

Abstract
Development of the endocrine compartment of the pancreas, as represented by the islets of Langerhans, occurs through a series of highly regulated events encompassing branching of the pancreatic epithelium, delamination and differentiation of islet progenitors from ductal domains, followed by expansion and three-dimensional organization into islet clusters. Cellular interactions with the extracellular matrix (ECM) mediated by receptors of the integrin family are postulated to regulate key functions in these processes. Yet, specific events regulated by these receptors in the developing pancreas remain unknown. Here, we show that ablation of the β1 integrin gene in developing pancreatic β-cells reduces their ability to expand during embryonic life, during the first week of postnatal life, and thereafter. Mice lacking β1 integrin in insulin-producing cells exhibit a dramatic reduction of the number of β-cells to only ∼18% of wild-type levels. Despite the significant reduction in β-cell mass, these mutant mice are not diabetic. A thorough phenotypic analysis of β-cells lacking β1 integrin revealed a normal expression repertoire of β-cell markers, normal architectural organization within islet clusters, and a normal ultrastructure. Global gene expression analysis revealed that ablation of this ECM receptor in β-cells inhibits the expression of genes regulating cell cycle progression. Collectively, our results demonstrate that β1 integrin receptors function as crucial positive regulators of β-cell expansion.
Keywords: β-cell development, β1 integrin, Cell adhesion, Extracellular matrix, Islets of Langerhans, Proliferation, Mouse
INTRODUCTION
Integrins comprise a family of α/β heterodimeric receptors that mediate the recognition of extracellular matrix (ECM) components and elicit the activation of bidirectional signaling from both the inside and the outside of cells. ‘Inside-out’ signaling occurs when intracellular biochemical signals induce integrins to bind to their matrix ligands (Aumailley et al., 2000; Liddington and Ginsberg, 2002), whereas ‘outside-in’ signaling results from the binding of a given ECM component by cognate integrin receptors (Hynes, 2002), which, in turn, promotes integrin association with the actin cytoskeleton and activates signaling pathways inside the cell (Giancotti and Ruoslahti, 1999). Whereas the α subunit regulates the specificity for recognition of ECM ligands, the β subunit functions as a signal transduction module. Through this dual partnership of ligand specificity and signaling properties, each α/β integrin heterodimer can transduce extracellular cues into distinct cellular responses as diverse as cell adhesion, migration, proliferation, differentiation and cell survival (Giancotti and Ruoslahti, 1999; Howe et al., 1998; Miyamoto et al., 1998; Schoenwaelder and Burridge, 1999). All of these functions are crucial during development and remain essential for the proper function of multiple cell types throughout postnatal life.
Several in vitro studies using embryonic pancreatic epithelium have shown that integrins regulate cell adhesion and migration (Cirulli et al., 2000; Kaido et al., 2004a; Yebra et al., 2011; Yebra et al., 2003), cell differentiation and proliferation (Kaido et al., 2004b; Kaido et al., 2006; Yebra et al., 2011), as well as secretory functions in pancreatic endocrine cells (Kaido et al., 2006; Parnaud et al., 2006). Specifically, whereas integrins αvβ3, αvβ5 and α6β4 regulate cell attachment to specific ECMs and the migration of undifferentiated pancreatic epithelial cells from ductal compartments (Cirulli et al., 2000; Yebra et al., 2003), β1 integrin functions encompass regulation of cell proliferation and differentiation (Kaido et al., 2004a; Kaido et al., 2006; Kaido et al., 2010; Yebra et al., 2011). A few studies have addressed the function of β1 integrins in the developing pancreas in vivo by targeting either collagen type I-producing cells (Riopel et al., 2011) or acinar cells (Bombardelli et al., 2010). However, virtually nothing is known about the requirement of β1 integrins in the development of the endocrine cell lineage, as represented by the islets of Langerhans (Orci and Unger, 1975) (P. Langerhans, PhD thesis, Friedrich-Wilhelms Universität, Berlin, Germany, 1869). Development of the endocrine compartment of the pancreas occurs through a series of highly regulated events involving branching of the pancreatic epithelium, specification and delamination of islet progenitors from ductal domains, followed by their differentiation, expansion and three-dimensional organization into islet clusters (Pan and Wright, 2011). Among these processes, mechanisms regulating islet cell expansion are crucial for the establishment of a suitable β-cell mass that will ensure adequate insulin secretion in response to normal and modified metabolic demands throughout life.
In this study, we investigated the function of β1 integrins in developing islet β-cells by targeting the deletion of exon 3 of the mouse β1 integrin gene (Itgb1) using a Cre-lox approach. Our results demonstrate that this class of integrin receptors is required for proper β-cell expansion during development and in postnatal life.
