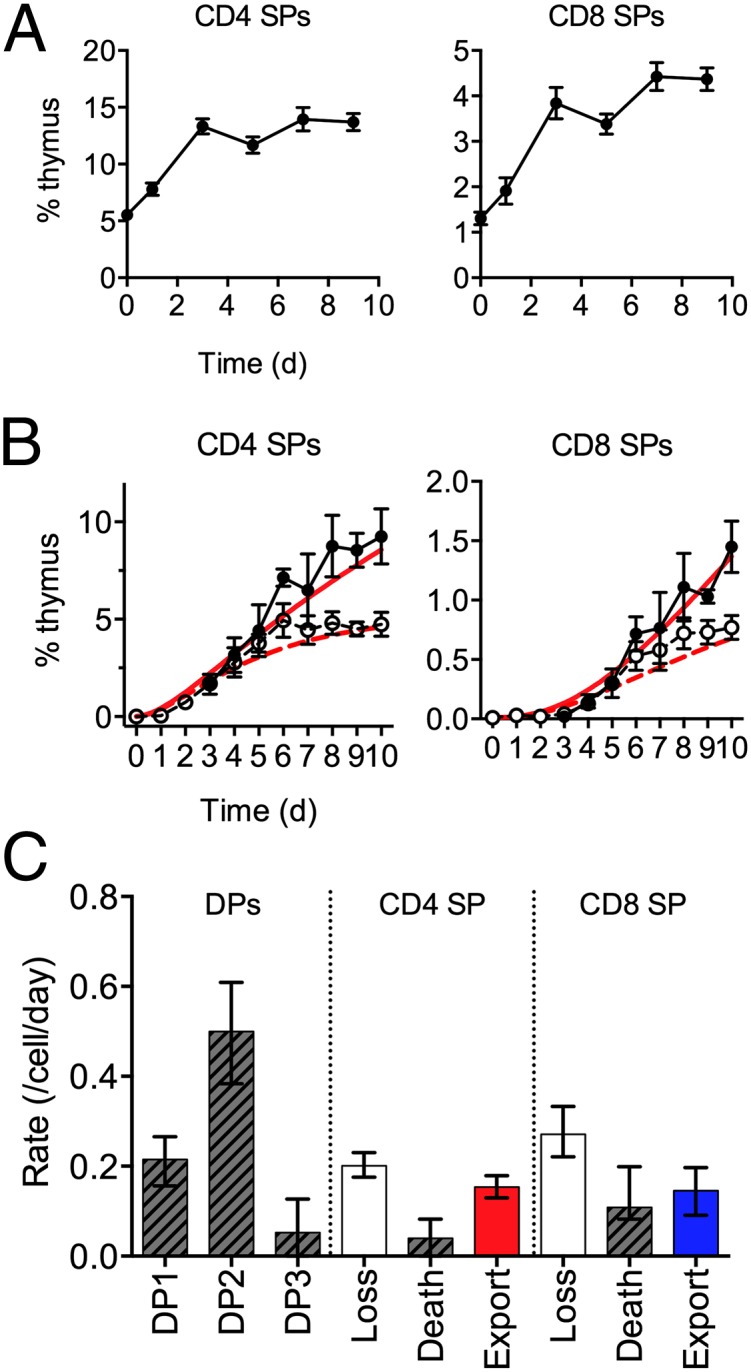
Estimating death of SP thymocytes in FTY720-treated TetZap70 mice. (A) Line graphs show a representation of TCRhi CD4 SP and TCRhi CD8 SP thymocytes with time following treatment of WT mice with FTY720. (B) Irradiation chimeras were generated using bone marrow from TetZap70 donors to reconstitute Rag1−/− hosts. Six weeks later, mice were fed doxycycline to induce Zap70 expression. Cohorts of mice were either injected with FTY720 or treated with PBS as a control. Groups of mice were taken at days 0–10, and frequencies of DP1, DP2, DP3, CD4 SP, and CD8 SP thymocytes among induced huCD2+ thymocytes were determined by fluorescence-activated cell sorting (n = 3–7 per day for FTY720 treatment). Graphs show the experimentally observed percentage of the total thymus of FTY720-treated mice (filled symbols) vs. controls (open symbols) for the indicated subset with time ±SD. Time course data from each host was used to identify best-fit parameter values for the model. Red lines show time courses of subset development as predicted by the best-fit model parameters from fits of control data (broken lines) and from model fits derived from analysis of FTY720-treated mice (solid lines). Data are pooled from three or more independent experiments. (C) Bar chart shows estimated death rates of DP subsets, original estimates of loss from SP compartments, and new estimates of death and export. Error bars indicate 95% CIs.
CD4:CD8 Lineage Bias Originates from Differential Death Rates Among Class I- and Class II-Restricted DP2 Thymocytes.
