

Abstract
Niemann–Pick disease, type C1 (NPC1), which arises from a mutation in the NPC1 gene, is characterized by abnormal cellular storage and transport of cholesterol and other lipids that leads to hepatic disease and progressive neurological impairment. Oxidative stress has been hypothesized to contribute to the NPC1 disease pathological cascade. To determine whether treatments reducing oxidative stress could alleviate NPC1 disease phenotypes, the in vivo effects of the antioxidant N-acetylcysteine (NAC) on two mouse models for NPC1 disease were studied. NAC was able to partially suppress phenotypes in both antisense-induced (NPC1ASO) and germline (Npc1−/−) knockout genetic mouse models, confirming the presence of an oxidative stress-related mechanism in progression of NPC1 phenotypes and suggesting NAC as a potential molecule for treatment. Gene expression analyses of NAC-treated NPC1ASO mice suggested NAC affects pathways distinct from those initially altered by Npc1 knockdown, data consistent with NAC achieving partial disease phenotype suppression. In a therapeutic trial of short-term NAC administration to NPC1 patients, no significant effects on oxidative stress in these patients were identified other than moderate improvement of the fraction of reduced CoQ10, suggesting limited efficacy of NAC monotherapy. However, the mouse model data suggest that the distinct antioxidant effects of NAC could provide potential treatment of NPC1 disease, possibly in concert with other therapeutic molecules at earlier stages of disease progression. These data also validated the NPC1ASO mouse as an efficient model for candidate NPC1 drug screening, and demonstrated similarities in hepatic phenotypes and genome-wide transcript expression patterns between the NPC1ASO and Npc1−/− models.
INTRODUCTION
Niemann–Pick disease, type C (NPC) is an autosomal recessive, neurovisceral disease stemming from abnormal intracellular transport and storage of cholesterol and lipids. NPC patients exhibit a broad range of clinical symptoms with variable presentation, including hepatosplenomegaly, vertical supranuclear gaze palsy, gelastic cataplexy, dysarthria, dystonia, cerebellar ataxia, seizures, and progressive dementia (1). Currently there are no FDA-approved treatments for NPC, and definitive NPC diagnosis typically occurs following neuronal damage.
The majority of NPC patients harbor mutations in the gene encoding NPC1, while a small subset of patients carry mutations in the gene encoding NPC2; these two genetically distinct disease groups are respectively defined as Niemann–Pick disease, type C1 (NPC1, MIM #257220) and Niemann–Pick disease, type C2 (NPC2, MIM #607625) (2,3). The protein encoded by NPC1 is a large, multipass transmembrane glycoprotein that localizes to the late endosome/lysosome (LEL) compartment, while NPC2 is a small, soluble protein localized to the lysosomal lumen. NPC1 and NPC2 both bind cholesterol and work together to facilitate LEL cholesterol export and to maintain proper intracellular cholesterol/lipid homeostasis, including the direct transfer of cholesterol from NPC2 to the N-terminal domain of NPC1 (4–6). Mutation of either NPC1 or NPC2 is associated with impaired intracellular trafficking and the accumulation of unesterified cholesterol and glycosphingolipids in LELs (reviewed in (7)). This defective trafficking initiates an intracellular pathological cascade that includes oxysterol deficiency (8), dysfunctional peroxisomes (9), perturbed sphingosine metabolism and calcium homeostasis (10,11), impaired synthesis of neurosteroids (12), neuroinflammation (13), and induction of apoptosis (14,15). In addition, increased oxidative stress has been identified as a potential pathological process in NPC1 disease (16,17).
Although a classical germline mouse model for NPC1 disease has been available to study for over 15 years (18), recently an Npc1 antisense oligonucleotide (ASO) knockdown model was created (19). In the spontaneous germline mutant BALB/cNctr-Npc1m1N/J (hereafter Npc1−/−), mutant mice harbor a mutation at the Npc1 locus that is predicted to result in no functional NPC1 protein (18). Npc1−/− homozygotes demonstrate liver phenotypes along with retarded growth, neurological abnormalities and defects in LEL trafficking of lipids and cholesterol paralleling those of human NPC1 patients (14,20–24). The Npc1−/− neurological symptoms initially present at 6–7 weeks of age as visually detectable ataxia or tremor and then progress to death at 11–12 weeks. Thus the Npc1−/− mice accurately model both neurological and hepatic aspects of NPC1 disease, but the severity of their neurological phenotypes greatly restricts their experimental usage. In contrast, the new Npc1 ASO knockdown model (hereafter NPC1ASO) is generated by transient intraperitoneal injection of 2′-O-methoxyethyl-modified ASOs, which block Npc1 expression in the liver but not other organs, thus replicating the NPC1 hepatic phenotype in a neurologically normal mouse. The NPC1ASO model can be rapidly generated in large numbers, and the absence of neurodegeneration allows hepatic phenotypic assessment in viable animals, a characteristic that could be readily exploited for candidate compound testing.
One such candidate molecule to study in NPC1 disease is N-acetylcysteine (NAC; C5H9NO3S, PubChem Compound CID 12035), the acetylated form of the amino acid cysteine. NAC has been proposed to be a direct reactive oxygen species (ROS) scavenger, to regulate redox reactions and consequently alter cell signaling, and to reduce inflammatory cytokines (25,26). Importantly, NAC undergoes deacetylation in vivo to yield cysteine, which is the rate-limiting precursor to the synthesis of the tripeptide glutathione (GSH; C10H17N3O6S, PubChem Compound CID 124 886), a molecule essential for regulation of intracellular oxidative stress via reduction of peroxides and other free radical-producing molecules. Systemic administration of NAC has the potential to increase cysteine availability and replenish intracellular GSH, thereby increasing the body's ability to manage oxidative stress. This function of NAC enables it to act in a variety of clinical applications where GSH has been severely depleted, including as a highly effective antidote for acetaminophen toxicity in the liver (27,28).
Several studies have found quantitative biochemical indicators of oxidative stress that result from the absence of normal NPC1 protein, suggesting that suppression of oxidative stress may be a reasonable therapeutic approach to slow NPC1 disease clinical progression. Cultured NPC1-deficient cells from human patients and mouse models demonstrated increased ROS in fibroblasts, and apoptosis secondary to increased ROS following treatment of primary cultured cortical neurons with U18666A, a drug that induces an NPC-like cellular phenotype (15,29–31). In vivo data from Npc1−/− mice showed elevated mitochondrial cholesterol levels and depletion of GSH in cultured hepatocytes, elevated cholesterol oxidation products in macrophages and plasma, and lower glutathione S-transferase levels in cerebellum (32–34). Human NPC1 patients may also exhibit a lower antioxidant capacity, as cerebrospinal fluid showed abnormal glutathione S-transferase and superoxide dismutase levels, and plasma measurements showed a reduced fraction of Coenzyme Q10 (CoQ10) and Trolox Equivalent Antioxidant Capacity (TEAC) along with elevated thiobarbituric acid-reactive species and carbonyl formation (16,34,35). Additional biochemical abnormalities are present that may reflect the unique biochemical intersection of excess intracellular cholesterol along with increased oxidative stress, as the non-enzymatically generated oxysterols 7-ketocholesterol (7-KC) and 3β,5α,6β-cholestanetriol (3β,5α,6β-triol) are significantly elevated in NPC1 patients as well as in Npc1−/− mice (36,37).
This paper demonstrated that NAC reduced oxidative stress and provided modest therapeutic benefit to both the NPC1ASO and Npc1−/− mouse models. Furthermore, these studies validated the NPC1ASO model and proved that it is useful for rapid, efficient screening of drug candidates for treating NPC1 disease. These NAC studies were extended to a crossover clinical trial of NPC1 patients; however, NAC treatment showed no measureable changes in a variety of blood-based biomarkers, including plasma levels of 7-KC or 3β,5α,6β-triol. These results suggest that further studies will be needed to determine whether the beneficial NAC effects seen in mouse NPC1 models are achievable in human NPC1 patients, potentially as part of combination therapy, in which individual treatments each address various aspects of NPC1 disease including oxidative stress.
RESULTS
NPC1ASO mice recapitulate pathological and biochemical changes consistent with induction of NPC1 disease pathology
To establish efficient screening of candidate compounds for NPC1 disease treatment, we generated an NPC1ASO mouse model by adding minor modifications to previously described NPC1ASO protocols (19). Adult BALB/c female mice injected with NPC1ASO demonstrated a large decrease in NPC1 protein expression after 2 weeks of injection, and undetectable expression after 4 or 8 weeks of injection (Fig. 1A and data not shown). NPC1ASO mice developed hepatomegaly and showed a significant and progressive increase in the liver/total body weight ratio after 4 and 8 weeks of therapy relative to control ASO-injected (CTLASO) mice (Fig. 1B). This change was not attributable to reduced total body weight, since there was no significant difference in body weight for CTLASO and NPC1ASO mice (data not shown).
Figure 1.

