

Abstract
Both Akt 2 and acid ceramidase (ASAH1) are found aberrantly overexpressed in cancer cells, but whether these two enzymes cooperate to induce malignant transformation is not known. We found that in immortalized, non-transformed cells, ectopic co-expression of Akt2 and ASAH1 is significantly more effective than expression of each gene alone at inducing cell invasion and at conferring resistance to apoptosis. Consistent with these observations, siRNA-mediated depletion of both Akt2 and ASAH1 is much more potent than depleting each alone at inhibiting cell viability/proliferation and cell invasion. Furthermore, pharmacological inhibitors of Akt (TCN or MK-2206) and ASAH1 (B13) synergize to inhibit cell viability/proliferation, and combinations of these drugs are more effective than single-agent treatments at inhibiting cell invasion. Taken together, the results suggest that these two enzymes cooperate to induce malignant transformation and warrant further preclinical studies to evaluate the potential of combining inhibitors of Akt and ASAH1 to treat cancer.
Keywords: Akt, acid ceramidase, cell invasion, apoptosis, synthetic lethal interaction, drug synergy, TCN, MK-2206, B13
Introduction
Cancer cells ensure their survival and sustain malignant transformation by harboring several aberrantly activated signaling pathways that are complementary and inter-dependent.1,2 Often suppression of only one of these pathways is insufficient to induce tumor cell death and to reverse malignant transformation. Therefore, understanding the relationship among such pathways can lead to a rational exploration of drug combinations that may be more effective.3-8 Two frequently hyperactive pathways in cancer are those regulated by the serine/threonine kinase Akt and acid ceramidase ASAH1 (N-acylsphingosine amidohydrolase 1). Although these two enzymes regulate distinct yet overlapping pathways that are important for survival and malignant transformation (see below), it is presently unknown whether some human tumors require both pathways for oncogenesis, and whether targeting these two enzymes simultaneously would be more beneficial than targeting each alone.
Akt is an important promoter of tumor cell survival, proliferation as well as migration and invasion.9-11 PI3K-catalyzed formation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), is required for Akt recruitment to the plasma membrane and subsequent activating phosphorylation at T308 and S473. Akt phosphorylates many substrates that mediate tumor progression.10 This has made Akt a major target for cancer drug discovery.12-15 There are three mammalian Akt isoforms that may play distinct but also overlapping roles in development, normal physiology and tumorigenesis. For example, in mice, loss of Akt1 function results in smaller body size and significant growth defects.16,17 Mice lacking Akt2 are unable to maintain glucose homeostasis and are diabetic,18 while Akt3-knockout mice have smaller brains but are otherwise normal.19 In cancer, Akt1 is frequently found persistently phosphorylated and hyperactivated. Furthermore, Akt2 is often overexpressed in human cancer, and its forced overexpression results in increased PI3K-dependent invasion and metastasis of breast and ovarian cancer cells.20 Increased Akt3 expression and loss of PTEN result in the development of melanoma, and Akt3 siRNA stimulates apoptosis and inhibits melanoma development.21
ASAH1 is a ubiquitously expressed enzyme that converts ceramide into sphingosine and free fatty acids.22-24 Ceramide is a major intracellular activator of apoptotic cell death, whereas sphingosine, after its conversion to sphingosine-1-phosphate by sphingosine-1 kinase (SPHK1), stimulates cell growth and proliferation.22-24 SPHK1 is known to activate Akt25 and is itself subject to activating phosphorylation.26 ASAH1 is upregulated in many cancers, particularly prostate cancer,27 and is believed to have an important role in tumor promotion. For instance, in prostate cancer cells, stable overexpression of ASAH1 stimulates cell proliferation and cell invasion and confers resistance to apoptosis.28 Therefore, ASAH1 has emerged as a promising cancer drug target (reviewed in refs. 29–31).
In this paper we have explored whether ASAH1 and Akt cooperate to induce, and whether combined inhibition of ASAH1 and Akt blocks, malignant transformation. Thus, we show here that co-expressing ASAH1 and Akt2 is more effective than expressing each enzyme alone at inducing cell invasion and at causing resistance to apoptosis. We also show that the concomitant knockdown of both ASAH1 and Akt by siRNA is more effective at suppressing cell viability/proliferation and cell invasion. These observations were confirmed by demonstrating that pharmacological inhibitors of ASAH1 and Akt synergistically inhibit cell viability/proliferation, and that the drug combination is more effective than single drugs at inhibiting cell invasion.
Results and Discussion
Akt2 and ASAH1 together are more effective than each alone at promoting cell invasion and inducing resistance to apoptosis in immortalized non-transformed cells.
Both ASAH1 and Akt2 have individually been implicated in cell invasion via distinct mechanisms,20,32-34 raising the question whether these two enzymes cooperate to induce cell invasion. To address this question, we transfected immortalized, non-transformed HPNE cells with ASAH1 and Akt2, either alone or together, and determined their effects on cell invasion as described in “Materials and Methods.” Figure 1A shows that HPNE cells express little ASAH1 and reasonable amounts of Akt2. Figure 1B shows that overexpression of ASAH1 in HPNE cells increased the ability of HPNE cells to invade over 10-fold, from 35 invading cells in empty vector-transfected cells to 364 invading cells in ASAH1-transfected cells. Figure 1B shows that overexpression of Akt2 resulted in even a greater increase in invasion from 35 to 580 (17-fold) invading cells. Overexpression of both ASAH1 and Akt2 resulted in much more than an additive effect, leading to 1,947 invading cells (55-fold increase relative to control). Therefore, the fact that ASAH1 or Akt2 stimulated invasion by 10- or 17-fold, respectively, but that together they promoted invasion by 55-fold, suggests that these two enzymes cooperate to stimulate invasion. These effects are most likely not due to enhanced proliferation rates, since overexpression of each enzyme alone increased the growth rate by 1.3-fold, and, more importantly, co-expression did not provide further growth advantage, and also only increased the growth rate by 1.3-fold (Fig. 2A).