MATERIALS AND METHODS
Animal studies
The use of animal subjects was reviewed and approved by the University of California San Diego and University of Washington Institutional Animal Care and Use Committee. RIP-Cre (RIP, rat insulin 2 promoter) transgenic mice (Herrera, 2000) were crossed with floxed β1 integrin mice (Raghavan et al., 2000) to generate conditional knockout mice lacking β1 integrin in pancreatic β-cells. Genotyping was performed by PCR using primers as previously described (Herrera, 2000; Raghavan et al., 2000) (supplementary material Table S1). For proliferation studies, adult mice were injected intraperitoneally with BrdU (Sigma-Aldrich) at 0.1 g/kg body weight every other day for 1 week before harvesting the pancreas. The glucose tolerance test was performed after an overnight fast by intraperitoneal injection of glucose (1 mg/kg body weight) and blood samples were obtained from the tail vein at different time points. Blood glucose was measured with a glucometer (LifeScan) and plasma insulin levels were measured by ELISA (Alpco Diagnostic).
FACS analysis
Pancreatic islets were dissociated into a cell suspension, fixed, permeabilized, and stained by two-color immunofluorescence with PE-conjugated anti-β1 integrin (Biolegend 102207) and Alexa 488-conjugated sheep anti-insulin antibodies, and analyzed using a FACSVantage cell sorter (Becton Dickinson).
Adhesion and proliferation assays
Islets were isolated by intraductal injection of 0.5 mg/ml Liberase (Roche), purified on a Ficoll gradient and either cultured overnight in RPMI containing 10% fetal calf serum (FCS) or dissociated into a single-cell suspension with a non-enzymatic dissociation medium (Sigma-Aldrich) and plated onto different ECMs as previously described (Yebra et al., 2011). After 1 hour, cells were fixed, stained for insulin or glucagon by indirect immunocytochemistry and positive cells counted under the microscope.
For in vitro proliferation assessment, whole islets or single-cell suspensions were plated onto 804G-coated coverslips in RPMI with 10% FCS supplemented with 20 ng/ml hepatocyte growth factor [HGF; also known as scatter factor (SF)] (Bosco et al., 2000; Hayek et al., 1995). Forty-eight hours after plating, cells were pulsed with 10 μM BrdU (Sigma-Aldrich) and cultured for an additional 24 hours. After staining for BrdU and insulin, double-positive cells (BrdU+/insulin+) were counted under a fluorescence microscope and results expressed as a percentage of total β-cells.
Immunofluorescence staining and morphometric analysis
Two- and three-color immunofluorescence and confocal analysis were performed on paraffin sections of fetal and adult mouse pancreas or isolated mouse pancreatic islet cells, as previously described (Yebra et al., 2003). Primary antibodies are listed in supplementary material Table S2. The species-specific fluorophore-labeled F(ab)2 secondary antibodies LRSC-donkey anti-rabbit and anti-mouse IgGs, FITC-donkey anti-rat and anti-sheep IgGs, and Cy5-donkey anti-sheep IgGs were from Jackson ImmunoResearch. Stained sections were viewed on a Zeiss Axiovert 35M microscope equipped with a laser scanning confocal attachment (MRC-1024, Bio-Rad) or on a fluorescence microscope (Nikon Eclipse 800), and morphometric measurement performed using Image Pro Plus software (Media Cybernetics). Measurements of insulin- and glucagon-positive areas were then expressed as a percentage of total pancreatic area. At least 50 non-consecutive sections (5 μm) at intervals of 100 μm were analyzed per adult pancreas (∼5% of the pancreas), collected from five to eight animals per group. For P4 pancreas, we analyzed 30 sections at intervals of 50 μm (∼10% of the pancreas), collected from six animals per group. For E17.5 pancreas, we analyzed 20 sections at intervals of 25 μm (∼20% of the pancreas), collected from eight embryos per group.
Western blotting, quantitative PCR (qPCR) and microarray analysis
Protein extracts from mouse islet lysates were resolved by PAGE, transferred to a PDVF membrane, blocked and incubated with the antibodies described in supplementary material Table S2. Membranes were incubated with HRP-conjugated secondary antibody and visualized by ECL (Pierce).
Total RNA from sorted β-cells was extracted using TRIzol reagent (Life Technologies) and 1 μg total RNA was reverse transcribed using random primers and SuperScript III reverse transcriptase (Life Technologies) following the manufacturer’s instructions. SYBR Green qPCR was performed using the ABI Prism 7900HT system (Life Technologies) and the primers listed in supplementary material Table S1.