We next wanted to map the emergence of the CD4:CD8 bias during development. TetZap70 thymocyte development in MHC-deficient hosts provided information regarding the lineage-specific maturation and death rates from DP2 onward under conditions of isolated development. Using these data, it was possible to calculate the efficiencies of selection of each lineage in the absence of the other (Fig. 3C). Notably, we identified significant and substantial differences in rates of death within DP2 for CD4 and CD8 lineage precursors [0.28 per day (95% CI: 0.00, 0.56) and 0.88 per day (95% CI: 0.71, 1.33), respectively], meaning the CD8 lineage in DP2 is more than threefold as susceptible to death as the CD4 lineage. Also, in isolation, the lineages matured at very different rates [0.21 per day (95% CI: 0.18, 0.26) for class II-restricted DP2 into CD4 SP and 0.07 per day (95% CI: 0.067, 0.09) for class I-restricted DP2 into DP3)]. Thus, the MHC-deficient data lead us to conclude that class II-restricted DP2 cells select much more efficiently than class I-restricted cells. These data also showed that the total input rates of the two lineages from DP1 from DP2 were very similar. This is because the steady-state DP1 compartment sizes are comparable in the two groups (Fig. 2) and the per-cell rates of death and maturation from DP1 are indistinguishable (Fig. 3). Taken together these observations and parameter estimates from MHC-deficient animals suggest that enrichment for the CD4 lineage first arises in DP2 and the differences in the efficiency of selection derive from differences both in the susceptibility of each lineage to death and in their rates of maturation out of DP2. We then sought to confirm experimentally that death rates among DP2 thymocytes were higher in class I-restricted cells than in class II-restricted cells. To do this, we measured survival of DP2 thymocytes purified from Mhc-II−/−, b2m−/−, or WT donors after transfer into WT mice. Survival of DP2 thymocytes from class II-deficient hosts was reduced, significantly so at day 1, compared with DP2 thymocytes from b2m−/− donors (Fig. 7A). The relative survival of DP2 thymocytes from class II-deficient hosts compared with DP2 thymocytes from b2m−/− mice was 1.73/3.14 = 0.55. This was in striking agreement with the survival ratio arising from the point estimates of DP2 death rates in the MHC-restricted time courses: exp(−0.88)/exp(−0.27) = 0.54.
Fig. 7.

Estimated efficiencies of class I- and class II-restricted thymocyte development. (A) Survival of the indicated thymocyte populations was measured following intrathymic transfer to CD45.1 WT hosts. Thymic subpopulations were purified by cell sorting, and together with cell dye-labeled thymocytes from Zap70−/− donors as an internal control, were transferred to CD45.1 WT hosts by intrathymic injection. Survival relative to Zap70−/− thymocytes was determined at the indicated days by cotransfer of DP2 thymocytes from either b2m−/− (n = 14 at day 1, n = 10 at day 2), Mhc-II−/− (n = 16 at day 1, n = 9 at day 2), or WT controls (n = 17 at day 1, n = 12 at day 2) with the Zap70−/− control cohort. *P < 0.05. (B) Point estimates of influxes and efficiencies of selection of each lineage in DP2 using the analysis of control data and estimated lineage-specific death rates and death, and export among SPs using point estimates from Fig. 6. (C) Pie charts show the fate of class I- and class II-restricted thymocytes that initiate selection, demonstrating the proportion of cells that die in DP2 (black), die in DP3 (dark gray), die in SP (light gray), or are exported from the thymus (red- or blue-filled).
Quantifying the Relative Contributions of Precursor Ratio and Differential Selection Efficiency to the CD4 Lineage Bias Emerging from DP2.
Despite this concordance between the model and experiment, it remained possible that our estimates of precursor ratios and selection efficiencies at each developmental stage do not strictly apply when both lineages select together in the same environment. In particular, the dynamics of thymocytes in the control setting could not be described using the MHC-restricted time courses additively. Most strikingly, the control steady-state DP2 compartment was smaller than the sum of those in the MHC-deficient animals, in which from DP2 onward, each of the lineages develops in the absence of the other (Fig. S3). This implied that there is interaction or competition between the lineages in DP1 and/or DP2 when selecting ligands for both are available. When both lineages are able to select out of DP1, class I- and class II-restricted precursors are mutually suppressive, although possibly to different degrees. Direct estimates of class I- and class II-restricted precursor behavior in the control time courses are not possible because in DP1 and DP2, the lineages cannot be directly distinguished experimentally. However, additional knowledge of the death rates of the two lineages within DP2 in controls is sufficient to back-calculate the precursor ratio emerging from DP1 and to estimate the selection efficiencies of both lineages in DP2 (SI Methods). We proceeded using the control group parameters and associated uncertainties but assumed that the death rates of each lineage in DP2 were equal to those estimated in the MHC-deficient animals. We considered this a reasonable assumption because the relative survival of DP2 thymocytes from Mhc-II−/− or b2m−/− donors in WT hosts was in such striking agreement with the prediction using these estimated death rates (Fig. 7A). Fig. 7B summarizes this analysis. The predicted 4:8 lineage ratio entering DP2 is 0.8 (95% CI: 0.4, 1.4), and within DP2, the CD4 lineage selects 2.3 (95% CI: 1.2, 4.5)-fold more efficiently than the CD8 lineage. Together, these figures account for the 4:8 lineage ratio of 1.9 (95% CI: 1.5, 2.2) emerging from DP2 in the control chimeras. Further, roughly 64% of cells in DP2 at steady state are of CD4 lineage, and, notably, in contrast to the predictions from the MHC-deficient and control groups, the lineage-specific rates of maturation from DP2 are comparable (0.22 vs. 0.20 per day). This is likely because the control DP2 maturation rates µ24 and µ23 (0.14 and 0.07), are averaged across DP2, and so must be smaller than the rates specific to each lineage; µ24 and µ23 are recovered from these rates by averaging over the 64% to 36% composition of DP2. This analysis lends further support to our conclusion that the dominant source of bias in the CD4:CD8 ratio indeed is at the DP2 stage and indicates that the lower selection efficiency of the CD8 lineage in DP2 compared with CD4 is, in fact, due largely to a greater susceptibility to death, and likely not to a substantially lower rate of maturation. This is further illustrated when the ultimate fate of class I- and class II-restricted cells that start selection from DP1 was calculated (Fig. 7C). Of cells starting selection, most death occurred at the DP2 stage for both lineages, and only a small proportion of cells in comparison were estimated to die in the SP compartment.