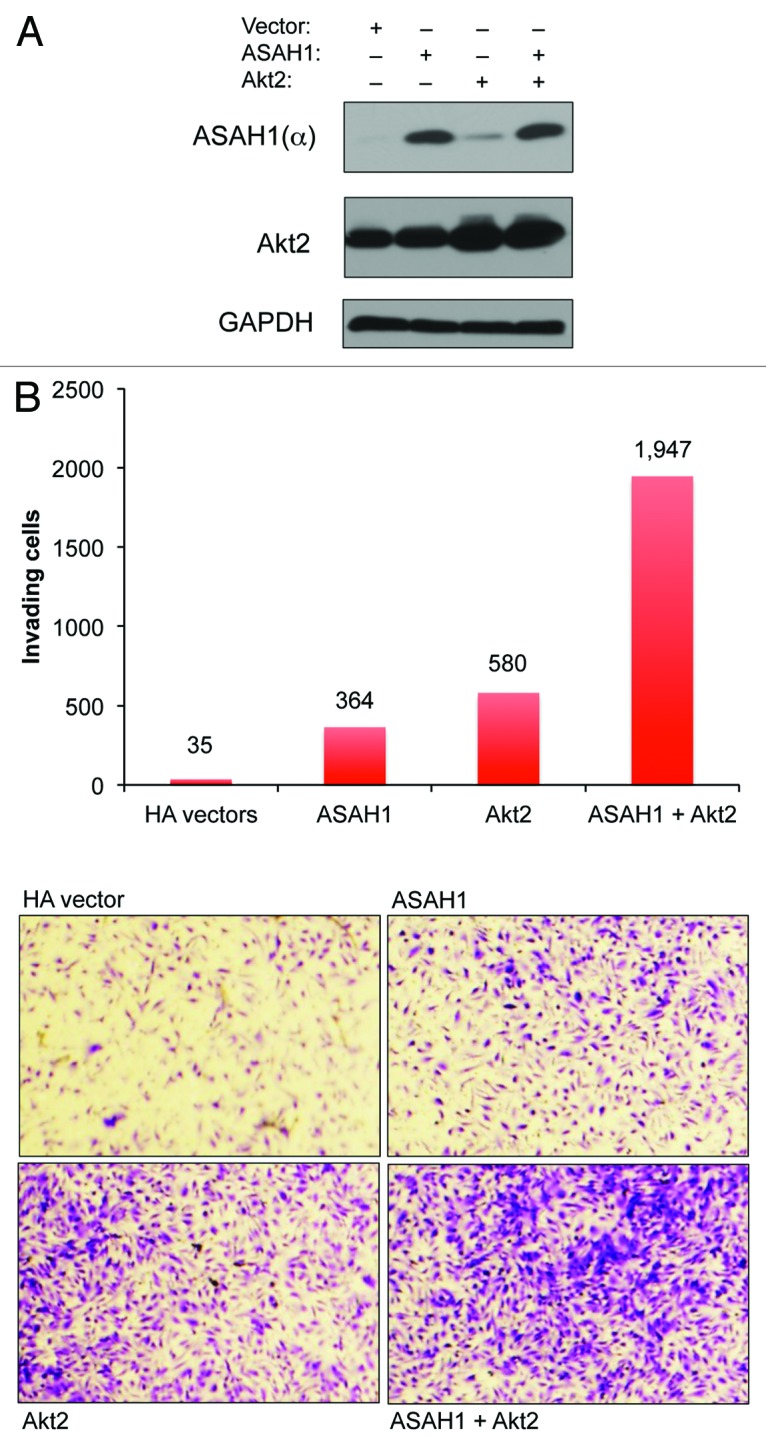
Figure 1. ASAH1 and Akt2 cooperate to stimulate cell invasion. HPNE cells were transfected with 0.4 µg of plasmids encoding ASAH1 and/or Akt2. (A) Cells were harvested 48 h post-transfection, and the resulting lysates were analyzed for the levels of ectopically expressed proteins by western blotting. (B) Cells were harvested 48 h post-transfection, counted, and 20,000 cells were incubated in invasion chambers for another 48 h. The number of invading cells is indicated above each column. The data are representative of three independent experiments.