The NPC1ASO mouse model shows physiological and pathological liver changes consistent with the absence of NPC1 protein. (A) Western blot analysis indicated that 2 weeks of ASO injections reduced NPC1 protein in the liver of NPC1ASO mice by 60–70% relative to CTLASO mice (left panel), and 4 weeks of ASO injections rendered NPC1 protein levels undetectable in NPC1ASO liver (right panel). The NPC1 antibody recognizes various glycosylated forms of NPC1, with prominent bands occurring at 220 kDa and 170 kDa. Expression of β-actin (42 kDa) was used to normalize the NPC1 signal. (B) NPC1ASO mice developed hepatomegaly, as indicated by the significant increase in the mean liver/body weight ratio of NPC1ASO mice (black bars) as treatment time increases, and also relative to the liver/body weight ratio of control ASO mice (white bars). (C) Comparison of histological sections from CTLASO (left) and NPC1ASO liver (right) showed activated macrophages with lipid storage as a result of NPC1ASO treatment. (D and E) Plasma ALT and AST levels in NPC1ASO mice (black bars) are both significantly increased above those in CTLASO mice (white bars) at 4 and 8 weeks of ASO treatment. For B, D and E, columns which do not share common letters (a, b, c) show significantly different mean values by two-way ANOVA with post-tests, P < 0.05; no difference was observed between columns with identical letters.
Liver histopathology after 4 weeks of NPC1ASO treatment showed histiocytosis and numerous activated, lipid-laden macrophages (Fig. 1C). Consistent with induction of a hepatic NPC1 phenotype, the livers of these mice also showed significantly elevated serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) relative to CTLASO mice (Fig. 1D and E). ALT activity was increased by 214% at 4 weeks and 469% at 8 weeks in NPC1ASO mice, and AST activity was increased by 132% at 4 weeks and 175% at 8 weeks in NPC1ASO mice. Collectively, these results showed that NPC1ASO-injected mice demonstrated pathological and biochemical changes consistent with NPC1 disease liver pathology.
NPC1ASO mice demonstrate similar gene expression changes to Npc1−/− mice
Transcriptome-wide microarray analysis was used to characterize gene expression changes in the liver of NPC1ASO mice. Comparison of NPC1ASO and CTLASO mice following 4 weeks of ASO treatment (n = 5 each for CTLASO and NPC1ASO) revealed 2209 transcripts with significantly different expression levels (FDR-corrected P < 0.05) (Supplementary Material, Table S1). As expected, one of these transcripts was Npc1, which showed significantly reduced expression in NPC1ASO mice (−7.4-fold). However, the majority of these transcripts were upregulated by NPC1ASO injection (1411/2209, or 64%), similar to previous reports in Npc1−/− mice (38–41).
We then compared liver gene expression in 12-week-old NPC1ASO mice which had received 4 weeks of ASO treatment with liver gene expression in Npc1−/− mice at various ages (1, 3, 5, 7, 9, 11 weeks) (38). Hierarchical clustering using the 2209 significantly altered transcripts in NPC1ASO mice (relative to CTLASO mice) showed that this subset of transcripts can successfully differentiate Npc1−/− mice from Npc1+/+ mice, and also indicated that the expression of these transcripts in the NPC1ASO mouse model most closely resembles the expression patterns observed in Npc1−/− mice at 3 and 5 weeks of age (Supplementary Material, Fig. S1). Direct comparison of the transcripts showing statistically significant changes in expression levels in NPC1ASO versus CTLASO (2209 transcripts) and Npc1−/− and Npc1+/+ at 3 weeks of age (2276 transcripts, Supplementary Material, Table S2) showed 675 common transcripts (30%, Fig. 2A). The altered expression of these 675 transcripts correlated in both the models, such that hierarchical clustering grouped CTLASO with Npc1+/+ and NPC1ASO with Npc1−/− (Fig. 2B). Furthermore, functional enrichment ontology analysis (GeneGo MetaCore software) with these 675 transcripts in both the ASO and the Npc1−/− experiments indicated NPC1 disease-related pathways and networks in both of these models. For example, functional ontology analysis of Disease Biomarkers, in which gene expression changes in an experimental dataset are mapped onto biomarker networks previously identified as changing in specific disease states, showed commonly affected pathways in both the NPC1ASO and Npc1−/− models that corresponded with NPC1 disease phenotypes, including inborn errors of metabolism, lysosomal storage and lipid metabolism (Fig. 2C). Additional ontology analyses were also consistent with the NPC1ASO and Npc1−/− models exhibiting parallel gene expression changes in pathways related to NPC1 disease phenotypes (Map Folders, Process Networks and Metabolic Networks; Supplementary Material, Fig. S2). To further validate that the NPC1ASO expression changes were similar to those of Npc1−/−, we compared the NPC1ASO data with previously published genome-wide expression data from Npc1−/− mice (39,41–43). The genes differentially expressed in the NPC1ASO model included many (10–100%) of the genes previously reportedly in large-scale expression analyses of Npc1−/− mutants with a high degree of correlation in the direction of expression change (27–100%; Supplementary Material, Table S3). Interestingly, 100% overlap and correlation occurred between the transcripts altered in NPC1ASO and 12 secretory genes identified as potential markers of disease progression in Npc1−/− (39), further suggesting strong parallels between the two models.
Figure 2.

Microarray analysis shows similar expression patterns in the NPC1ASO and 3-week-old Npc1−/− mouse models. (A) A Venn diagram illustrates that 675 transcripts have statistically significant expression changes in both CTLASO versus NPC1ASO (2209 total transcripts) and Npc1+/+ versus Npc1−/− at 3 weeks of age (2276 total transcripts). (B) Hierarchical clustering with the 675 common transcripts shared between NPC1ASO versus CTLASO and Npc1+/+ versus Npc1−/− at 3 weeks of age. Similar transcript expression patterns grouped CTLASO (solid red) with Npc1+/+ (diagonally striped red) and NPC1ASO (solid blue) with Npc1−/− (diagonally striped blue), consistent with correlated gene expression patterns in the two mouse models. (C) Functional enrichment ontology analysis (GeneGo MetaCore) was performed to identify significantly altered pathways common to both the NPC1ASO and the Npc1−/− models. Shown here are disease biomarkers analysis results, indicating strong correlation with NPC1-associated disease phenotypes for both the NPC1ASO and the 3-week old Npc1−/− models (blue and orange histograms, respectively), illustrated by histograms of FDR-corrected P-values for the association of the experimental gene expression changes in each dataset with the indicated disease. Additional MetaCore results are shown in Supplementary Material, Fig. S2.
Hierarchical clustering of the remaining significant transcripts unique to either model (NPC1ASO versus CTLASO = 1534 transcripts, and 3-week Npc1−/− versus Npc1+/+ = 1601 transcripts, Fig. 2A) did not group NPC1ASO and Npc1−/− in the same branch of the dendogram, as expected (Supplementary Material, Fig. 3). These dissimilar expression changes likely reflect inherent differences in the two mouse models, such as adult rather than germline alteration of Npc1 expression, as well as the physiological alterations caused by ASO injection. However, taken together, the collective phenotypic and gene expression data on NPC1ASO mice demonstrate that NPC1ASO injection establishes a mouse model relevant to the study of NPC1 hepatic disease.
N-acetylcysteine treatment suppresses a subset of NPC1 pathologies in NPC1ASO mice
Because NAC may reduce oxidative stress in vivo, we next used the NPC1ASO mouse model to test the potential efficacy of NAC in the reduction of NPC1 pathology. As a proof of principle and positive control, we treated 12-week NPC1ASO mice with 4000 mg/kg 2-hydroxypropyl-β-cyclodextrin (HPβCD), a compound previously reported to reduce disease phenotypes in Npc1−/− mice (44–47). This HPβCD treatment significantly reduced both unesterified and total cholesterol in NPC1ASO liver by ∼50% (P < 0.01, Fig. 3A and B), and also reduced the elevated mRNA expression of the cholesterol synthetic pathway gene Srebp2 and the macrophage marker Itgax in NPC1ASO liver (P < 0.01, Fig. 3C). These data were consistent with HPβCD liberating late endosomal/lysosomal cholesterol in NPC1ASO mice in a similar manner as HPβCD-treated Npc1−/− mice, thus demonstrating that the NPC1ASO model can be used as an effective tool for testing therapeutic efficacy.
Figure 3.

HPβCD treatment reverses lipid storage and alters gene expression in NPC1ASO liver. (A) HPβCD treatment reduced the significantly elevated unesterified cholesterol in NPC1ASO liver from 13.6 ± 0.8 to 6.8 ± 0.5 mg/g. (B) The significantly elevated total cholesterol in NPC1ASO liver was decreased from 14.0 ± 0.9 to 7.8 ± 0.5 mg/g after HPβCD treatment. (C) The significantly elevated mRNA expression of Srebp2 and Itgax in NPC1ASO liver was reduced by HPβCD treatment, consistent with reductions in cholesterol synthesis and inflammation. The expression levels of mRNA are the relative fold change over untreated CTLASO. Data analyzed by two-way ANOVA with post-tests; column lettering (a, b, c) indicates significance (P < 0.05), as described in Figure 1.