Applying Apoptotic Stress to Thymocytes Preferentially Perturbs Class I-Restricted Thymocyte Development.
Our analysis of thymocyte development in TetZap70 mice revealed the critical role thymocyte death has in determining the relative efficiency of CD4 and CD8 lineage development. In particular, both modeling and experimentation showed that apoptosis was unexpectedly high among DP2 thymocytes and that class I-restricted thymocytes were most susceptible. We therefore wished to investigate the mechanisms regulating cell death in developing thymocytes and to test directly the conclusion that the 4:8 ratio is influenced strongly by distinct susceptibilities of class I- and class II-restricted thymocytes to death during selection. Apoptosis of thymocytes is regulated by Bcl2 family members. We therefore analyzed gene expression of Bcl2 family members in different DP populations from WT mice (Fig. 8A). Key multidomain BH3 executioner molecules expressed in thymocytes are Bax and Bak. Both were consistently and highly expressed throughout thymic development. Among antiapoptotic genes, Mcl1 was expressed at a consistently high level throughout development. In contrast, Bcl-Xl was only highly expressed in DP1 thymocytes, because its expression was rapidly lost as cells entered selection. Conversely, Bcl2 was not expressed by DP1 thymocytes at all, but its expression increased as cells progressed through selection. Among BH3-only family members, which induce death indirectly by sequestering antiapoptotic proteins, Bad, Bik, Bid, and Puma were expressed at low levels, whereas Bmf was expressed in DP1s but lost rapidly thereafter. Significantly, Bim, which is implicated in thymocyte apoptosis during negative selection, was also substantially induced in DP2, DP3, and recently selected heat-stable antigen (HSA)hi CD4 SP thymocytes during selection but reduced in mature SPs. Taken together, these data show that DP2 thymocytes had an expression profile of low Bcl2, low Bcl-Xl, and high Bim that could contribute to their increased susceptibility to apoptosis.
Fig. 8.

Increased apoptotic stress preferentially impairs CD8 lineage development. (A) Thymocyte populations from WT mice were purified, and gene expression was determined by RNA-sequencing analysis. Bar charts show mRNA expression level [number of reads per kilobase of exon per million sequenced reads (nRPKM)] of the indicated antiapoptotic (Bcl2, BclXl, and Mcl1), BH3-only (Bim, Bad, Bid, and Bmf), and multidomain proapoptotic (Bax and Bak1) factors in the indicated thymocyte subsets. Only Bcl2 family members with expression >1 nRPKM are shown. For each subset, three biological replicates were sequenced. (B–D) Mice expressing Bax transgene under control of huCD2 expression elements were analyzed at 6–8 wk of age. (B) Thymocytes were analyzed by flow cytometry. Density plots are (from left to right) of CD4 vs. CD8 by total live thymocytes, CD5 vs. TCR by DP-gated thymocytes, CD4 vs. CD8 by HSAhiTCRhi thymocytes, and CD4 vs. CD8 by HSAloTCRhi thymocytes from either Bax transgenic mice (Lower) or transgene-negative littermate controls (Upper). (C) Total lymph node cells were analyzed by flow cytometry. Density plots are of (from left to right) HSA vs. Qa2 by CD44loTCRhi cells, CD4 vs. CD8 by recent thymic emigrants (RTE), and CD4 vs. CD8 by mature naive T cells. Gates on plots of HSA vs. Qa2 are those used to identify HSAhiQa2lo RTE and HSAloQa2hi mature naive T cells. (D) Bar chart shows the CD4:CD8 ratio among HSAhi SP thymocytes (HSAhi) and HSAhiQa2lo RTE in lymph node cells in Bax transgenic (red bars, n = 3) or WT controls (black bars, n = 4). Data are representative of two independent experiments.
Our data suggest that class I-restricted cells are more susceptible to apoptosis than class II-restricted cells, especially at the DP2 stage. To test this directly, we analyzed T-cell development in mice overexpressing a transgene for the proapoptotic factor Bax, controlled by huCD2 expression elements (BaxhuCD2), that induce similar transgene expression levels throughout thymocyte development (12). Endogenous Bax is expressed at a similar level throughout thymocyte development (Fig. 8A). Therefore, Bax overexpression would be expected to apply a uniform apoptotic stress to developing thymocytes. Analysis of BaxhuCD2 mice confirmed previous reports of a reduction of ∼50% in thymus size (13), consistent with impaired survival of DP1 and DN precursors due to Bax expression. The relative size of the DP2 compartment was also reduced ∼2.5-fold in BaxhuCD2 thymus (Fig. 8B). This may reflect increased apoptosis of DP2 thymocytes and/or be the result of reduced recruitment of selecting thymocytes from DP1 due to the reduced t1/2 of this precursor population. Strikingly, however, the DP3 subset was dramatically reduced by >10-fold in BaxhuCD2 mice compared with controls (Fig. 8B). Overall, the number of cells completing selection was reduced in BaxhuCD2 mice, because total SP thymocyte frequencies were less than in controls. Analysis of a 4:8 ratio in HSAhi SP thymocytes, which are the direct progeny of selecting DP precursors, was increased twofold in BaxhuCD2 mice, suggesting that class I-restricted development was most perturbed by Bax expression. The increased 4:8 ratio, compared with WT controls, was maintained in mature HSAlo SPs (Fig. 8B). Measuring the 4:8 ratio among recent thymic emigrants in the periphery revealed that the increased 4:8 ratio observed in BaxhuCD2 thymocytes was maintained at an identical level in newly exported cells (Fig. 8 C and D). The mature naive compartment of BaxhuCD2 mice had a greater representation of CD4 T cells than controls. However, although BaxhuCD2 transgene expression is maintained throughout development and in mature T cells, the preferential loss of CD8 lineage T cells only occurred in DP thymocytes and no further skewing toward a CD4 lineage was observed at any subsequent stage of development.