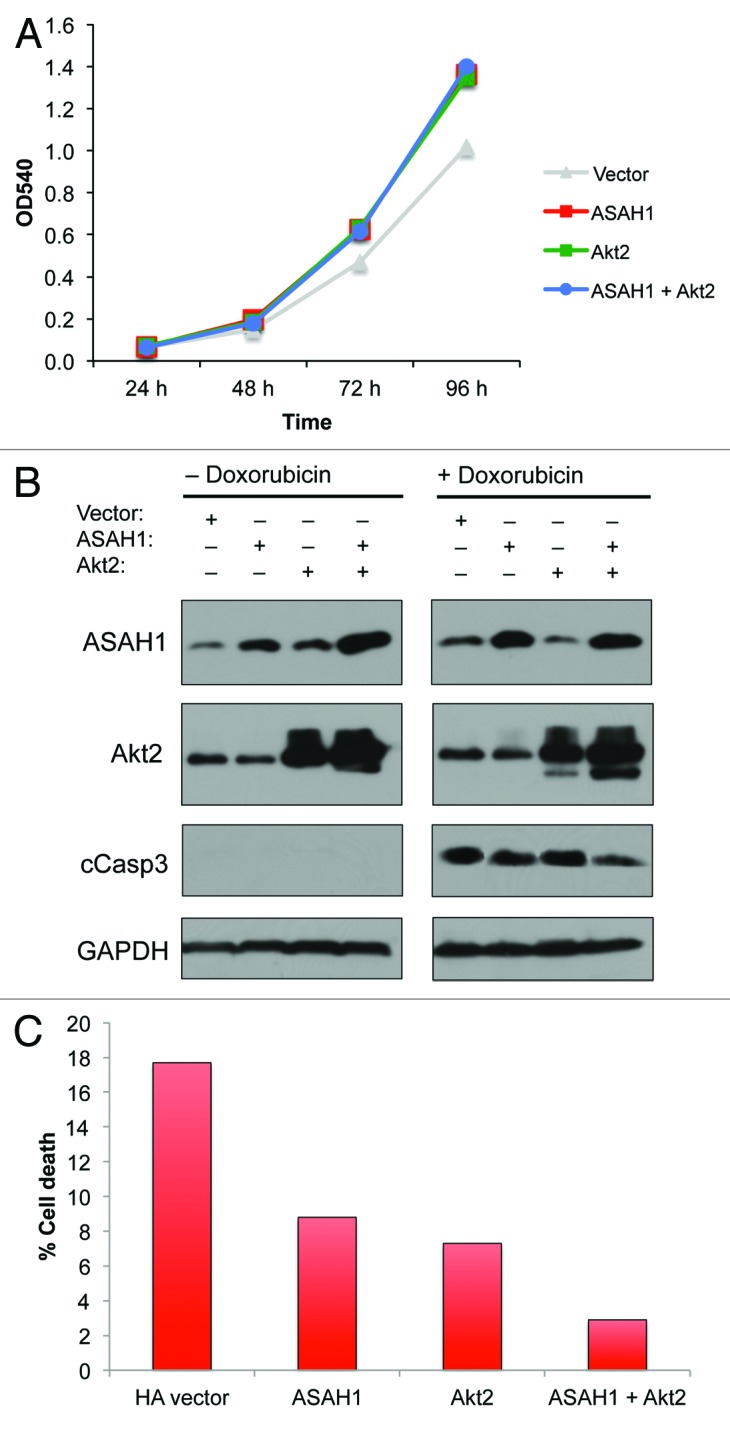
Figure 2. ASAH1 and Akt2 cooperate to evade drug-induced apoptosis, but not to stimulate cell proliferation. HPNE cells were transfected with 0.4 µg of plasmids encoding ASAH1 and/or Akt2. (A) Cell proliferation/viability: cells were harvested 48 h post-transfection, counted and seeded at 1,000 cells per well in 96-well plates. Cell proliferation/viability was assessed by MTT assay for up to 96 h. (B) Apoptosis: western blots are showing the overexpression of ASAH1 and Akt2 and the extent of caspase-3 cleavage following exposure to doxorubicin for 24 h. (C) Cell death: 48 h post-transfection, cells were exposed to 1.0 µM doxorubicin for 24 h, cells were harvested, and the amount of cell death was quantified by determining the number of cells incorporating trypan blue. The data are representative of two independent experiments.
Since both ASAH1 and Akt also regulate cell survival, we investigated whether co-expressing these two enzymes is more effective than expressing each alone at protecting against drug-induced apoptosis. To this end, we expressed ASAH1 and Akt2 alone or together in HPNE cells and determined their effects on doxorubicin-induced apoptosis by measuring the ability of doxorubicin to activate caspase 3. Figure 2B shows that in the absence of doxorubicin, cleaved caspase 3 was not detected. In contrast, treatment with doxorubicin resulted in activation of caspase 3. Furthermore, overexpression of either ASAH1 or Akt2 alone had little effect on the ability of doxorubicin to activate caspase 3. In contrast, co-expression of both ASAH1 and Akt2 inhibited the ability of doxorubicin to activate caspase 3 (Fig. 2B). Similar results were also obtained by determining the effects of these two enzymes on cell death as measured by trypan blue, as described in “Materials and Methods.” Figure 2C shows that doxorubicin induced 18, 9 and 7% cell death in empty vector-, ASAH1- and Akt2-transfected cells, respectively. In contrast, Doxorubicin’s ability to induce cell death was reduced to 3% when both enzymes were concomitantly expressed. Since co-expression of ASAH1 and Akt2 did not result in a growth advantage (Fig. 2A), the enhanced invasion, resistance to apoptosis and cell death following co-expression of these two enzymes was most likely not due to increased cell proliferation. Together, these data suggest that ASAH1 and Akt may cooperate to induce or maintain malignant transformation, raising the question whether inhibition of both of these pathways could be beneficial to human cancer patients.