For microarray analysis the Illumina mouse-6 v2 Expression BeadChip array was used as previously described (Yebra et al., 2011). Briefly, biotinylated cRNA was prepared using the Illumina RNA Amplification Kit (catalog number1 L1791; Ambion, Austin, TX, USA) according to the manufacturer’s directions starting with 100 ng total RNA. Hybridization of labeled cRNA to the BeadChip, and washing and scanning were performed according to the Illumina BeadStation 500× manual. The arrays were scanned on the Illumina BeadArray Reader, a confocal-type imaging system with 532 (cye3) nm laser illumination. Data analysis and QC was carried out using the BeadStudio software (Illumina). Data presented are MIAME compliant and raw data have been deposited in the EBI ArrayExpress Database (accession number: E-MEXP-3736).
Statistics
The statistical significance of differences in data values was validated by analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test, or by two-tailed Student’s t-test, using the Prism 4 statistical package (GraphPad), with significance limit set at P<0.05.
RESULTS
Cre-mediated deletion of β1 integrin in pancreatic β-cells
The β1 integrin subunit is expressed in virtually all cell types (Giancotti and Ruoslahti, 1999). In the pancreas, β1 integrin has been documented to mediate important functions ranging from adhesion and migration of progenitor cell populations (Yebra et al., 2011; Yebra et al., 2003), to cell survival and secretory functions of adult islet cells (Bombardelli et al., 2010; Bosco et al., 2000; Kaido et al., 2004a; Kaido et al., 2004b; Kaido et al., 2006; Kaido et al., 2010; Parnaud et al., 2006; Riopel et al., 2011; Saleem et al., 2009).
In situ immunolocalization reveals that the β1 integrin subunit is broadly expressed in both the developing and postnatal pancreas (Fig. 1). In E17.5 pancreas, the brightest levels of β1 integrin-specific immunoreactivity are in ductal cells (Fig. 1A-D, cyan arrows) and in vascular structures colonizing developing islet cell clusters (Fig. 1A-D, cyan arrowheads). Significant β1 integrin-specific immunoreactivity is readily detected at cell-cell and cell-matrix boundaries of developing β-cells (Fig. 1A-D, yellow arrowheads). This expression pattern remains relatively unaltered in postnatal life at P4 (Fig. 1E-H), when β1 integrin remains expressed at high levels in the ductal epithelium (cyan arrows) and in endothelial cells (cyan arrowheads), and at lower levels in β-cells (yellow arrowheads) and in acinar cells, as determined by both qualitative assessment (Fig. 1A-H) and quantitative measurement of the pixel intensity of β1 integrin-specific immunoreactivity (Fig. 1I).
Fig. 1.
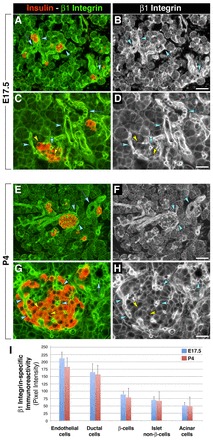
Expression pattern of β1 integrin in the developing and postnatal mouse pancreas. (A-H) Immunolocalization of β1 integrin and insulin in E17.5 (A-D) and P4 (E-H) pancreas. (B,D,F,H) Monochromatic images to better visualize cell-cell and cell-matrix boundaries immunostained for β1 integrin. The brightest β1 integrin-specific immunoreactivity is detected in ductal cells (cyan arrows) and in endothelial cells (cyan arrowheads). Significant β1 integrin expression is also detected at cell-cell and cell-matrix boundaries of developing β-cells (yellow arrowheads). (I) Quantitative assessment of the pixel intensity of β1 integrin-specific immunoreactivity in E17.5 and P4 pancreas. Values are presented as mean±s.e.m. Scale bars: 50 μm in B,F; 25 μm in D,H.
Building on these results, and based on the notion that the signaling properties of integrin receptors are dictated primarily by their state of activation rather than by their level of expression (Giancotti and Ruoslahti, 1999; Liddington and Ginsberg, 2002), we investigated the functional requirement of β1 integrin during β-cell development by means of a conditional gene knockout approach. We chose RIP-Cre mice, which have been used previously to successfully target floxed DNA sequences in pancreatic β-cells (Herrera, 2000). Fig. 2A shows X-Gal staining of islets of Langerhans in a pancreas from a heterozygous RIP-Cre;129S-Gt(ROSA)26Sortm1Sor/J mouse (Soriano, 1999). Following this control experiment demonstrating Cre activity in islet cells, we crossed RIP-Cre mice with homozygous B6;129-Itgb1tm1Efu/J mice that harbor loxP sites flanking exon 3 of the β1 integrin gene (Raghavan et al., 2000). Fig. 2B shows the PCR genotyping of β1 integrin heterozygous/RIP-Cre+ (ht/Cre+) and β1 integrin homozygous/RIP-Cre+ (hm/Cre+, henceforth referred to as RIP-Cre/β1KO). RIP-Cre/β1KO mice were viable and were used for biochemical and morphological characterization.