Discussion
Using a straightforward model describing progression through the canonical stages of CD4 and CD8 T-cell development, we identified characteristics of thymic development that account for the CD4 bias in thymic selection. Using detailed time courses of thymocyte development, our analysis specifically identified an unexpectedly high death rate at the DP2 stage of selection, as well as death rates that were significantly greater for class I- than class II-restricted cells within this population. Importantly, these key predictions of the model were confirmed experimentally. Transcriptomic analysis of developing thymocyte populations revealed changes in the expression of Bcl2 family members that could account for the specific increase in susceptibility of DP2 thymocytes to cell death. Applying apoptotic stress to developing thymocytes by ubiquitous Bax expression preferentially killed class I-restricted cells. Together, our results indicate that the lineage bias in thymocyte development derives from lineage-specific susceptibility to apoptosis at the earliest stages of selection and that regulation of apoptotic susceptibility is an important parameter affecting thymic output.
In principle, there are two possible explanations for a bias toward CD4 lineage development. The first is a greater abundance of functionally class II-restricted thymocytes, such that more class II- than class I-restricted thymocytes are triggered into development. We found that this is not likely a significant contributor to the bias; support for this came independently from (i) the observation that the total flux of cells from DP1 to DP2 was very similar between class I- and class II-deficient TetZap70 mice, strongly suggesting that class I-restricted precursors are not limiting for selection in the randomly generated TCR repertoire, and (ii) the estimation of the precursor ratio using DP2 dynamics in a control setting. The second possibility is that development of class I-restricted cells is less efficient than development of class II-restricted cells. We found clear evidence that this was the case. Analysis of developmental time courses and empirical tests of thymocyte survival in vivo both revealed an unexpected high death rate among DP2 thymocytes. Significantly, modeling and empirical validation indicated that class I-restricted DP2 cells had a higher death rate than class II-restricted DP2 cells. The poor efficiency with which class I-restricted cells are selected was also reflected in the estimates of cells starting selection compared with those completing it. The model estimates that 25% or more of thymocytes enter the selection process but that, across all lineages, ∼74% fail (Fig. 3C). Using estimated death rates in DP2 for each lineage, we estimated that of the 25% of DP1 thymocytes entering selection, ∼64% are class I-restricted, but these cells are substantially less efficient at selecting; ∼7% survive to CD8 SP vs. 35% of class II-restricted cells that survive to CD4 SP (Fig. 7C).
The high death rate of selecting thymocytes was unexpected and seemed to be almost exclusively restricted to the DP2 stage; the DP3 and SP stages were found to have much lower death rates both in the models and measured empirically in vivo. Analysis of gene expression in thymocyte populations identified key changes in the expression of Bcl2, Bcl-Xl, and Bim that could contribute to the susceptibility of DP2 thymocytes to apoptosis. Bcl2 and Bcl-Xl are paralogs that are able to serve similar functions in regulating apoptosis (14). Therefore, the loss of Bcl-Xl and coincident induction of Bcl-2 triggered by the onset of thymic selection may represent a “paralog switch,” specifically to create a hiatus in antiapoptotic Bcl2 family member expression in DP2 thymocytes. In combination with Bim up-regulation, we speculate that such a switch functions by sensitizing DP2 thymocytes to negative selection. It is evident that Bcl2 and Bcl-Xl are limiting for thymocyte survival, because overexpression of either factor greatly improves its survival (13, 15–17), as does loss of Bim expression (18, 19). However, it is interesting to note that absence of Bim or overexpression of Bcl2/Bcl-Xl does not phenocopy the severe autoimmune pathology that occurs when negative selection is abrogated from loss of Aire expression (20). In fact, recent evidence suggests that additional loss of NOXA and PUMA is a requirement for autoimmune manifestation in Bim-deficient thymocytes (21), implying redundancy between BH3-only proteins during negative selection. We see little detectable expression of NOXA and PUMA in DP2 thymocytes in our system. It will be interesting to test whether Bim up-regulation acts unanimously in all DP2 thymocytes as a negative selection-sensitizing step, whereas negative selection may be specified to high-avidity clones via PUMA and NOXA up-regulation. Ongoing work will also address how the paralog switch from Bcl-Xl to Bcl2 expression is regulated in selecting thymocytes. We speculate that the TCR-dependent selection signal is likely to play a central role in directing expression of these factors. It is possible that repression of Bcl-Xl and induction of Bcl2 during selection are regulated at different signaling thresholds. Therefore, the distinct TCR signaling required for CD4 vs. CD8 lineage specification may also regulate the paralog switch from Bcl-Xl to Bcl2 differently in class I- and class II-restricted thymocytes in such a way that renders them differentially susceptible to apoptosis.