To assess NAC treatment in NPC1ASO mice, 8-week-old CTLASO and NPC1ASO mice were given 1% NAC orally for 4 weeks, concurrent with their ASO injections. NAC was able to suppress the hepatomegaly and elevated ALT and AST in NPC1ASO mice to levels indistinguishable from those of CTLASO mice (Fig. 4A–C). These improved phenotypes associated with NAC therapy were not due to NAC interfering with NPC1 knockdown, as NPC1 protein levels remained undetectable (Fig. 4D). Liver GSH was elevated in NPC1ASO mice in response to NAC treatment (3.8 ± 1.3 and 7.8 ± 2.2 µmol GSH/g tissue for NPC1ASO and NPC1ASO + NAC, respectively); however, this increase was not statistically significant (Fig. 4E). Of note, the mean GSH level in NPC1ASO was significantly lower than that of CTLASO (P < 0.05; Fig. 4E), consistent with oxidative stress in NPC1ASO liver. Histopathological evaluation of liver sections from untreated and NAC-treated NPC1ASO mice showed no suppression of activated macrophages or histiocytosis in the NAC-treated liver (Fig. 4F), suggesting that NAC-mediated effects in NPC1ASO liver lie downstream of the primary defect in lipid accumulation. Consistent with the histopathological findings, NAC treatment did not suppress either the 7-fold increase in unesterified cholesterol or the 6.7-fold increase in total cholesterol present in NPC1ASO livers (Fig. 5A and B). Of note, esterified cholesterol was dramatically elevated from 0.38 to 4.24 mg/g tissue in NPC1ASO mice following NAC treatment (P < 0.01; Fig. 5C), suggesting that although the effect of NAC on cholesterol in these mice is limited, it may cause some alteration of their abnormal cholesterol homeostasis.
Figure 4.

NAC treatment ameliorates a subset of abnormal phenotypes in NPC1ASO liver. (A) NAC reduced the significantly increased liver/body weight ratio in NPC1ASO mice to that of CTLASO mice. (B and C) NAC reduced the significantly elevated plasma levels of ALT and AST in NPC1ASO mice to CTLASO levels. (D) NPC1 protein remained undetectable in NAC-treated NPC1ASO mice, demonstrating that NAC treatment did not interfere with ASO-induced knockdown of NPC1. (E) Without NAC treatment (left two bars), NPC1ASO mice showed a significantly lower GSH level compared to CTLASO mice, indicative of oxidative stress. NAC treatment of NPC1ASO mice increased the GSH level in liver, but this increase was not statistically significant. (F) Comparison of histological sections from untreated (left) and NAC-treated (right) NPC1ASO mice showed that NAC treatment did not reduce the activated macrophages and histiocytosis in NPC1ASO liver. All mice received ASO treatment for 4 weeks, and NAC-treated mice concurrently received NAC for 4 weeks. Data analyzed by two-way ANOVA with post-tests; column lettering (a, b) indicates significance (P < 0.05), as described in Figure 1. Scale bar in F = 5 µm. n = 4–5 for each treatment group.
Figure 5.

NAC treatment increases esterified cholesterol in NPC1ASO mice. (A and B) NAC treatment had no effect on the elevated levels of unesterified (A) and total cholesterol (B) in NPC1ASO mice. (C) Esterified cholesterol in NAC-treated NPC1ASO mice was significantly elevated relative to untreated NPC1ASO or CTLASO mice. Data analyzed by two-way ANOVA with post-tests; column lettering (a, b) indicates significance [P < 0.001for (A and B), and P < 0.01 for (C)], as described in Figure 1. n = 4–5 for each treatment group.
To gain insight into the biological pathways altered in NAC-treated NPC1ASO mice, changes in liver gene expression were identified using microarray analysis. Analysis of differential gene expression between NAC-treated NPC1ASO mice and untreated NPC1ASO mice identified 490 significantly altered transcripts (Supplementary Material, Table S4), with the majority showing downregulation resulting from NAC treatment (338/490, or 69%). These 490 transcripts altered by NAC treatment included only 92 of the 2209 transcripts (4%) significantly altered by NPC1ASO injection (Fig. 6A), indicating that NAC primarily affected genes and pathways distinct from those altered by NPC1ASO injection. In support of this, functional enrichment ontology analysis using these 92 overlapping transcripts showed distinct differences between the effects of NAC treatment and NPC1ASO injection. For example, Disease Biomarker analysis did not identify the lysosomal- and lipid-related disease pathways previously identified by the gene expression changes associated with NPC1ASO-mediated knockdown of Npc1 (Fig. 2C), but instead identified cancer-related pathways indicative of cell cycle perturbation (Fig. 6C). Additional ontology analysis (Map Folders, Process Networks and Metabolic Networks; Supplementary Material, Fig. S4) also showed little overlap between NAC-altered pathways and those altered by NPC1ASO injection. Of note, hierarchical clustering of samples using the 92 common transcripts (Fig. 6B) grouped NPC1ASO + NAC in the same branch of the dendogram as CTLASO. This suggested that for this transcript group, NAC treatment restored abnormal NPC1ASO transcript expression towards normal expression. Interestingly, these 92 transcripts showed few expression differences between CTLASO and CTLASO + NAC (Fig. 6B), suggesting that the NAC-induced changes in NPC1ASO mice are specific to the context of NPC1 knockdown. Collectively, the biochemical and gene expression data for NAC-treated NPC1ASO liver demonstrated that NAC treatment caused partial suppression of NPC1 disease phenotypes, but affected gene expression pathways distinct from those affected by NPC1ASO treatment.
Figure 6.

Microarray analysis of NPC1ASO and NAC-treated NPC1ASO liver shows that NAC treatment induces expression changes distinct from those of NPC1ASO alone. (A) A venn diagram illustrates that 92 transcripts have statistically significant expression changes in both CTLASO versus NPC1ASO (2209 total transcripts) and NPC1ASO versus NPC1ASO + NAC (490 total transcripts). (B) Hierarchical clustering with the 92 common transcripts shared between CTLASO versus NPC1ASO and NPC1ASO versus NPC1ASO + NAC. Similar transcript expression patterns grouped NPC1ASO + NAC (vertically striped blue) with CTLASO (solid red) in the dendogram, consistent with NAC restoring some normal gene expression in NPC1ASO. When data from CTLASO + NAC were included (vertically striped red), few expression differences were seen between CTLASO and CTLASO + NAC, suggesting that the observed NAC-induced changes in NPC1ASO mice are specific to the NPC1 knockdown conditions. (C) Functional ontology enrichment analysis (GeneGo MetaCore) was performed to identify significantly altered pathways with the 92 common transcripts. Shown here are the results of disease biomarkers analysis, showing numerous cancer-related pathways that suggest cell cycle alterations. These differ from the lysosomal- and lipid-related pathways identified by analysis of the transcripts altered in NPC1ASO treatment (see Fig. 2C). The histograms indicate FDR-corrected P-values for the association of the indicated disease pathways with experimental gene expression changes in CTLASO versus NPC1ASO (blue) or NPC1ASO versus NPC1ASO + NAC (orange). Additional MetaCore results are shown in Supplementary Material, Fig. S4.
Effect of N-acetylcysteine treatment on Npc1−/− mice
Given that NAC therapy moderately suppressed hepatic disease phenotypes in the NPC1ASO mouse, we administered NAC to the Npc1−/− mouse, to determine whether NAC could reduce markers of oxidative stress in the brain as well as liver, and also suppress the adverse neurological symptoms, weight loss and reduced lifespan that occur in this genetic model. Since overt neurological symptoms in Npc1−/− mice begin at 5 weeks of age, oral NAC administration was initiated immediately prior to the symptom onset, at 4 weeks of age, and/or soon after symptom onset, at 6 weeks of age (for weight loss, tremor and lifespan analyses), to determine whether age-dependent effects occurred.
For liver and brain biochemical analyses, 250 mg NAC / kg body weight was administered beginning at 4 weeks of age, then after 2 weeks or 4 weeks of NAC therapy (ages 6 and 8 weeks, respectively) brains, livers, and/or plasma were analyzed for GSH, 7-KC and 3β,5α,6β-triol. At these ages, untreated Npc1−/− mice showed significantly lower GSH levels in liver (P = 0.02) and brain (P = 0.007) relative to Npc1+/+ littermates (Fig. 7), consistent with previously reported GSH levels in Npc1−/− hepatocytes (32), and also consistent with GSH levels in NPC1ASO mice (Fig. 4E). NAC-treated Npc1−/− mice showed significantly increased GSH levels in liver (P = 0.007) and brain (P = 0.004) relative to Npc1−/− mice, rendering the GSH levels in NAC-treated Npc1−/− liver and brain statistically indistinguishable from those of Npc1+/+ mice (P = 0.08 and 0.39, respectively) (Fig. 7). However, NAC treatment did not alter Npc1−/− plasma levels of either 7-KC or 3β,5α,6β-triol at 8 weeks (data not shown), and although slight reduction of liver levels of both of these oxysterols occurred, these were not statistically significant (Fig. 8A). Similarly, a modest but insignificant reduction of the elevated 3β,5α,6β-triol levels in the brain occurred in NAC-treated Npc1−/− mice relative to untreated Npc1−/− mice at 8 weeks (Fig. 8B). NAC treatment did significantly decrease brain 7-KC levels in both Npc1+/+ and Npc1−/− mice (∼30%; P < 0.0001; Fig. 8C), indicating NAC acted to reduce levels of this oxysterol regardless of genotype. In summary, these biochemical data suggest that 250 mg/kg NAC administered from 4–8 weeks of age achieves measureable but modest effects on reducing oxidative stress in Npc1−/− mice.