Depletion of both Akt and ASAH1 together is more effective than depletion of each alone at inhibiting cell survival/proliferation and cell invasion
Figures 1 and 2 demonstrated that ASAH1 and Akt2 cooperate to stimulate invasion and inhibit apoptosis. Therefore, we reasoned that some human cancer cells might be dependent on ASAH1 and/or Akt for cell survival and invasion. To this end, we first assessed the requirement of Akt and ASAH1 alone or together on the survival/proliferation of human cancer cells by depleting Akt alone, ASAH1 alone or both in 13 human cancer cell lines of different lineages. Of the three Akt isoforms, we focused on Akt1 and Akt2, because Akt3’s role in cancer is not as well established as that of the other two isoforms. Following transfection with 20 nM siRNA targeting each enzyme, cells were incubated for 48, 72 and 96 h, and their viability/proliferation was examined by MTT assay as described under “Materials and Methods.” Figure S1 shows that transfection with the siRNAs was effective in depleting the corresponding proteins in all 13 cell lines. Figure 3A shows that, 96 h post-transfection with the indicated siRNAs, the survival/proliferation of 7 out of the 13 cell lines (RXF-393, MCF-7, PANC-1, 786-O, AsPC-1, U-87-MG and MDA-MB-231) was not significantly affected by depletion of Akt1/2 and ASAH1, either alone or together, suggesting that 54% of the human cancer cell lines tested do not depend on Akt1/2 and/or ASAH1 for survival/proliferation. In contrast, three cell lines (23%, PC-3, MDA-MB-468 and DU-145) depended on combined, but not individual, Akt1/2 and ASAH1 functions for survival/proliferation. Similar effects were seen 48, 72 and 96 h post-transfection with siRNAs (Fig. 3B; Fig. S2). Figure 3A also shows that one cell line (LnCaP) was dependent on either Akt1/2 or ASAH1 individually. Others, such as SW-1990 and SkOV-3 were dependent on either Akt1/2 or ASAH1 alone, respectively. These results suggested that 6 out of 13 human cancer cell lines tested (PC-3, MDA-MB-468, DU-145, LnCaP, SW-1990, SkOV-3) depend on Akt1/2 and/or ASAH1 for survival/proliferation. In three out of these six cell lines (PC-3, MDA-MB-468, DU-145), loss of either Akt1/2 or ASAH1 had only a minor to moderate effect, whereas the combined loss of both resulted in dramatic loss of viability, perhaps suggesting a synthetic lethal interaction. These data suggest that the effects of ASAH1 or Akt depletion on cell viability are highly cell type-dependent. For Akt, this is consistent with studies by Koseoglu et al.35 who showed that MDA-MB-468, BT-474 and LnCaP cells, but not U-87-MG or PC-3 cells, become apoptotic upon knockdown of Akt1/2/3. With the exception of MDA-MB-468 cells, our results confirmed these data. This discrepancy may be due to the fact that these authors depleted in MDA-MB-468 cells all three, rather than just two, Akt isoforms.

Figure 3. Combined silencing of Akt and acid ceramidase is synthetically lethal in a subset of human cancer cells. Thirteen human cancer cell lines were transfected with 20 nM siRNA targeting Akt1/Akt2 and ASAH1, either alone or in combination for 48, 72 or 96 h. Cell proliferation/viability was then determined by MTT assay. Cell viability was calculated as percent of cells treated with the appropriate concentration of non-targeting siRNA. The graphs show representative data of at least two independent experiments. (A) Ninety-six hours post-transfection, seven cell lines did not depend on either ASAH1 or Akt1/2, and three cell lines depend on Akt1/2 or ASAH1 or either for proliferation/viability. (B) Three out of 13 cell lines tested are significantly affected by combined, but not individual, depletion of ASAH1 and Akt1/2.The knockdown efficiency associated with these experiments and the proliferation/viability data for all three time points are shown in Figures S1 and S2, respectively. The data are representative of at least two independent experiments.
Figure 1 demonstrated that ASAH1 and Akt2 cooperate to stimulate invasion. Therefore, we next evaluated the requirements of Akt and ASAH1 alone and together on cell invasion as described under “Materials and Methods.” As shown in Figure 4A, in MDA-MB-468 cells, siRNA to ASAH1 inhibited invasion by 13% relative to NT siRNA, whereas siRNA to Akt2 inhibited by 45%. Combined depletion of ASAH1 and Akt2 resulted in 73% inhibition of invasion. Similar results were obtained with PC3 cells where the inhibition of invasion was 44, 64 and 87% for ASAH1, Akt2 and ASAH1 + Akt2, respectively (Fig. 4A). Figure 4B shows that the inhibition of invasion was associated with very efficient knockdown of both enzymes. Together with the results from Figure 1, the data suggest that ASAH1 and Akt2 cooperate to promote invasion, and that inhibition of both enzymes may be required to suppress invasion.