Consistent with the greater death rate of selecting class I-restricted cells, their development does appear to be particularly sensitive to manipulations of thymocyte survival. It has long been recognized that reducing apoptotic stress by overexpression of antiapoptotic factors (15–17) can apparently increase CD8 SP development. Here, we also found the converse to be true. Increasing apoptotic stress on thymocytes by ubiquitous overexpression of Bax resulted in the preferential death of class I-restricted thymocytes specifically at the DP2 stage. An explanation as to why class I-restricted thymocytes should be more susceptible to apoptosis may lie in the dynamics of selection. Class I- and class II-restricted T-cell development has different signaling requirements and occurs with distinct kinetics. In addition, thymic selection must purge the repertoire of autoreactive thymocytes at the same time in a process possibly facilitated by the paralog switch from Bcl-Xl to Bcl2 expression, which creates a window of apoptotic vulnerability to Bim induction. Therefore, reduced signaling in class I-restricted cells combined with slower kinetics of development, both features of CD8 lineage specification, also increases their exposure to apoptotic stress in the DP2 stage. It seems to be an inevitable compromise between competing demands of positive and negative selection, which specifically conspire to reduce the survival of class I-restricted thymocytes.
Our data also revealed that the proportion of thymocytes signaled into selection is surprisingly high, at 25%. It had hitherto been presumed that the proportion of thymocytes undergoing selection in vivo was much lower, at around 5%, on the basis of SP thymocyte generation and steady-state expression of markers, such as CD69 (22). In vitro studies have previously suggested that this figure might, in fact, be higher (23), and studies of reaggregate organ cultures show that the extent of positive selection depends on the availability of thymic epithelial cells (24). Competition for cortical thymic epithelial cells may underlie the competition between class I- and class II-restricted cells we detected within the DP2 population. In addition, the high proportion of thymocytes with TCRs that can trigger selection seems to be rather more than would be expected if TCR structure generation were truly random and supports the argument for germ-line–encoded recognition of MHC by TCR genes, as debated and suggested by others (25, 26). Our study did not make any assumptions about cause of thymocyte death. A recent study of Bim-deficient Nur77-EGFP reporter mice revealed that the extent of negative selection in DP thymocytes is far greater than previously appreciated (27), and this very likely contributes to the high death rate we detected in DP2 thymocytes. However, given the weight of evidence suggesting that CD8 lineage selection is associated with weaker/intermittent TCR signaling, it is also possible that the increased death of class I-restricted thymocytes is not solely accounted for by increased negative selection. Consistent with this, defective negative selection in Bim-deficient mice is not associated with increased selection of CD8 T cells (28). Rather, given the large fraction of the TCR repertoire that triggers selection and the high failure rate among class I-restricted thymocytes, it is possible that the paralog switch from Bcl-Xl to Bcl2 expression may also serve as an additional stringency test to induce apoptosis in thymocytes whose TCRs may recognize MHC too weakly. Therefore, our data suggest that the repertoire of TCRs that trigger selection is both broader than previously thought and subject to greater quality control at both ends of the spectrum of self-recognition.
In conclusion, the results here suggest that the relatively inefficient generation of CD8 thymocytes is an undesirable but necessary consequence of a developmental process with conflicting interests, rather than the alternative (i.e., that the adaptive immune system simply does not need as many CD8 as CD4 T cells). In this respect, it may be significant that the t1/2 of naive CD8 T cells in the periphery is longer than that of CD4 T cells (29–31) and, consequently, the naive CD4:CD8 ratio is lower in the periphery than it is among newly generated T cells in the thymus. Naive CD8 T cells can use IL-15 survival signals that naive CD4 T cells do not and die more rapidly in the absence of either IL-7 or TCR-dependent survival signals (30). These observations suggest that naive CD8 T cells have greater utilization of and dependence on homeostatic survival signals than do their CD4+ counterparts, which may represent a specific adaptation to overcome their reduced generation during thymopoiesis.
Methods
Mice.
b2m−/− (32) and B6.129-H-2 <dlAb1-Ea> [Mhc-II−/− (33)] mice were bred with Rag1−/−-deficient mice (34) to generate b2m−/−Rag1−/− and Mhc-II−/−Rag1−/− mice, respectively. F5 Rag1−/− (35), F5 Rag1−/−b2m−/−, OT-I Rag1−/− (36), Zap70−/− (37), Baxα25 (38), and tetracycline-inducible Zap70 mice bearing a huCD2 reporter gene (TetZap70) (9) have been described previously. Mice were bred in a specific pathogen-free colony at the National Institute for Medical Research. TetZap70 mice were fed 3 mg/g of doxycycline-containing diet to induce continuous Zap70 expression. Radiation bone marrow chimeras were generated by transferring 5 × 106 TetZap70 bone marrow cells i.v. to sublethally irradiated (500 rad per 5 Gy) b2m−/−Rag1−/− or Mhc-II−/−Rag1−/− recipient mice, which were allowed to reconstitute for more than 4 wk. Animal experiments were approved by National Institute for Medical Research Ethical Review Panel and under UK Home Office Project License 80.2506.
Flow Cytometry.