Figure 7.

NAC treatment restores normal GSH levels in Npc1−/− mice. From 4 to 8 weeks of age, untreated Npc1−/− mice (open circles) had significantly decreased GSH levels compared with Npc1+/+ mice (triangles) in liver (A) and brain (B) (P < 0.05). When NAC was administered to Npc1−/− mice from 4 to 8 weeks of age (filled circles), significant increases in both liver (A) and brain (B) GSH were seen relative to untreated Npc1−/− mice (P < 0.01). These increased GSH levels achieved in NAC-treated Npc1−/− mice were statistically similar to those of Npc1+/+ (P ≥ 0.08). NAC-treated Npc1−/− mice received NAC at 250 mg/kg NAC beginning at 4 weeks of age (P28). Significance determined by two-way ANOVA. n = 4–5 for each treatment group.
Figure 8.

NAC treatment shows little effect on oxysterol levels in Npc1−/− mice. (A) In liver, both 7-KC and 3β,5α,6β-cholestanetriol (3β,5α,6β-triol) levels showed statistically insignificant reductions following NAC treatment of Npc1−/− mice (unpaired t-test, P > 0.3; n = 11 for Npc1−/−, n = 9 for NAC-treated Npc1−/− mice). (B) In brain, the significantly higher level of 3β,5α,6β-triol of Npc1−/− mice relative to Npc1+/+ (P < 0.0001) was not reduced following NAC treatment (P = 0.12). (C) In brain, Npc1+/+ and Npc1−/− mice showed similar 7-KC levels (P = 0.3), and NAC treatment uniformly reduced 7-KC levels in both genotypes (P < 0.0001). For (B) and (C): n = 4–6 for each group; data analyzed by two-way ANOVA with Tukey's post-test; column lettering (a, b) indicates significance, as described in Figure 1. NAC-treated mice received 250 mg/kg NAC for 4 weeks, beginning at 4 weeks of age (P28). Means ± SD are shown.
To determine whether NAC treatment could suppress weight loss, tremor onset and premature death in Npc1−/− mice, NAC doses of both 125 and 250 mg/kg body weight were each administered from 4 to 12 and from 6 to 12 weeks of age. NAC treatment resulted in a moderate but statistically insignificant improvement of the weight loss phenotype, as indicated by a later mean age at which peak weight was reached (Npc1−/− mice = 6.25 days, while all NAC-treated Npc1−/− mice ranged from 6.5 to 6.875 days) (Fig. 9A). However, after reaching this weight gain peak, NAC-treated mice showed precipitous weight loss at a rate similar to or greater than that of untreated Npc1−/− mice (Fig. 9A). Similarly, NAC therapy had a modest effect on the tremor phenotype that is present in Npc1−/− mice (22). Tremor amplitude at 11 Hz, a frequency with relatively high tremor amplitude peaks in Npc1−/− mice (data not shown), was lower at most timepoints (5–10 weeks) in all NAC-treated mice relative to untreated Npc1−/− mice (Fig. 9B), which was reflected in significantly different line elevations of tremor amplitude over time (P < 0.0001). However, the slopes of tremor amplitude over time did not differ among NAC-treated and Npc1−/− mice (P = 0.18), indicating that NAC treatment did not alter tremor acquisition rates. Interestingly, significant increases in the lifespan of NAC-treated Npc1−/− mice occurred relative to untreated Npc1−/− mice when NAC therapy was initiated at 4 weeks, but NAC treatment initiated at 6 weeks showed less effect (Fig. 9C). While Npc1−/− mice exhibited a mean lifespan of only 77.5 days, the lifespan of Npc1−/− mice receiving 125 mg/kg NAC beginning at 4 weeks was significantly extended to 82.5 days (P = 0.0065), and those receiving 250 mg/kg NAC beginning at 4 weeks showed a significantly longer lifespan of 84 days (P = 0.0003). Overall, no dosage dependence was apparent when comparing the 125 and 250 mg/kg dosages. In summary, NAC appeared to have moderate, beneficial effects on the phenotypes of weight loss, tremor and premature death in Npc1−/− mice, and delaying initial NAC administration until after the onset of symptoms appeared to lessen these modest benefits.
Figure 9.
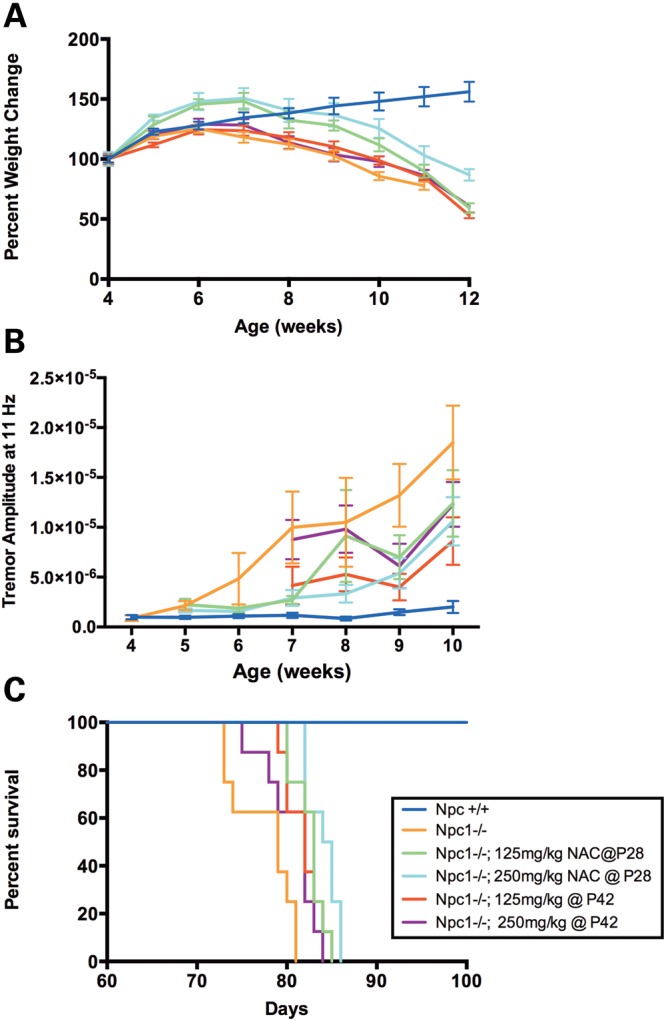
NAC facilitates moderate improvement of the Npc1−/− mouse phenotypes of weight loss, tremor and shortened lifespan. (A) NAC-treated Npc1−/− groups (green, teal, red, and purple) reached their peak weight at a slightly later age than untreated Npc1−/− (orange), but this difference was not statistically significant (Kruskal–Wallis test, P = 0.2259). Following the peak of weight gain, NAC-treated Npc1−/− groups showed a rapid weight loss at a rate similar to or greater than that of untreated Npc1−/− mice. (B) Analysis of the tremor amplitude at 11 Hz in Npc1−/− mice indicated that at most timepoints (from 5–10 weeks) the tremor amplitude was lower in NAC-treated Npc1−/− groups relative to untreated Npc1−/− (line elevations by linear regression, P < 0.0001), but the rate of tremor acquisition did not differ among NAC-treated Npc1−/− and Npc1−/− groups (slopes by linear regression, P = 0.18). Means ± SD are shown. (C) NAC treatment significantly increased the lifespan of NAC-treated Npc1−/− mice relative to that of Npc1−/− at both the 125 mg/kg and 250 mg/kg dosages when initiated at 4 weeks (P = 0.0065 and 0.0003, respectively, log-rank test with Bonferroni's correction). Initiation of NAC treatment at the later timepoint of 6 weeks provided a modest increase of lifespan at the 125 mg/kg dosage, but no increase at the 250 mg/kg dosage relative to that of Npc1−/− (P = 0.03 and 0.1, respectively). The six experimental groups (n = 8 per group) were: untreated Npc1+/+, untreated Npc1−/−, Npc1−/− treated with 125 mg/kg NAC beginning at 4 weeks (P28), Npc1−/− treated with 250 mg/kg NAC beginning at 4 weeks, Npc1−/− treated with 125 mg/kg beginning at 6 weeks (P42), and Npc1−/− treated with 250 mg/kg NAC beginning at 6 weeks.
Effect of N-acetylcysteine treatment in individuals with NPC1 disease
Patients with a diagnosis of NPC1 disease by biochemical testing or mutation analysis were enrolled in a double-blind, placebo-controlled, crossover trial to evaluate the effectiveness of oral NAC treatment. Of the 35 patients enrolled (18 male, 17 female; mean age 16.9 ± 13 years) 30 completed the trial and 5 were withdrawn from the study. Two patients were withdrawn due to increased serum transaminase levels while receiving NAC; in both cases liver enzymes decreased after discontinuation of the drug. The remaining three patients were withdrawn during the placebo phase due to hematuria, aspiration pneumonia requiring hospitalization and non-compliance. The primary outcome measures for the study were reduction of the abnormally elevated plasma levels of 7-KC or 3β,5α,6β-triol (37). Secondary outcome measures related to decreased oxidative stress included improved plasma / total CoQ10 ratio and increased TEAC, two measures of antioxidant capacity that have previously been shown to be abnormally low in NPC1 patients (16). Plasma GSH, glutathione disulfide (GSSG), ALT and AST levels were also measured. Secondary subjective measures of clinical improvement included the PedsQLTM Pediatric Quality of Life Inventory and the PedsQLTM Multidimensional Fatigue Scale.