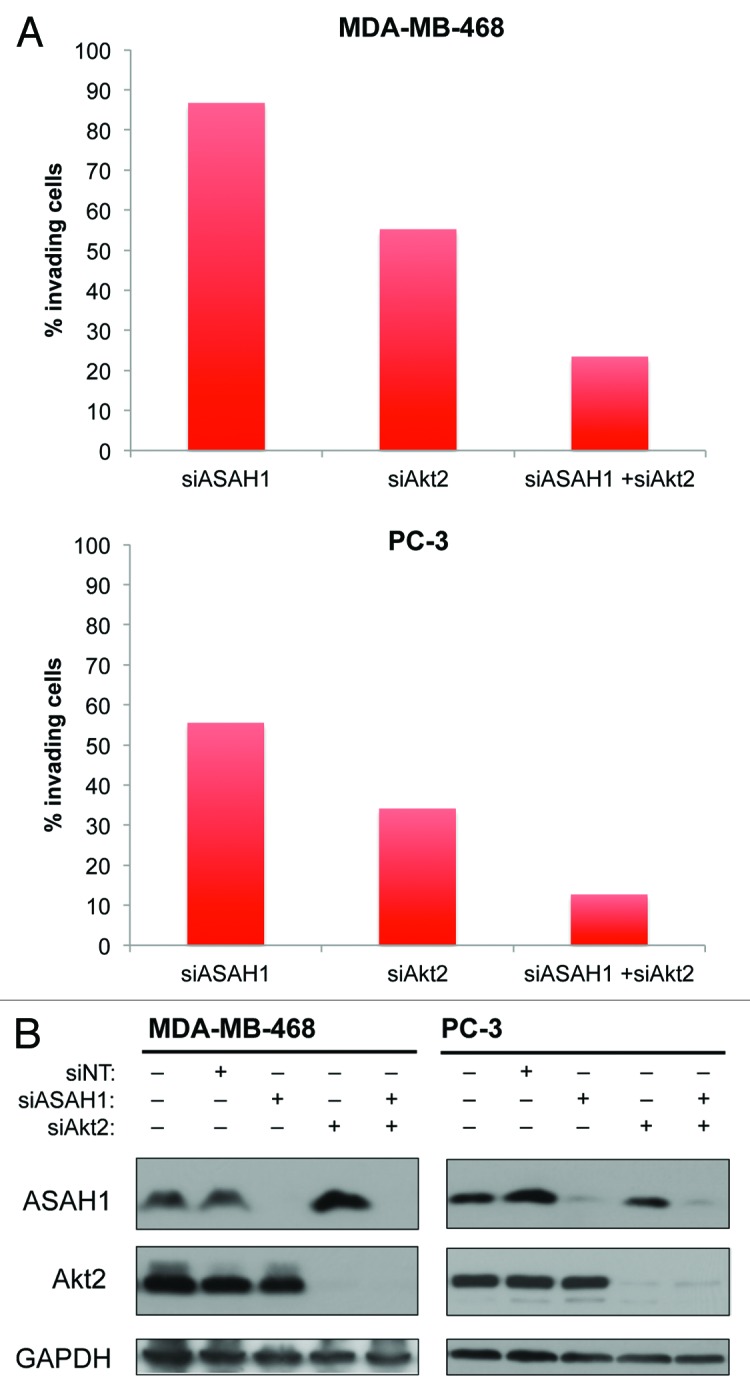
Figure 4. Depletion of ASAH1 and Akt2 is more potent at inhibiting cell invasion than depletion of ASAH1 or Akt2 alone. MDA-MB-468 or PC-3 cells were transfected with 12.5 nM siASAH1 and/or 12.5 nM siAkt2. (A) Cells were harvested 48 h post-transfection and used in invasion assays. The graphs show invading cells (in % of NT siRNA). (B) Western blots show the knockdown efficiencies of siASAH1 and siAkt2 associated with this experiment. The data are representative of three independent experiments.
The Akt inhibitors TCN or MK2206 and the ASAH1 inhibitor B13 act synergistically to inhibit proliferation/survival and suppress cell invasion
The results from Figures 3 and 4 demonstrated that depletion of ASAH1 and Akt together is more effective at suppressing cell survival/proliferation and invasion in some human cancer cells and prompted us to determine whether combining Akt and ASAH1 pharmacological inhibitors is more effective than single-agent treatment. First, we determined the effect of combining the Akt inhibitors TCN36 or MK-220637 with the ASAH1 inhibitor B1338 on cell viability/proliferation. We investigated the effects of these two drugs employing eight different dose combinations at a fixed ratio of 1:1. TCN, MK-2206 and B13 inhibited cell survival/proliferation in a concentration-dependent manner in both MDA-MB-468 and PC3 cells (data not shown). Using Calcusyn software and the Chou and Talalay method, we then determined whether these two drug combinations have synergistic effects.39,40Figure 5 and Table S1 show that in MDA-MB-468 and PC-3 cells, 8 out of 8 combination indices (CIs) were below 1, which indicates synergistic interaction between these compounds. For example, in MDA-MB-468 and PC3 cells, the average CIs for the eight TCN/B13 combinations were 0.20 and 0.42, respectively, indicating a highly synergistic interaction. Similarly, the combinations of MK2206 and B13 were also synergistic with average CIs of 0.55 and 0.37 for MDA-MB-468 and PC3 cells, respectively. Therefore, using two structurally distinct Akt inhibitors, TCN and MK-2206, we demonstrated that inhibition of Akt and ASAH1 is significantly more effective than inhibition of each alone to suppress cell survival/proliferation. These results obtained with pharmacological inhibitors corroborate the results of our genetic depletion approach.

Figure 5. Pharmacological inhibitors of Akt and ASAH1 act synergistically to inhibit cell viability/proliferation. Human cancer cells were treated in 96-well plates with either TCN and/or B13 (A) or MK-2206 and/or B13 (B) for 24 h. The concentrations of the drugs used in the combination treatments were at a ratio of 1:1. The viability of cells was then determined by MTT assay with six replicates per condition, and the data was processed using CalcuSyn software for combination index (CI) analysis to determine synergy (defined as CI values below 1) as described in “Materials and Methods.” The graphs display CI values vs. the fraction affected (Fa) by each combination treatment (effect). The effect ranges from 0 (no inhibition) to 1 (complete inhibition). The data are representative of two independent experiments.