Flow cytometric analysis was performed with 2–5 × 106 thymocytes as previously described (39). Briefly, surface staining was performed with saturating concentrations of antibody in PBS containing BSA (0.1%) and 1 mM azide (PBS-BSA-azide) for 1 h at 4 °C at a cell density of ≤5 × 107 cells per milliliter. The following primary antibodies were used in this study: FITC-conjugated antibodies against CD5, CD24 (HSA), Ly5.1 (CD45.1), and Ly5.2 (CD45.2); phycoerythrin (PE)-conjugated antibodies against CD4, HSA (CD24), or huCD2; Pe-Cy5–conjugated antibodies against TCRβ; Pe-Cy7–conjugated antibodies against CD8α; allophycocyanin-conjugated antibodies against TCRβ or CD8α; Efluor450-conjugated antibodies against CD4; and biotinylated antibody against Qa-2, Ly5.1, or Ly5.2. Antibodies were purchased from eBioscience or Biolegend unless otherwise indicated. For detection of biotinylated primary antibodies, cells were washed and subsequently stained with streptavidin bound to PE-Texas Red or Pacific Orange (Invitrogen). For detection of Bcl-2, cells were fixed with Intracellular fixation buffer (eBioscience) on ice for 10 min, permeablized with 0.1% Nonidet P-40 (Igepal; Sigma), and stained for 1 h at 4 °C with PE-conjugated antibodies against Bcl-2 (BD Pharmingen). Flow cytometric analysis was performed on a BD FACSCanto II (Becton Dickinson) or CyAn ADP (Beckman Coulter) instrument. Cell sorting was performed on a BD FACSAriaII (Becton Dickinson) or MoFlo XDP (Beckman Coulter) instrument, with sorted populations being enriched to ≥90%. Flow cytometric data were analyzed using FlowJo software (V9.4.11; TreeStar).
Mathematical Modeling of Kinetic Data.
An ordinary differential equation model was generated to characterize the progression of immature thymocytes through five transient stages of development: DP1 (X1), DP2 (X2), DP3 (X3), CD4 SP (Y4), and CD8 SP (Y8). The model assumes a strict precursor relationship between transitional compartments: DP1→DP2→CD4 SP or DP1→DP2→DP3→CD8 SP. Input into the early DP1 compartment is assumed to be constant (λ cells per day); all remaining transitions and cell loss are modeled as exponential processes. The model is depicted in Fig. 1A, as described formally below:
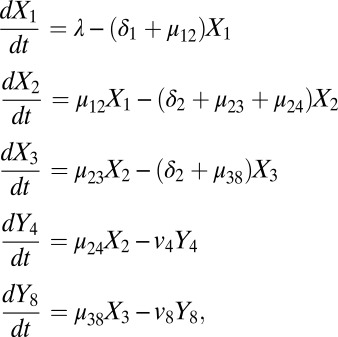 |
where, µ12, µ23, µ24, and µ38 represent per-cell rates of maturation from DP1 to DP2, DP2 to DP3, DP2 to CD4 SP, and DP3 to CD8 SP, respectively; δ1, δ2, and δ3 represent per-cell rates of death in the DP1, DP2, and DP3 compartments, respectively; and v4 and v8 represent combined rates of loss due to export and death of CD4 SP and CD8 SP cells, respectively (per day). The population behaviors are assumed to obey first-order kinetics with the independent rate constants defined above; that is, cells have fixed probabilities in any small interval of time to die or to mature. These rates and probabilities are connected. If r is a death rate, for example, the probability of death occurring within 1 d is 1 − exp(−r), which is approximately the rate constant r itself if r
 1. Importantly for our analysis, these rates or probabilities are population averages, because thymocytes have diverse TCR specificities and potentials to select. For example, at any time, DP1 will contain subpopulations that are MHC class I- or class II-restricted and capable (but not guaranteed) of maturing to DP2 with potentially different rates.
1. Importantly for our analysis, these rates or probabilities are population averages, because thymocytes have diverse TCR specificities and potentials to select. For example, at any time, DP1 will contain subpopulations that are MHC class I- or class II-restricted and capable (but not guaranteed) of maturing to DP2 with potentially different rates.
The observation that the thymic populations and, in particular, the CD4 SP and CD8 SP compartments approach equilibrium by day 8 allowed rates of CD4 and CD8 loss (v4 and v8) to be constrained as functions of input into the CD4 SP and CD8 SP compartments, leaving seven free rate constants. Further, it was assumed that input into the DP1 compartment (λ) was identical in all mice, whereas all other parameters were allowed to vary between strains.
A maximum-likelihood approach was then used to identify free parameters, using the FME package in R. Data from control and MHCI and MHCII KO mice were fitted simultaneously, and a logit transformation was applied to normalize residuals. We generated bootstrap distributions of parameter estimates and 95% CIs by creating 10,000 new datasets by sampling (with replacement) from the observed experimental dataset and repeating the fitting procedure.
Data from FTY720-treated mice were used to identify per-cell rates of death in the CD4 SP and CD8 SP compartments: v4_FTY and v8_FTY, respectively. It was assumed that the rates of loss and maturation in the DP1, DP2, and DP3 compartments, including rates of recruitment into the CD4 SP and CD8 SP compartments, were unaltered by FTY720 treatment. A maximum-likelihood approach was used to identify v4_FTY and v8_FTY, where the remaining parameters were the rates identified from control mice. Rates of egress of CD4 SP and CD8 SP (Ω4 and Ω8, respectively) were calculated with the assumption that cell death in the SP compartments was identical in control and FTY720-treated mice; that is, v4_CTRL = v4_FTY + Ω4 and v8_CTRL = v8_FTY + Ω8. Bootstrap distributions of the parameter estimates were generated by sampling (with replacement) from the observed FTY720 dataset and the distribution of parameter estimates (derived from control mice) and by repeating the fitting procedure 10,000 times.