Intrasubject plasma levels of 7-KC (Fig. 10A–C) and 3β,5α,6β-triol (Fig. 10D–F) did not change significantly from baseline levels when either placebo or NAC were administered, suggesting no improvement in circulating oxysterol levels in this NPC1 patient cohort as a result of NAC administration. The 7-KC and 3β,5α,6β-triol levels in patients who were simultaneously taking miglustat, an inhibitor of glycosphingolipid synthesis that has been shown to lessen some phenotypes of NPC1 disease (48), showed no significant differences from those not taking miglustat (data not shown). Similarly, the secondary outcome measurements showed little change as a result of NAC treatment. Intrasubject measurements of both GSH/GSSG (Fig. 10G–I) and TEAC (data not shown) were unchanged over the placebo or NAC treatment phases, and the elevated levels of ALT or AST were unaffected (data not shown). Total and reduced forms of CoQ10 did not change significantly from baseline in either placebo- or NAC-treated datasets (data not shown); however, the fraction of reduced CoQ10 during the NAC treatment phase showed a small but statistically significant increase compared with that of the placebo treatment phase (Fig. 10J–L). In the NAC treatment phase, the mean reduced CoQ10 percentages were maintained at 93%, but in the placebo phase they dropped from 94% at baseline to 91% after treatment (P = 0.02, Fig. 10L). Neither the PedsQLTM Pediatric Quality of Life Inventory nor the PedsQLTM Multidimensional Fatigue Scale revealed any significant difference between placebo and NAC treatment phases. In summary, this cohort of NPC1 patients did not exhibit reduction of oxidative stress from NAC treatment.
Figure 10.

Administration of NAC to NPC1 patients in a crossover clinical trial shows little evidence of phenotypic rescue. A, D, G, and J show individual patient measurements at baseline and end of the placebo phase (each connected by a line) for 7-ketocholesterol (7-KC), 3β,5α,6β-cholestanetriol (3β,5α,6β-triol), reduced to oxidized glutathione (GSH/GSSG) ratio, and reduced CoQ10 percentage, respectively. B, E, H and K show individual patient measurements at baseline and end of the NAC treatment phase for these same biochemical measurements. The box-and-whisker plots in C, F, I, and L show the change (Δ) in the same biochemical measurements for each patient during placebo and NAC phases. (A–F) Plasma levels of 7-KC (A–C) and 3β,5α,6β-triol (D–F) showed no significant changes resulting from NAC treatment in NPC1 patients. The mean 7-KC levels (in ng/mL) were as follows: baseline placebo = 245, end placebo = 235, baseline NAC treatment = 265, and end NAC treatment = 239. Mean 3β,5α,6β-triol levels (in ng/mL) were as follows: baseline placebo = 91.9, end placebo = 88.9, baseline NAC treatment = 101, and end NAC treatment = 94.1. (G–I) GSH/GSSG levels were unchanged in NAC-treated NPC1 patients when compared with placebo levels. Mean GSH/GSSG ratios were as follows: baseline placebo = 7.2, end placebo = 12.2, baseline NAC treatment = 13.8, and end NAC treatment = 9.4. (J–L) The percentage of reduced CoQ10 in NPC1 patients was maintained at a slightly higher level during NAC treatment when compared to placebo treatment, and the Δ reduced CoQ10 percentage was significantly different between NAC and placebo (panel L, P = 0.02). Mean reduced CoQ10 percentages were as follows: baseline placebo = 93.5, end placebo = 90.9, baseline NAC treatment = 93.2, and end NAC treatment = 93.0. Data analyzed by Wilcoxon-matched pairs signed rank-test.
DISCUSSION
In this study, we assessed the impact of pharmacological intervention with the antioxidant NAC in alleviating oxidative stress and disease symptoms in two distinct NPC1 disease mouse models and individuals with NPC1 disease, since oxidative stress in NPC1 disease has been suggested in Npc1−/− mice and NPC1 patients (16,29–31,37,49). Our data showed a number of distinct effects of NAC, demonstrating that NAC reduced biochemical, molecular and phenotypic changes in liver tissue of both the NPC1ASO and Npc1−/− mouse models and had small but beneficial neurological effects in the Npc1−/− mouse model. Although NAC treatment in NPC1 patients did not show measureable positive effects other than minimal improvement of the fraction of reduced CoQ10, the mouse model data suggest NAC holds potential as a treatment for NPC1 disease and confirm the existence of a mechanism involving oxidative stress in the NPC1 pathological cascade.
NAC has been widely used in a variety of clinical trials and treatments. In humans, NAC is used to treat acetaminophen overdose, based on its ability to replenish GSH levels, and to relieve bronchial obstruction based on mucolytic effects (27,28). NAC can cross the blood–brain barrier, and prior studies have suggested that NAC therapy may be beneficial in human neurodegenerative disorders such as Alzheimer disease (50,51) and Parkinson disease (52,53). Additionally, NAC has been shown to improve neuron survival in the CA1 region of the hippocampus following ischaemic brain injury (54). NAC has also been shown to have some benefit in acute liver failure and chronic hepatitis C (55,56). Mouse models also suggest beneficial effects of NAC, as NAC reverses memory impairment and diminishes brain oxidative stress in age-accelerated SAMP8 mice (57), and rescues neurons from apoptotic death by activation of the Ras-ERK pathway (58).
Similar to what has been shown previously (19), the NPC1ASO mouse model replicated phenotypic aspects of NPC1 hepatic disease. These included increased liver size, presence of lipid inclusions in hepatic cells and increased serum transaminase levels. Using gene expression array analysis, we extended the characterization of this model, showing that gene expression patterns in NPC1ASO mice share many similarities to gene expression patterns observed in 3week old Npc1−/− mice. Pathway analysis showed that significantly altered pathways common to both mouse models corresponded to NPC1 disease-affected pathways. Our studies not only demonstrated that the NPC1ASO model recapitulates the hepatic phenotypes and gene expression changes of Npc1 null animals, but also proved with HPβCD and NAC treatments that the NPC1ASO mouse can be used to quickly and effectively screen for drug treatments to alleviate NPC1 hepatic phenotypes. Future studies to examine NPC1 disease drug candidates in NPC1ASO mice rather than Npc1−/− mice can be performed economically and efficiently, due to the ease of creating these mice via administration of the ASO to healthy inbred strains and avoidance of the interruption of severe neurological symptoms present in Npc1−/− mice.
In NPC1ASO mice, NAC treatment ameliorated some abnormal phenotypes, causing significant decreases in hepatomegaly as well as serum ALT and AST levels. However, other liver phenotypes were unchanged by NAC treatment. NAC therapy increased GSH levels in liver tissue from NPC1ASO mice, but these were small, statistically insignificant changes. However, this could be a result of a later age of NAC administration (from 8 to 12 weeks of age); administration at younger ages might result in greater GSH changes, as was seen in the Npc1−/− model when NAC given from 4 to 8 weeks of age. NAC therapy also did not significantly reduce total or unesterified cholesterol levels, nor did it provide improvement of liver histopathology. Interestingly, esterified cholesterol levels were increased in liver tissue from NAC-treated NPC1ASO mice; this phenomenon has been observed previously in Npc1−/− mice in response to HPβCD treatment (45). This could suggest partial restoration of cholesterol flux in NPC1ASO cells by NAC, inhibition of lysosomal acid lipase A (LIPA), which removes the fatty acid from cholesterol esters in the endolysosomal system, or increased sterol O-acyltransferase 1 (SOAT1), which esterifies intracellular cholesterol. However, the genome-wide expression data did not indicate significant alterations in gene expression for either Lipa or Soat1 following NAC treatment of NPC1ASO mice, so further biochemical analyses will be needed to determine the causes for this esterified cholesterol increase.
Hierarchical analyses of the expression profile of NAC-treated NPC1ASO mouse liver suggested that NAC conferred gene expression changes that differed from those changed by NPC1 knockdown, and also that these changes occurred in an NPC1 disease-specific manner. The small number of transcripts (92) that were common to both NPC1ASO and NPC1ASO + NAC was primarily changed to resemble control expression levels in the NPC1ASO + NAC samples, suggesting that NAC acted to restore some normal gene expression. Overall, the gene expression data were consistent with the concept that NAC therapy did not address the primary biochemical defect in NPC1ASO mice, but rather treated one aspect of the NPC1 pathological cascade.
In Npc1−/− mice, NAC treatment increased both liver and brain GSH levels and additionally showed modest improvement in the more severe, neurologically derived symptoms of this model. Early initiation of NAC therapy in Npc1−/− mice had transitory but beneficial effects in maintaining weight, decreasing tremor and increasing survival. These results, taken collectively with the data from NAC-treated NPC1ASO mice, indicate that oral supplementation of NAC partially improved liver function and moderately reduced neurologic symptoms. Quantitative in vivo measurements showing restoration of GSH levels suggest that these NAC-induced effects are achieved by reduction of oxidative stress. Recent studies examining NAC's effect in Npc1−/− cerebellum and liver (43) or on Npc1−/− primary neurons (15) have also suggested that NAC could alleviate portions of oxidative stress-related phenotypes, thus correlating well with our results.