Next, we studied the effects of these Akt and ASAH1 pharmacological inhibitors on cell invasion. To this end, we treated MDA-MB-468 cells with TCN (0.3, 1.0 and 3.0 µM), MK2206 (0.3, 1.0 and 3.0 µM) or B13 (1.0, 3.0 and 10 µM), either alone or in combination (TCN/B13 and MK2206/B13). Figure 6 shows that in MDA-MB-468 cells all the TCN/B13 and MK2206/B13 combinations (0.1/3.0, 1.0/3.0 and 3.0/10 µM) were much more effective than the single-agent treatments, with the highest combination (3.0/10 µM) resulting in complete inhibition of invasion. Similar results were obtained with PC3 cells, where the combination of either TCN/B13 or MK2206/B13 was much more effective at inhibiting invasion than treatment with each drug alone. These results are consistent with the siRNA data of Figure 4, where depletion of both ASAH1 and Akt2 was more effective than the single depletion at inhibiting invasion.

Figure 6. Pharmacological inhibition of ASAH1 and Akt is more effective at inhibiting cell invasion than single-agent treatment. MDA-MB-468 and PC-3 cells were mixed with the indicated drug concentrations and then replated to determine the extent of cell invasion as described in “Material and Methods.” The data are representative of three independent experiments.
Thus, in this paper we demonstrate that ASAH1 and Akt cooperate to induce malignant transformation, and that their combined depletion is more effective in suppressing malignant transformation. Furthermore, a synergy between agents targeting these two proteins was demonstrated. These important findings may be due to the fact that ASAH1 and Akt affect distinct pathways that regulate proliferation, cell death and invasion. Indeed, Akt stimulates cell survival and proliferation by acting on a number of proteins (Forkhead, IKK, Mdm2, TSC1/2) that are apparently not affected by ASAH1.9-11 Furthermore, Akt was shown to promote invasiveness by upregulating the expression of IGF-1 receptor, β1-integrin and MMP-9, respectively,20,33,34 whereas ASAH1 was shown to promote proliferation and survival by affecting retinoblastoma protein (RB) phosphorylation41 and Bax levels,42 and to stimulate invasion by upregulating cathepsin B.32 In addition, there may be crosstalk between ASAH1 and Akt-regulated pathways. For example, ASAH1 increases cellular sphingosine-1 phosphate, which was previously shown to activate Akt.25 Furthermore, a major role of ceramide is to activate the protein phosphatases PP2A and PP1,43-46 and the upregulation of ASAH1 often seen in cancer would decrease ceramide and, therefore, PP1/PP2A activities. PP2A is known to act as a tumor suppressor via dephosphorylating T308 in Akt,47,48 RalA,49 SPHK1,50 but also anti-apoptotic Bcl-251 and pro-apoptotic Bax.52 Similarly, PP1 plays anti-proliferative and pro-apoptotic roles via dephosphorylating RB,53-55 Bad,56 Bax,57 caspase-958 and T450 in Akt.59 Therefore, inhibition of both Akt and ASAH1 would be predicted to result in maximal dephosphorylation of key regulators of the malignant phenotype.
In summary, our results suggest a novel strategy for cancer therapy that would exploit the fact that certain cancer cells depend on both Akt and ASAH1 function for cell survival. Further investigations are needed to delineate the mechanism(s) by which the combined inhibition of these two enzymes is beneficial in a subset of cancer cells.
Materials and Methods
Cells and reagents
The following cells were used in this study and purchased from the American Type Culture Collection: breast cancer cell lines MCF-7, MDA-MB-468 and MDA-MB-231, brain cancer cell line U-87-MG, kidney cancer cell lines 786-O and RXF-393, ovarian cancer cell line SkOV-3, pancreatic cancer cell lines AsPc-1, PANC-1 and SW-1990, prostate cancer cell lines DU-145, LnCaP and PC-3 and the immortalized pancreatic duct epithelial cells HPNE. TCN was obtained from the National Cancer Institute, and MK-2206 was purchased from Chemietek(#CT-MK2206). The ASAH1 inhibitor B13 was from Cayman Chemical (#10006305).
Antibodies were purchased from Cell Signaling (Akt1, #2938; Akt2, #3063; cleaved caspase-3, #9664), BD Biosciences (ASAH1 α subunit, #612302), Covance (GAPDH, #MMS-580S) and Sigma-Aldrich (vinculin, #V9131). Peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch (goat anti-mouse IgG, #115-035-003 and goat anti-rabbit IgG, #111-035-003).
siGENOME SMARTpool siRNA reagents targeting Akt1 (#M-003000-03-0005), Akt2 (#M-003001-02-0005) and ASAH1 (#M-049456-01-0005) as well as the non-targeting siRNA #2 (#D-001206-14-05) were purchased from Dharmacon.
RNAi-mediated silencing
Cells were seeded in either 6-well or 96-well plates and allowed to adhere overnight. The next day, cells were transfected with a mixture of siRNA and transfection reagent lipofectamine RNAiMAX (Invitrogen, #13778150) in OptiMEM (Invitrogen, #31985) at a ratio of 0.1 µl per pmol of siRNA.
Ectopic expression of Akt2 and ASAH1
HA-ASAH1 was amplified from a cDNA clone of acid ceramidase 1 (Invitrogen, Clone ID 3923451) using PCR with HA-pcDNA3 as a vector as described for RhoB by us.60 The forward and reverse primers used for PCR were ATGCCGGGCCGGAGTTGCGTCGCCTTAGT and TCACCAACCTATACAAGGGTCAGGGCA GTCCCGC, respectively. The PCR product was digested with BamH1 and XhoI, cloned into HA-pcDNA3 vector and further confirmed by sequencing. The pLXSN-HA-Akt2 construct was provided by Dr Jin Cheng. Cells were seeded in 6-well plates using media free of antibiotics, and the next day the growth media was replaced by 2 ml OptiMEM, to which was added a mixture of Lipofectamine 2000 (Invitrogen, #11668019) and plasmid constructs in 0.5 ml OptiMEM. Six h later, the OptiMEM was replaced by 2 ml complete growth media.