Fig. 2.
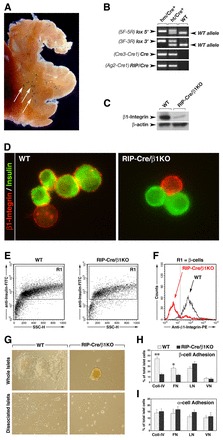
Efficient deletion of β1 integrin in pancreatic β-cells. (A) Whole-mount of X-Gal-stained (blue) adult pancreas from a RIP-Cre/ROSA26 mouse demonstrating targeting of Cre-mediated recombination to pancreatic islet clusters (arrows). (B) PCR genotyping of tail DNA showing the presence of the loxP sequences flanking exon 3 of Itgb1 (lox 5′ and lox 3′); Cre3-Cre1 primers were used to detect the presence of the Cre gene band and Ag2-Cre1 to detect the correct integration of the Cre gene with the RIP sequence. (C) Western blot analysis of isolated pancreatic islets from WT or RIP-Cre/β1KO mice probed for β1 integrin. β-actin was used as loading control. (D) Immunofluorescence analysis of dissociated islet cells from WT or RIP-Cre/β1KO pancreas stained for β1 integrin (red) and insulin (green). (E) Flow cytometry analysis of islet cells isolated from WT or RIP-Cre/β1KO pancreas. (F) Levels of β1 integrin-specific immunoreactivity in insulin-positive cells from the R1 gate in E. (G) Long-term adhesion and spreading assay of whole islets (top row) or dissociated islet cells (bottom row). (H,I) Quantitative assessment of β-cell and α-cell adhesion to the ECM proteins collagen IV (Coll-IV), fibronectin (FN), laminin (LN) and vitronectin (VN). Values are expressed as a percentage ± s.d. of total number of cells seeded. (H) n=5, *P<0.001, **P<0.05.
Western blotting analysis of islet protein extracts from RIP-Cre/β1KO mice demonstrates efficient ablation of β1 integrins (Fig. 2C), with only a faint β1 integrin-specific immunoreactive band in the RIP-Cre/β1KO sample, which is likely to be due to the non-insulin-expressing endocrine cells or other non-endocrine cells present within the islets. Fig. 2D shows immunofluorescent staining of isolated islet cells for β1 integrin (red) and insulin (green). Note that whereas significant β1 integrin-specific immunoreactivity is detected in wild-type (WT) insulin-positive and insulin-negative cells (Fig. 2D, left), in RIP-Cre/β1KO mice the β-cells (green) lack β1 integrin-specific staining (Fig. 2D, right). Flow cytometry analysis on islet cells immunostained for insulin and β1 integrin demonstrates ∼95% recombination in RIP-Cre/β1KO mice (Fig. 2E,F). Collectively, these experiments demonstrate that β1 integrin is efficiently deleted in islet β-cells of RIP-Cre/β1KO mice.
Defective β-cell adhesive properties in RIP-Cre/β1KO mice
The ability of integrin receptors to support cell adhesion depends on the recognition of specific ECM ligands present in the extracellular environment. To determine whether pancreatic β-cells from RIP-Cre/β1KO mice exhibit impaired cell adhesion properties, intact islet clusters or isolated islet cells were tested for their ability to adhere to a cell-assembled basal membrane-like matrix (804G) (Bosco et al., 2000; Hayek et al., 1995) or to select ECM components. We found that islet cell clusters isolated from WT mice promptly adhered and spread onto 804G matrix, and established monolayer colonies within 72 hours in culture (Fig. 2G, top left). By contrast, RIP-Cre/β1KO islet cells, cultured under the same conditions, remained loosely attached to the 804G matrix and failed to establish cell monolayers (Fig. 2G, top right). When islets were dissociated into a single-cell suspension and plated on the 804G matrix, WT islet cells adhered and established small colonies (Fig. 2G, bottom left), whereas islet cells isolated from RIP-Cre/β1KO mice were unable to do so (Fig. 2G, bottom right).