Concomitant with the NPC1 mouse model analyses, we performed a placebo-controlled, crossover clinical trial studying orally administered NAC in NPC1 patients. We previously hypothesized that increased plasma levels of the disease-specific biomarkers 7-KC and 3β,5α,6β-triol in NPC1 disease are due to the combination of intralysosomal accumulation of unesterified cholesterol and increased oxidative stress (37), thus the primary outcome measures for this study were plasma 7-KC and 3β,5α,6β-triol levels. NAC therapy did not have a beneficial effect on the plasma levels of these oxysterols, mirroring the data on plasma oxysterols in the Npc1−/− mouse model and suggesting that 7-KC and 3β,5α,6β-triol plasma levels may not be sensitive outcome measures for NAC treatment. These patients did show slight improvement in the fraction of reduced CoQ10; however, this small change is unlikely to be clinically significant. Given the additional results that neither plasma GSH/GSSG nor total serum antioxidant capacity were altered, it appears that this therapeutic regimen of NAC did not significantly impact the degree of oxidative stress in these patients.
It is possible that antioxidant therapy may be limited by inherent characteristics of NPC1 disease. These include the fact that oxidative stress arises late in the NPC1 disease pathogenic cascade (37,38), or in the case of NAC, that mitochondrial membrane changes within NPC1 mutant cells limit GSH subcellular transport and thus restrict the efficiency of GSH antioxidant activity, as proposed by Vazquez et al. (17). However, the Npc1−/− mouse model data showed greater efficacy if NAC was administered prior to neurological symptom onset, suggesting that NAC antioxidant therapy might benefit NPC1 patients if initiated earlier in disease progression, a scenario that would require a reduction in the currently long lag time between the symptom onset and definitive diagnosis (59). Furthermore, future studies using different diagnostic markers of oxidative stress, such as enzymes that function to regulate lipid peroxidation, may provide more details regarding potential antioxidant effects following NAC treatment in NPC1 patients. In support of this, a recent study on the glycosphingolipid synthesis inhibitor miglustat showed that elevated GSH-peroxidase activity in erythrocytes from NPC1 patients was significantly lessened following miglustat treatment, but reduced GSH in erythrocytes did not differ among controls, untreated NPC1 patients or migulstat-treated NPC1 patients (35).
It is important to note that two NPC1 patients had to be withdrawn from the trial while receiving NAC, due to increased serum transaminase levels. Although mild transaminase elevations are common in NPC1 patients, retrospective analysis of their medical histories showed that these individuals had higher than typical transaminase elevations. This was unanticipated and underscores the fact that safety of supplements in rare diseases should not be assumed and, like any pharmacological therapy, should be evaluated in controlled studies. Should an NPC1 patient elect to initiate antioxidant therapy based on the data from NAC-treated NPC1 disease mouse models, we would strongly recommend that serum transaminase levels be monitored.
Although the human trial results did not support NAC as a treatment for NPC1 disease, the data collected provided valuable biochemical data from the NPC1 patient human subject population. Repeated measures of plasma oxysterol species and other biomarkers will be a useful foundation for future biomarker-driven NPC1 trials. Additionally, the mouse data support the idea that NAC could play a role as one component of a multi-drug, combinatorial therapy that would address multiple aspects of the NPC1 disease pathological cascade. Since most currently available therapeutic interventions only correct a subset of pathological defects, a combination therapy that targets multiple NPC1 pathological aspects may be required. Future studies in NPC1 disease mouse models such as the NPC1ASO model could efficiently determine whether therapeutic benefits exist from an increased NAC dose, a combination of antioxidants or NAC administered in combination with other drugs that could potentiate antioxidant therapy or simultaneously address other aspects of the pathological cascade. These results paired with improved diagnostic methods for NPC1 disease in human patients (36) will make more effective NPC1 disease treatment possible in the future.
MATERIALS AND METHODS
Mouse care and treatment
Protocols for experiments on ASO mice were approved by the National Human Genome Research Institute Animal Care and Use Committee and were in compliance with the NIH Guide for the Care and the Use of Laboratory Animals. Eight-week-old female Balb/cJ mice (The Jackson Laboratory, Bar Harbor, ME, USA) were injected intraperitoneally twice per week with either an Npc1 mRNA-targeted ASO (5′-CCCGATTGAGCTCATCTTCG-3′) or a mismatched CTLASO (5′-CCTTCCCTGAAGGTTCCTCC-3′) as previously described (19). Both CTL and NPC1 ASOs were obtained from Isis Pharmaceuticals, Inc. For HPβCD treatment, mice that had been injected for 4 weeks with CTLASO and NPC1ASO were given a single intraperitoneal injection of HPβCD at a 4000 mg/kg dosage, then after 24 h, tissues were analyzed. For NAC treatment, mice were treated with 1% NAC (Sigma-Aldrich, St Louis, MO, USA) administered in drinking water for 4 weeks, concurrent with ASO injections. NAC water was changed twice per week.
Protocols for experiments on Npc1+/+ and Npc1−/− mice were approved by the Washington University Animal Care and Use Committee. NAC was administered in drinking water that was changed every other day. Dosing was based on water consumption (7 ml/day), which was determined experimentally in control groups before initiation of the trial. The dose was adjusted weekly based on weights of the animals, allowing for accurate NAC dose delivery.
For animal survival studies, the general clinical condition of the mice was monitored daily. Death was defined as either being found dead in the cage or the inability to eat or drink on their own, necessitating euthanasia.
Mouse biochemical, histological and tremor analyses
For western blot analysis, 20 µg of individual mouse liver protein preparations were separated by 4–12% NuPAGE Tris-Acetate gels, transferred to nitrocellulose membranes using iBlot, and then chemiluminescence detection was performed using WesternBreeze kits (Life Technologies/Invitrogen, Grand Island, NY). Primary antibodies and dilutions were: rabbit-derived NPC1 polyclonal antibody NB400-148 (Novus Biologicals, Littleton, CO, USA) at 1:2000, and mouse-derived β-actin monoclonal antibody (Bio-Rad Laboratories, Hercules, CA, USA) at 1:10 000. Band intensity was quantified with Quantity One software (Bio-Rad Laboratories) and normalized to β-actin.
For serum chemistry evaluation, blood was obtained from the retro-orbital sinus and centrifuged at 14 000 rpm for 10 min to isolate serum. ALT and AST levels were measured by the Department of Laboratory Medicine, Clinical Center, NIH. Sterol and oxysterol analyses were performed by GC/MS and LC/MS, respectively, as previously described (36,60). Approximately 30 and 100 mg of tissue were used for sterol and oxysterol measurements, respectively. Total GSH levels were measured using the Glutathione Assay kit (Cayman Chemical Company, Ann Arbor, MI, USA).
Liver tissue was fixed in 10% formalin. Sectioning, H&E staining and pathology reporting were performed by Histoserv, Inc. (Germantown, MD, USA).
Tremor was characterized in Npc1−/− mice using a San Diego Instruments tremor monitor (San Diego, CA, USA) according to manufacturer's instructions (10). Each animal was allowed a 5 min acclimation period in the chamber, followed by a 256 s data acquisition period.
Microarray analyses
Microarray experiments were performed using Affymetrix-recommended protocols (Affymetrix, Inc., Santa Clara, CA, USA). Briefly, RNA quality and quantity were evaluated using a Bioanalyzer (Agilent, Inc., Santa Clara, CA, USA) and NanoDrop (Thermo Scientific, Inc., Waltham, MA, USA). Two hundred nanograms of total RNA were used for labeling with the Affymetrix-recommended protocol. The hybridization cocktail containing fragmented, labeled complementary DNA (cDNAs) was hybridized to whole transcriptome Affymetrix Mouse GeneChip 1.0 ST arrays, which were subsequently washed and stained using Affymetrix Fluidics Station standard protocols. The arrays were stained with streptavidin phycoerythrin solution (Molecular Probes, Carlsbad, CA, USA) enhanced with a 0.5 mg\ml biotinylated anti-streptavidin antibody solution (Vector Laboratories, Burlingame, CA, USA). Arrays were scanned with an Affymetrix GeneChip Scanner 3000 and gene expression intensities were calculated using Affymetrix GeneChip Command Console software. Affymetrix .CEL files were normalized using the Robust Multi-Array Analysis algorithm within Partek Genomics Suite software, version 6.5 (Partek Inc., St Louis, MO, USA). Analysis of variance (ANOVA) and linear contrasts were used to identify differentially expressed genes. Type I error was controlled using the Benjamini-Hochberg False Discovery Rate (FDR) correction for multiple testing (61). All microarray-associated P-values reported herein were subjected to FDR correction. Hierarchical clustering was carried out using Partek, and pathway analysis was carried out using MetaCore software (GeneGo, Inc., St Joseph, MI, USA). Full datasets available at GEO Accession number GSE33467.