MTT cell viability assay
Cells were seeded in 96-well plates and allowed to adhere overnight. Following transfection or drug treatment for the times indicated in the figure legends, ½ volume of MTT reagent (Calbiochem, #475989) (3 mg/ml filtered PBS) was added to each well and incubated for ≥ 3 h at 37°C. The supernatant-containing medium and MTT reagent was then aspirated and replaced with 100 µl DMSO. The plates were placed on an orbital shaker for 5 min, and the absorbance at 540 nm was determined with a µQUANT multiplate reader (Bio-Tek Instruments).
Cell invasion assay
Invasion assays were performed using BioCoat Matrigel invasion chambers (BD Biosciences, #354480). For studies involving siRNA or recombinant DNA, cells were transfected with the indicated concentrations of ASAH1 or Akt2 siRNAs/cDNAs and allowed to grow for 48 h on 6-well plates. The cells were then trypsinized, resuspended in serum-free medium and counted by the trypan-blue exclusion method. An equal number of live cells were then plated in the inserts at the indicated concentrations. The bottom chamber contained 10% FBS as the “chemoattractant.” After 48 h at 37°C, the inserts were treated as described in the manufacturer’s instructions, and invading cells were fixed in methanol and stained with 0.2% crystal violet. After an overnight drying period, the inserts were photographed with an Olympus Stereomicroscope, and invading cells were counted using ImagePro 6.2 software.
Synergy analysis
Calcusyn (Biosoft), a software based on the Chou-Talalay combination index (CI) method,39,40 was used to determine the effects of the drug combinations. In the CI method, which is based on the median-effect equation, a derivation from the mass-action law, Drug1 is combined with Drug2 at a constant ratio (IC50)Drug1/(IC50)Drug2, and their effects on proliferation are determined by MTT assay. The resulting data, along with the data obtained from single drug treatments, were entered into Calcusyn to determine a CI value for each combination point, which quantitatively defines additivity (CI = 1), synergy (CI < 1) and antagonism (CI > 1). The resulting values were used to construct a plot of CI values over a range of fractions affected (Fa-CI plot).39,40
Various methods
Estimation of protein concentration and western blotting was performed by using standard laboratory procedures as described by us previously.36
Supplementary Material
Acknowledgments
This work was funded in part by NIH grant RO1-CA098473. We also thank the Microscopy Core at the Moffitt Cancer Center for their cooperation and expertise.
Glossary
Abbreviations:
- ASAH1
N-acylsphingosine amidohydrolase 1, acid ceramidase
- CI
combination index
- DMSO
dimethyl sulfoxide
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PCR
polymerase chain reaction
- PI3K
phosphoinositide-3 kinase
- PTEN
phosphatase and tensin homolog
- SPHK1
sphingosine kinase 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25043
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25043
References
- 1.Kamb A. What’s wrong with our cancer models? Nat Rev Drug Discov. 2005;4:161–5. doi: 10.1038/nrd1635. [DOI] [PubMed] [Google Scholar]
- 2.Kamb A, Wee S, Lengauer C. Why is cancer drug discovery so difficult? Nat Rev Drug Discov. 2007;6:115–20. doi: 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- 3.Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5:649–59. doi: 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- 4.Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33:407–20. doi: 10.1053/j.seminoncol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Klein S, Levitzki A. Targeting the EGFR and the PKB pathway in cancer. Curr Opin Cell Biol. 2009;21:185–93. doi: 10.1016/j.ceb.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130–7. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kodadek T. Rethinking screening. Nat Chem Biol. 2010;6:162–5. doi: 10.1038/nchembio.303. [DOI] [PubMed] [Google Scholar]
- 8.Fedorov O, Müller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol. 2010;6:166–9. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 9.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 10.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle. 2008;7:2991–6. doi: 10.4161/cc.7.19.6784. [DOI] [PubMed] [Google Scholar]
- 12.Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–45. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci. 2007;12:4011–9. doi: 10.2741/2367. [DOI] [PubMed] [Google Scholar]
- 14.Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, et al. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137–44. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 15.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–8. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 18.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu QW, Crenshaw EB, 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 19.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–78. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 21.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 22.Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002;1585:114–25. doi: 10.1016/S1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 23.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–16. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 24.Taha TA, Mullen TD, Obeid LM. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim Biophys Acta. 2006;1758:2027–36. doi: 10.1016/j.bbamem.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morales-Ruiz M, Lee MJ, Zöllner S, Gratton JP, Scotland R, Shiojima I, et al. Sphingosine 1-phosphate activates Akt, nitric oxide production, and chemotaxis through a Gi protein/phosphoinositide 3-kinase pathway in endothelial cells. J Biol Chem. 2001;276:19672–7. doi: 10.1074/jbc.M009993200. [DOI] [PubMed] [Google Scholar]