Intrathymic Injections.
Intrathymic injections were performed by blind injection into the thoracic cavity as described previously (40). Briefly, mice were anesthetized through administration of the inhalation anesthetic isofluorane (Isoflow; Abbott). An incision of ∼1 cm was made in the skin, along the midline overlying the lower cervical and upper thoracic regions. Cell suspensions (10 μL, 2–3 × 106 cells) were injected into the anterior superior portion of each thymic lobe using a 1-mL microfine insulin syringe (Becton Dickinson) mounted on a Tridak stepper pipette. Following injection, the incision was treated with the local anesthetic bupivacaine hydrochloride (0.25%, Marcain Polyamp; AstraZeneca) and closed with 9-mm wound clips (Becton Dickinson).
Measurement of Relative Thymocyte Survival.
Total thymocytes from Zap70−/− mice were stained with CD8a antibodies conjugated to magnetic microbeads (Miltenyi) and enriched for DP cells using microbead activated cell separation large size separation columns according to manufacturer’s instructions. Enriched DP cells were labeled with CellTrace violet (CTV, 5 mM) in PBS containing 0.05% (vol/vol) BSA for 10 min at 37 °C at a density of 1 × 107 cells per milliliter. Excess CTV was removed by washing cells twice in two volumes of PBS containing 0.1% (vol/vol) BSA. CTV-labeled Zap70−/− thymocytes were mixed with labeled populations obtained by cell sorting, and the ratio of these cell populations was determined by flow cytometry. Mixed cell populations were next cotransferred to congenically marked hosts for 1 or 2 d and reisolated, and the relative ratio of Zap70−/− and test populations was reassessed by flow cytometry. Relative survival rates were calculated as (output ratio)/(input ratio), where (input ratio) denotes the ratio of test and Zap70−/− populations before transfer and (output ratio) denotes the ratio of test and Zap70−/− populations subsequent to reisolation.
RNA-Sequencing Analysis.
Indicated cellular populations were lysed with TRIzol (Invitrogen), and RNA was prepared according to the manufacturer’s instructions. RNA-sequencing libraries were prepared using the mRNA-sEq. 8-sample preparation kit (Illumina) and the Illumina duplex-specific nuclease protocol (41) according to the manufacturer’s instructions. Samples were sequenced at the Medical Research Council National Institute for Medical Research high-throughput sequencing facility using an Illumina Genome Analyzer IIx, and 36-bp single-end reads were obtained using the Illumina pipeline. Reads were aligned to the Mus musculus genome (mm9 assembly) using CLC Genomic Workbench (V5) software with standard settings. Aligned reads were mapped to the RefSeq database and were normalized by means of the DESeq method (42) using Avadis NGS (V1.3.1) software. Following DESeq normalization, gene expression was displayed as the number of reads per kilobase of exon per million sequenced reads (nRPKM), using a previously described method (43).
Statistics.
Statistical analyses were performed using R and GraphPad Prism 5 (v5.0d) software.
Supplementary Material
Acknowledgments
We thank S. Tung and the Biological Services staff for assistance with mouse breeding and typing. We thank Becca Asquith for useful discussion. We are also grateful to the Medical Research Council (MRC) National Institute for Medical Research flow cytometry and high-throughput sequencing core facilities. This work was supported by the MRC (United Kingdom) under Programme Code U117573801 and by National Institutes for Health Grant R01AI093870.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304859110/-/DCSupplemental.
References
- 1.Reinherz EL, Kung PC, Goldstein G, Schlossman SF. Separation of functional subsets of human T cells by a monoclonal antibody. Proc Natl Acad Sci USA. 1979;76(8):4061–4065. doi: 10.1073/pnas.76.8.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouillet P, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415(6874):922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- 3.Hernández-Hoyos G, Anderson MK, Wang C, Rothenberg EV, Alberola-Ila J. GATA-3 expression is controlled by TCR signals and regulates CD4/CD8 differentiation. Immunity. 2003;19(1):83–94. doi: 10.1016/s1074-7613(03)00176-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, et al. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat Immunol. 2008;9(10):1122–1130. doi: 10.1038/ni.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato T, et al. Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity. 2005;22(3):317–328. doi: 10.1016/j.immuni.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Park JH, et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat Immunol. 2010;11(3):257–264. doi: 10.1038/ni.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woolf E, et al. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc Natl Acad Sci USA. 2003;100(13):7731–7736. doi: 10.1073/pnas.1232420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. 2007;204(8):1945–1957. doi: 10.1084/jem.20070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saini M, et al. Regulation of Zap70 expression during thymocyte development enables temporal separation of CD4 and CD8 repertoire selection at different signaling thresholds. Sci Signal. 2010;3(114):ra23. doi: 10.1126/scisignal.2000702. [DOI] [PubMed] [Google Scholar]