Relative quantification of RNA transcripts
Total RNA was extracted from 30 mg of liver tissue using an RNAeasy kit (Qiagen, Germantown, MD, USA), and cDNA libraries were prepared using an Invitrogen High Capacity cDNA Reverse Transcription kit. Realtime-PCR was performed with a TaqMan® Universal PCR Master Mix and Itgax/CD11c (Mm00498698_m1) and Srebp2 (Mm01306297) probes (all from Applied Biosystems, Carlsbad, CA, USA). All probes were validated, and the results were evaluated by the comparative CT method using Gapdh as the internal control. NPC1ASO gene expression levels were relative to control samples, reflecting the fold change compared with control.
NPC1 patient treatment and biochemical analyses
NPC1 patients were evaluated at the National Institutes of Health (NIH) in Bethesda, MD from September, 2009 to August, 2010 in a crossover clinical trial (NCT00975689) approved by the Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Written consent, and when feasible assent, was obtained. A total of 35 patients were enrolled: 18 male and 17 female, with a mean age of 16.9 ± 13 years (s.d.), an age range of 1–53 years at the time of enrollment and a mean NPC1 disease severity score of 20 ± 13 points (s.d.), with a range 1–43 points (62). NAC was provided under IND #106,112. This trial was not formally randomized for miglustat; however, 16 patients in this cohort were receiving miglustat off-label prior to the trial, and therefore continued a constant dose 2 months prior to and throughout the trial. After a pretrial 4-week washout period during which all dietary supplements were discontinued, patients were randomized to receive either PharmaNAC effervescent tablets (BioAdvantex Pharma, Mississauga, ON, Canada) or placebo for an 8-week period. Patients were given an initial dose of 15 mg/kg/day for 1 week, then 30 mg/kg/day for the second week and then advanced to 60 mg/kg/day for the remainder of the phase (maximum dose of 900 mg per day); these were divided into three doses spaced throughout the day. Upon successful completion of the first phase, patients underwent a second 4-week washout, followed by a second 8-week period of receiving NAC (if placebo in Phase 1) or placebo (if NAC in Phase 1). The treatment order was determined by block randomization. For all datasets, no order effects were observed, other than a modest order effect for the fraction of reduced CoQ10 (data not shown), thus all data were combined for statistical analyses. Patients were evaluated at the NIH at baseline (Weeks 0 and 12) and phase conclusion (Weeks 8 and 20) time points, in addition to interval blood draws obtained at home every 1 to 2 weeks to monitor liver enzymes. Blood was collected in BD Vacutainer® serum collection tubes. The primary outcome measures were reduction of 7-KC or 3β,5α,6β-triol plasma levels, and these were measured using LC/MS. Secondary outcome measures were reduced/total CoQ10 ratio, GSH/GSSG ratio and increased total serum antioxidant capacity (TEAC). Total and reduced serum CoQ10 levels were measured by the Mayo Clinic Department of Laboratory Medicine and Pathology. GSH/GSSG ratio and TEAC were measured using Glutathione Assay and Trolox Equivalent Assay kits from Cayman Chemical Company (Ann Arbor, Michigan) and performed as per manufacturer's protocols. Subjective measures of clinical improvement were also collected as an exploratory outcome measure using the PedsQL™ Pediatric Quality of Life Inventory and PedsQL™ Multidimensional Fatigue Scale.
Statistical analysis
All graphical data present mean ± SEM unless otherwise indicated. Differences between the mean values were tested for statistical significance (P < 0.05) using two-tailed t-tests for two groups, and either one-way or two-way ANOVA (with Bonferroni's correction when appropriate) for three or more groups, followed by Newman–Keuls, Bonferroni, Tukey or Dunnett's post-tests. Significant differences in survival curves were determined by log-rank analysis (63). Significant differences for human crossover trial data were determined by Wilcoxon-matched-pairs signed-rank test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the intramural research program of the National Human Genome Research Institute, National Institutes of Health (W.J.P., A.I., D.E.W-C., L.L.B.); Dana's Angels Research Trust (D.S.O, R.S., S.F.); the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (F.D.P, C.A.W., N.M.Y.); the Ara Parseghian Medical Research Foundation (D.O., F.D.P., N.M.Y.); and a Bench to Bedside award from the Office of Rare Diseases and the National Institutes of Health Clinical Center.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Abdel Elkhalhoun and the NHGRI microarray core facility for microarray experiments, Bhavesh Borate, Niraj Trivedi and the NHGRI bioinformatics core for help with the microarray analysis, and the contribution of the caretakers, patients and their families who participated in this study.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Vanier M.T. Niemann-Pick disease type C. Orphanet. J. Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B., et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 3.Naureckiene S., Sleat D.E., Lackland H., Fensom A., Vanier M.T., Wattiaux R., Jadot M., Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 4.Sleat D.E., Wiseman J.A., El-Banna M., Price S.M., Verot L., Shen M.M., Tint G.S., Vanier M.T., Walkley S.U., Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl Acad. Sci. USA. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramanian K., Balch W.E. NPC1/NPC2 function as a tag team duo to mobilize cholesterol. Proc. Natl Acad. Sci. USA. 2008;105:15223–15224. doi: 10.1073/pnas.0808256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Infante R.E., Wang M.L., Radhakrishnan A., Kwon H.J., Brown M.S., Goldstein J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl Acad. Sci. USA. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenbaum A.I., Maxfield F.R. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011;116:789–795. doi: 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frolov A., Zielinski S.E., Crowley J.R., Dudley-Rucker N., Schaffer J.E., Ory D.S. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 2003;278:25517–25525. doi: 10.1074/jbc.M302588200. [DOI] [PubMed] [Google Scholar]
- 9.Schedin S., Sindelar P.J., Pentchev P., Brunk U., Dallner G. Peroxisomal impairment in Niemann–Pick type C disease. J. Biol. Chem. 1997;272:6245–6251. doi: 10.1074/jbc.272.10.6245. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd-Evans E., Morgan A.J., He X., Smith D.A., Elliot-Smith E., Sillence D.J., Churchill G.C., Schuchman E.H., Galione A., Platt F.M. Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 11.Shen D., Wang X., Li X., Zhang X., Yao Z., Dibble S., Dong X.-P., Yu T., Lieberman A.P., Showalter H.D., et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin L.D., Gong W., Verot L., Mellon S.H. Niemann–Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 13.Baudry M., Yao Y., Simmons D., Liu J., Bi X. Postnatal development of inflammation in a murine model of Niemann–Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y.-P., Mizukami H., Matsuda J., Saito Y., Proia R.L., Suzuki K. Apoptosis accompanied by up-regulation of TNF-alpha death pathway genes in the brain of Niemann–Pick type C disease. Mol. Genet. Metab. 2005;84:9–17. doi: 10.1016/j.ymgme.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Klein A., Maldonado C., Vargas L.M., Gonzalez M., Robledo F., Perez de Arce K., Muñoz F.J., Hetz C., Alvarez A.R., Zanlungo S. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann–Pick type C neurons. Neurobiol. Dis. 2011;41:209–218. doi: 10.1016/j.nbd.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Fu R., Yanjanin N.M., Bianconi S., Pavan W.J., Porter F.D. Oxidative stress in Niemann–Pick disease, type C. Mol. Genet. Metab. 2010;101:214–218. doi: 10.1016/j.ymgme.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vázquez M.C., Balboa E., Alvarez A.R., Zanlungo S. Oxidative stress: a pathogenic mechanism for niemann-pick type C disease. Oxid. Med. Cell Longev. 2012;2012:205713. doi: 10.1155/2012/205713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loftus S.K., Morris J.A., Carstea E.D., Gu J.Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M.A., Tagle D.A., et al. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- 19.Rimkunas V.M., Graham M.J., Crooke R.M., Liscum L. In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C- associated liver disease. Hepatology. 2008;47:1504–1512. doi: 10.1002/hep.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Võikar V., Rauvala H., Ikonen E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 2002;132:1–10. doi: 10.1016/s0166-4328(01)00380-1. [DOI] [PubMed] [Google Scholar]
- 21.Pentchev P.G., Boothe A.D., Kruth H.S., Weintroub H., Stivers J., Brady R.O. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 1984;259:5784–5791. [PubMed] [Google Scholar]
- 22.Morris M.D., Bhuvaneswaran C., Shio H., Fowler S. Lysosome lipid storage disorder in NCTR-BALB/c mice. I. Description of the disease and genetics. Am. J. Pathol. 1982;108:140–149. [PMC free article] [PubMed] [Google Scholar]
- 23.Xie C., Burns D.K., Turley S.D., Dietschy J.M. Cholesterol is sequestered in the brains of mice with Niemann–Pick type C disease but turnover is increased. J. Neuropathol. Exp. Neurol. 2000;59:1106–1117. doi: 10.1093/jnen/59.12.1106. [DOI] [PubMed] [Google Scholar]