- 26.Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22:5491–500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seelan RS, Qian C, Yokomizo A, Bostwick DG, Smith DI, Liu W. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes Cancer. 2000;29:137–46. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1018>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AE, et al. The functional effects of acid ceramidase overexpression in prostate cancer progression and resistance to chemotherapy. Cancer Biol Ther. 2007;6:1455–60. doi: 10.4161/cbt.6.9.4623. [DOI] [PubMed] [Google Scholar]
- 29.Park JH, Schuchman EH. Acid ceramidase and human disease. Biochim Biophys Acta. 2006;1758:2133–8. doi: 10.1016/j.bbamem.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Zeidan YH, Jenkins RW, Korman JB, Liu X, Obeid LM, Norris JS, et al. Molecular targeting of acid ceramidase: implications to cancer therapy. Curr Drug Targets. 2008;9:653–61. doi: 10.2174/138945008785132358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Elojeimy S, Turner LS, Mahdy AE, Zeidan YH, Bielawska A, et al. Acid ceramidase inhibition: a novel target for cancer therapy. Front Biosci. 2008;13:2293–8. doi: 10.2741/2843. [DOI] [PubMed] [Google Scholar]
- 32.Beckham TH, Lu P, Cheng JC, Zhao D, Turner LS, Zhang X, et al. Acid ceramidase-mediated production of sphingosine 1-phosphate promotes prostate cancer invasion through upregulation of cathepsin B. Int J Cancer. 2012;131:2034–43. doi: 10.1002/ijc.27480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res. 2001;61:589–93. [PubMed] [Google Scholar]
- 34.Zhang J, Han L, Zhang A, Wang Y, Yue X, You Y, et al. AKT2 expression is associated with glioma malignant progression and required for cell survival and invasion. Oncol Rep. 2010;24:65–72. doi: 10.3892/or_00000829. [DOI] [PubMed] [Google Scholar]
- 35.Koseoglu S, Lu Z, Kumar C, Kirschmeier P, Zou J. AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol Ther. 2007;6:755–62. doi: 10.4161/cbt.6.5.3995. [DOI] [PubMed] [Google Scholar]
- 36.Berndt N, Yang H, Trinczek B, Betzi S, Zhang Z, Wu B, et al. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010;17:1795–804. doi: 10.1038/cdd.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 38.Selzner M, Bielawska A, Morse MA, Rüdiger HA, Sindram D, Hannun YA, et al. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001;61:1233–40. [PubMed] [Google Scholar]
- 39.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 40.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 41.López-Marure R, Ventura JL, Sánchez L, Montaño LF, Zentella A. Ceramide mimics tumour necrosis factor-alpha in the induction of cell cycle arrest in endothelial cells. Induction of the tumour suppressor p53 with decrease in retinoblastoma/protein levels. Eur J Biochem. 2000;267:4325–33. doi: 10.1046/j.1432-1327.2000.01436.x. [DOI] [PubMed] [Google Scholar]
- 42.von Haefen C, Wieder T, Gillissen B, Stärck L, Graupner V, Dörken B, et al. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–19. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- 43.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–30. [PubMed] [Google Scholar]
- 44.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem. 1999;274:20313–7. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 45.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, et al. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–95. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 46.Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res. 2004;45:496–506. doi: 10.1194/jlr.M300347-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Andjelković M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci USA. 1996;93:5699–704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, et al. Protein phosphatase 2A negatively regulates Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–89. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969–82. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barr RK, Lynn HE, Moretti PA, Khew-Goodall Y, Pitson SM. Deactivation of sphingosine kinase 1 by protein phosphatase 2A. J Biol Chem. 2008;283:34994–5002. doi: 10.1074/jbc.M804658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274:20296–300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- 52.Xin M, Deng X. Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J Biol Chem. 2006;281:18859–67. doi: 10.1074/jbc.M512543200. [DOI] [PubMed] [Google Scholar]
- 53.Ludlow JW, Glendening CL, Livingston DM, DeCarprio JA. Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol Cell Biol. 1993;13:367–72. doi: 10.1128/mcb.13.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berndt N, Dohadwala M, Liu CWY. Constitutively active protein phosphatase 1α causes Rb-dependent G1 arrest in human cancer cells. Curr Biol. 1997;7:375–86. doi: 10.1016/S0960-9822(06)00185-0. [DOI] [PubMed] [Google Scholar]
- 55.Wang R-H, Liu CWY, Avramis VI, Berndt N. Protein phosphatase 1α-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20:6111–22. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- 56.Ayllón V, Martínez-A C, García A, Cayla X, Rebollo A. Protein phosphatase 1α is a Ras-activated Bad phosphatase that regulates interleukin-2 deprivation-induced apoptosis. EMBO J. 2000;19:2237–46. doi: 10.1093/emboj/19.10.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flores-Delgado G, Liu CWY, Sposto R, Berndt N. A limited screen for protein interactions reveals new roles for protein phosphatase 1 in cell cycle control and apoptosis. J Proteome Res. 2007;6:1165–75. doi: 10.1021/pr060504h. [DOI] [PubMed] [Google Scholar]
- 58.Dessauge F, Cayla X, Albar JP, Fleischer A, Ghadiri A, Duhamel M, et al. Identification of PP1α as a caspase-9 regulator in IL-2 deprivation-induced apoptosis. J Immunol. 2006;177:2441–51. doi: 10.4049/jimmunol.177.4.2441. [DOI] [PubMed] [Google Scholar]
- 59.Xiao L, Gong LL, Yuan D, Deng M, Zeng XM, Chen LL, et al. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010;17:1448–62. doi: 10.1038/cdd.2010.16. [DOI] [PubMed] [Google Scholar]
- 60.Wang D-A, Sebti SM. Palmitoylated cysteine 192 is required for RhoB tumor-suppressive and apoptotic activities. J Biol Chem. 2005;280:19243–9. doi: 10.1074/jbc.M411472200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.