- 10.Egerton M, Scollay R, Shortman K. Kinetics of mature T-cell development in the thymus. Proc Natl Acad Sci USA. 1990;87(7):2579–2582. doi: 10.1073/pnas.87.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yagi H, et al. Immunosuppressant FTY720 inhibits thymocyte emigration. Eur J Immunol. 2000;30(5):1435–1444. doi: 10.1002/(SICI)1521-4141(200005)30:5<1435::AID-IMMU1435>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 12.de Boer J, et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33(2):314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 13.Williams O, Norton T, Halligey M, Kioussis D, Brady HJ. The action of Bax and bcl-2 on T cell selection. J Exp Med. 1998;188(6):1125–1133. doi: 10.1084/jem.188.6.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37(3):299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chao DT, et al. Bcl-XL and Bcl-2 repress a common pathway of cell death. J Exp Med. 1995;182(3):821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67(5):879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 17.Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67(5):889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- 18.Hu Q, Sader A, Parkman JC, Baldwin TA. Bim-mediated apoptosis is not necessary for thymic negative selection to ubiquitous self-antigens. J Immunol. 2009;183(12):7761–7767. doi: 10.4049/jimmunol.0902181. [DOI] [PubMed] [Google Scholar]
- 19.Pellegrini M, et al. Loss of Bim increases T cell production and function in interleukin 7 receptor-deficient mice. J Exp Med. 2004;200(9):1189–1195. doi: 10.1084/jem.20041328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 21.Gray DH, et al. The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity. 2012;37(3):451–462. doi: 10.1016/j.immuni.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamashita I, Nagata T, Tada T, Nakayama T. CD69 cell surface expression identifies developing thymocytes which audition for T cell antigen receptor-mediated positive selection. Int Immunol. 1993;5(9):1139–1150. doi: 10.1093/intimm/5.9.1139. [DOI] [PubMed] [Google Scholar]
- 23.Merkenschlager M, et al. How many thymocytes audition for selection? J Exp Med. 1997;186(7):1149–1158. doi: 10.1084/jem.186.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkenschlager M, Benoist C, Mathis D. Evidence for a single-niche model of positive selection. Proc Natl Acad Sci USA. 1994;91(24):11694–11698. doi: 10.1073/pnas.91.24.11694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Laethem F, Tikhonova AN, Singer A. MHC restriction is imposed on a diverse T cell receptor repertoire by CD4 and CD8 co-receptors during thymic selection. Trends Immunol. 2012;33(9):437–441. doi: 10.1016/j.it.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia KC. Reconciling views on T cell receptor germline bias for MHC. Trends Immunol. 2012;33(9):429–436. doi: 10.1016/j.it.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stritesky GL, et al. Murine thymic selection quantified using a unique method to capture deleted T cells. Proc Natl Acad Sci USA. 2013;110(12):4679–4684. doi: 10.1073/pnas.1217532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouillet P, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 29.Polic B, Kunkel D, Scheffold A, Rajewsky K. How alpha beta T cells deal with induced TCR alpha ablation. Proc Natl Acad Sci USA. 2001;98(15):8744–8749. doi: 10.1073/pnas.141218898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seddon B, Zamoyska R. TCR signals mediated by Src family kinases are essential for the survival of naive T cells. J Immunol. 2002;169(6):2997–3005. doi: 10.4049/jimmunol.169.6.2997. [DOI] [PubMed] [Google Scholar]
- 31.den Braber I, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36(2):288–297. doi: 10.1016/j.immuni.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science. 1990;248(4960):1227–1230. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- 33.Madsen L, et al. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci USA. 1999;96(18):10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mombaerts P, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 35.Mamalaki C, et al. Positive and negative selection in transgenic mice expressing a T-cell receptor specific for influenza nucleoprotein and endogenous superantigen. Dev Immunol. 1993;3(3):159–174. doi: 10.1155/1993/98015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hogquist KA, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 37.Negishi I, et al. Essential role for ZAP-70 in both positive and negative selection of thymocytes. Nature. 1995;376(6539):435–438. doi: 10.1038/376435a0. [DOI] [PubMed] [Google Scholar]
- 38.Brady HJ, Gil-Gómez G, Kirberg J, Berns AJ. Bax alpha perturbs T cell development and affects cell cycle entry of T cells. EMBO J. 1996;15(24):6991–7001. [PMC free article] [PubMed] [Google Scholar]
- 39.Sinclair C, Saini M, Schim van der Loeff I, Sakaguchi S, Seddon B. The long-term survival potential of mature T lymphocytes is programmed during development in the thymus. Sci Signal. 2011;4(199):ra77. doi: 10.1126/scisignal.2002246. [DOI] [PubMed] [Google Scholar]
- 40.de la Cueva T, Naranjo A, de la Cueva E, Rubio D. Refinement of intrathymic injection in mice. Lab Anim (NY) 2007;36(5):27–32. doi: 10.1038/laban0507-27. [DOI] [PubMed] [Google Scholar]
- 41.Christodoulou DC, Gorham JM, Herman DS, Seidman JG. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr Protoc Mol Biol. 2011 doi: 10.1002/0471142727.mb0412s94. Chapter 4:Unit4.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 44.Thomas-Vaslin V, Altes HK, de Boer RJ, Klatzmann D. Comprehensive assessment and mathematical modeling of T cell population dynamics and homeostasis. J Immunol. 2008;180(4):2240–2250. doi: 10.4049/jimmunol.180.4.2240. [DOI] [PubMed] [Google Scholar]
- 45.McCaughtry TM, Wilken MS, Hogquist KA. Thymic emigration revisited. J Exp Med. 2007;204(11):2513–2520. doi: 10.1084/jem.20070601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rooke R, Waltzinger C, Benoist C, Mathis D. Targeted complementation of MHC class II deficiency by intrathymic delivery of recombinant adenoviruses. Immunity. 1997;7(1):123–134. doi: 10.1016/s1074-7613(00)80515-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.