- 24.Zervas M., Dobrenis K., Walkley S.U. Neurons in Niemann–Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 25.Dean O., Giorlando F., Berk M. N-Acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J. Psychiatry Neurosci. 2011;36:78–86. doi: 10.1503/jpn.100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parasassi T., Brunelli R., Costa G., De Spirito M., Krasnowska E., Lundeberg T., Pittaluga E., Ursini F. Thiol redox transitions in cell signaling: a lesson from N-acetylcysteine. Sci. World J. 2010;10:1192–1202. doi: 10.1100/tsw.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanter M.Z. Comparison of oral and i.v. acetylcysteine in the treatment of acetaminophen poisoning. Am. J. Health Syst. Pharm. 2006;63:1821–1827. doi: 10.2146/ajhp060050. [DOI] [PubMed] [Google Scholar]
- 28.Millea P.J. N-acetylcysteine: multiple clinical applications. Am. Fam. Physician. 2009;80:265–269. [PubMed] [Google Scholar]
- 29.Cenedella R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 2009;44:477–487. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- 30.Koh C.H.V., Whiteman M., Li Q.-X., Halliwell B., Jenner A.M., Wong B.S., Laughton K.M., Wenk M., Masters C.L., Beart P.M., et al. Chronic exposure to U18666A is associated with oxidative stress in cultured murine cortical neurons. J. Neurochem. 2006;98:1278–1289. doi: 10.1111/j.1471-4159.2006.03958.x. [DOI] [PubMed] [Google Scholar]
- 31.Zampieri S., Mellon S.H., Butters T.D., Nevyjel M., Covey D.F., Bembi B., Dardis A. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J. Cell. Mol. Med. 2009;13:3786–3796. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marí M., Caballero F., Colell A., Morales A., Caballeria J., Fernandez A., Enrich C., Fernandez-Checa J.C., García-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J.R., Coleman T., Langmade S.J., Scherrer D.E., Lane L., Lanier M.H., Feng C., Sands M.S., Schaffer J.E., Semenkovich C.F., et al. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J. Clin. Invest. 2008;118:2281–2290. doi: 10.1172/JCI32561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cologna S.M., Jiang X.-S., Backlund P.S., Cluzeau C.V.M., Dail M.K., Yanjanin N.M., Siebel S., Toth C.L., Jun H.-S., Wassif C.A., et al. Quantitative proteomic analysis of niemann-pick disease, type c1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS ONE. 2012;7:e47845. doi: 10.1371/journal.pone.0047845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribas G.S., Pires R., Coelho J.C., Rodrigues D., Mescka C.P., Vanzin C.S., Biancini G.B., Negretto G., Wayhs C.A.Y., Wajner M., et al. Oxidative stress in Niemann-Pick type C patients: a protective role of N-butyl-deoxynojirimycin therapy. Int. J. Dev. Neurosci. 2012 doi: 10.1016/j.ijdevneu.2012.07.002. 10.1016/j.ijdevneu.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Jiang X., Sidhu R., Porter F.D., Yanjanin N.M., Speak A.O., Vruchte,te D.T., Platt F.M., Fujiwara H., Scherrer D.E., Zhang J., et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann–Pick C1 disease from human plasma. J. Lipid Res. 2011;52:1435–1445. doi: 10.1194/jlr.D015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter F.D., Scherrer D.E., Lanier M.H., Langmade S.J., Molugu V., Gale S.E., Olzeski D., Sidhu R., Dietzen D.J., Fu R., et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann–Pick C1 disease. Sci. Transl. Med. 2010;2:56ra81. doi: 10.1126/scitranslmed.3001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cluzeau C.V.M., Watkins-Chow D.E., Fu R., Borate B., Yanjanin N., Dail M.K., Davidson C.D., Walkley S.U., Ory D.S., Wassif C.A., et al. Microarray expression analysis and identification of serum biomarkers for Niemann–Pick disease, type C1. Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds193. 10.1093/hmg/dds193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alam M.S., Getz M., Safeukui I., Yi S., Tamez P., Shin J., Velázquez P., Haldar K. Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS ONE. 2012;7:e48273. doi: 10.1371/journal.pone.0048273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy J.V., Ganley I.G., Pfeffer S.R. Clues to neuro-degeneration in Niemann–Pick type C disease from global gene expression profiling. PLoS ONE. 2006;1:e19. doi: 10.1371/journal.pone.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao G., Wen Z., Irizarry K., Huang Y., Mitsouras K., Darmani M., Leon T., Shi L., Bi X. Abnormal gene expression in cerebellum of Npc1-/- mice during postnatal development. Brain Res. 2010;1325:128–140. doi: 10.1016/j.brainres.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez M.E., Klein A.D., Hong J., Dimbil U.J., Scott M.P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 2012;21:2946–2960. doi: 10.1093/hmg/dds126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vázquez M.C., del Pozo T., Robledo F.A., Carrasco G., Pavez L., Olivares F., González M., Zanlungo S. Alteration of gene expression profile in Niemann–Pick type C mice correlates with tissue damage and oxidative stress. PLoS ONE. 2011;6:e28777. doi: 10.1371/journal.pone.0028777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davidson C.D., Ali N.F., Micsenyi M.C., Stephney G., Renault S., Dobrenis K., Ory D.S., Vanier M.T., Walkley S.U. Chronic cyclodextrin treatment of murine Niemann–Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu B., Turley S.D., Burns D.K., Miller A.M., Repa J.J., Dietschy J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl Acad. Sci. USA. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu B., Ramirez C.M., Miller A.M., Repa J.J., Turley S.D., Dietschy J.M. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 2010;51:933–944. doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez C.M., Liu B., Taylor A.M., Repa J.J., Burns D.K., Weinberg A.G., Turley S.D., Dietschy J.M. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann–Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 2010;68:309–315. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patterson M.C., Vecchio D., Jacklin E., Abel L., Chadha-Boreham H., Luzy C., Giorgino R., Wraith J.E. Long-term miglustat therapy in children with Niemann–Pick disease type C. J. Child Neurol. 2010;25:300–305. doi: 10.1177/0883073809344222. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M., Strnatka D., Donohue C., Hallows J.L., Vincent I., Erickson R.P. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1-/- mice. J. Neurosci. Res. 2008;86:2848–2856. doi: 10.1002/jnr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tucker S., Ahl M., Bush A., Westaway D., Huang X., Rogers J.T. Pilot study of the reducing effect on amyloidosis in vivo by three FDA pre-approved drugs via the Alzheimer's APP 5′ untranslated region. Curr. Alzheimer Res. 2005;2:249–254. doi: 10.2174/1567205053585855. [DOI] [PubMed] [Google Scholar]
- 51.Adair J.C., Knoefel J.E., Morgan N. Controlled trial of N-acetylcysteine for patients with probable Alzheimer's disease. Neurology. 2001;57:1515–1517. doi: 10.1212/wnl.57.8.1515. [DOI] [PubMed] [Google Scholar]
- 52.Clark J., Clore E.L., Zheng K., Adame A., Masliah E., Simon D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS ONE. 2010;5:e12333. doi: 10.1371/journal.pone.0012333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muñoz A.M., Rey P., Soto-Otero R., Guerra M.J., Labandeira-Garcia J.L. Systemic administration of N-acetylcysteine protects dopaminergic neurons against 6-hydroxydopamine-induced degeneration. J. Neurosci. Res. 2004;76:551–562. doi: 10.1002/jnr.20107. [DOI] [PubMed] [Google Scholar]
- 54.Cuzzocrea S., Mazzon E., Costantino G., Serraino I., Dugo L., Calabrò G., Cucinotta G., De Sarro A., Caputi A.P. Beneficial effects of N-acetylcysteine on ischaemic brain injury. Br. J. Pharmacol. 2000;130:1219–1226. doi: 10.1038/sj.bjp.0703421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koch A., Trautwein C. N-acetylcysteine on its way to a broader application in patients with acute liver failure. Hepatology. 2010;51:338–340. doi: 10.1002/hep.23484. [DOI] [PubMed] [Google Scholar]
- 56.Beloqui O., Prieto J., Suárez M., Gil B., Qian C.H., García N., Civeira M.P. N-acetyl cysteine enhances the response to interferon-alpha in chronic hepatitis C: a pilot study. J. Interferon Res. 1993;13:279–282. doi: 10.1089/jir.1993.13.279. [DOI] [PubMed] [Google Scholar]
- 57.Farr S.A., Poon H.F., Dogrukol-Ak D., Drake J., Banks W.A., Eyerman E., Butterfield D.A., Morley J.E. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 58.Yan C.Y., Greene L.A. Prevention of PC12 cell death by N-acetylcysteine requires activation of the Ras pathway. J. Neurosci. 1998;18:4042–4049. doi: 10.1523/JNEUROSCI.18-11-04042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson M.C., Mengel E., Wijburg F.A., Muller A., Schwierin B., Drevon H., Vanier M.T., Pineda M. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J. Rare Dis. 2013;8:12. doi: 10.1186/1750-1172-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wassif C.A., Krakowiak P.A., Wright B.S., Gewandter J.S., Sterner A.L., Javitt N., Yergey A.L., Porter F.D. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith–Lemli–Opitz syndrome fibroblasts. Mol. Genet. Metab. 2005;85:96–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 61.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 62.Yanjanin N.M., Vélez J.I., Gropman A., King K., Bianconi S.E., Conley S.K., Brewer C.C., Solomon B., Pavan W.J., Arcos-Burgos M., et al. Linear clinical progression, independent of age of onset, in Niemann–Pick disease, type C. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010;153B:132–140. doi: 10.1002/ajmg.b.30969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J.-S., Nam H.-J., Seo M., Han S.K., Choi Y., Nam H.G., Lee S.-J., Kim S. OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS ONE. 2011;6:e23525. doi: 10.1371/journal.pone.0023525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.