To examine the adhesive property of β-cells and non-β-cell types present in islet cell preparations, islet cells isolated from either WT or RIP-Cre/β1KO mice were tested for their ability to adhere to select ECM proteins using a short-term adhesion assay under serum-free conditions, as previously described (Yebra et al., 2011; Yebra et al., 2003). At the end of the assay, adherent cells were fixed, stained for insulin or glucagon by immunocytochemistry to identify α- and β-cells, respectively, and counted. Fewer β-cells from RIP-Cre/β1KO islets adhered to collagen IV and fibronectin when compared with WT cell preparations (Fig. 2H). By contrast, β-cell adhesion to laminin and vitronectin was unaffected, which was likely to be due to the ability of other integrin receptors, such as α6β4 and αvβ5, to mediate cell adhesion to these ECMs (Belkin and Stepp, 2000; Cirulli et al., 2000). Glucagon-producing α-cells, which were not targeted in RIP-Cre/β1KO mice, displayed comparable adhesion to all ECM substrates tested in both animal groups (Fig. 2I).
Reduced β-cell numbers in RIP-Cre/β1KO mice
Pancreatic sections from 8-week-old WT or RIP-Cre/β1KO mice were used for immunofluorescence and morphometric studies to determine whether the deletion of β1 integrin affected the phenotype and architecture of the islets of Langerhans. These experiments revealed that islet clusters are significantly smaller in RIP-Cre/β1KO mice when compared with WT (Fig. 3A-D). A representative example of this phenotype is shown in Fig. 3A, where immunostaining for insulin and glucagon identifies islet clusters. Within islets of RIP-Cre/β1KO mice, β1 integrin-specific immunoreactivity is only detected in α-cells (red) and in capillary endothelial cells (red arrows), but not in β-cells. Extensive morphometric measurements revealed that the area occupied by β-cells is reduced by ∼80% in RIP-Cre/β1KO mice compared with either WT or control RIP-Cre transgenics (Fig. 3B), whereas the number of α-cells is unaffected (Fig. 3C). In addition, analysis of the islet size distribution revealed that whereas the frequency of small islet clusters (50-100 cells) does not differ significantly between WT and RIP-Cre/β1KO mice, the number of larger islets (>100 cells) is dramatically reduced in RIP-Cre/β1KO mice, suggesting that β-cells, following their specification and differentiation, are unable to expand in the absence of β1 integrin (Fig. 3D). Morphometric assessment of apoptotic events as a possible mechanism for the reduction in β-cell numbers revealed no differences between RIP-Cre/β1KO and WT pancreata (data not shown). Further phenotypic characterization revealed that β-cells from RIP-Cre/β1KO mice exhibit a normal expression pattern of the transcription factors MAFA, PAX6, NKX6.1 and PDX1 (Fig. 3E), suggesting that loss of β1 integrin does not affect their ability to express these markers of endocrine differentiation.
Fig. 3.
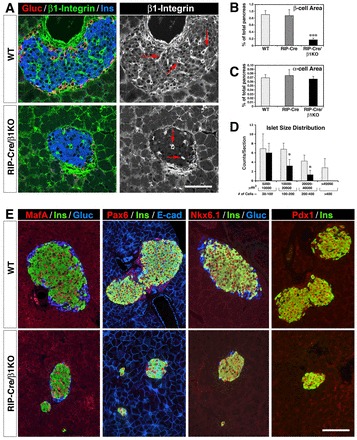
Characterization of β1 integrin-deficient pancreatic islets. (A) Representative confocal images of pancreatic sections from 8-week-old WT (top row) and RIP-Cre/β1KO mice (bottom row) immunostained for β1 integrin (green), glucagon (red) and insulin (blue). Red arrows in monochromatic images point to intra-islet endothelial cells strongly immunoreactive for β1 integrin. (B,C) Quantification of insulin-positive (B) and glucagon-positive (C) area in WT (n=5), RIP-Cre (n=3) and RIP-Cre/β1KO (n=5) pancreas. Values are expressed as a percentage of total pancreatic area ± s.d. ***P<0.001. (D) Islet cell number and size distribution in WT and RIP-Cre/β1KO mice. Values are expressed as mean ± s.d. of the number of islets per section (n=3 animals). *P<0.05. (E) Immunolocalization of transcription factors MAFA, PAX6, NKX6.1 and PDX1 in WT and RIP-Cre/β1KO pancreas. Scale bars: 50 μm in A; 70 μm in E.
Loss of β1 integrin does not affect the differentiation or function of β-cells
To fully define the phenotype of RIP-Cre/β1KO β-cells we performed ultrastructural studies by transmission electron microscopy (TEM). Representative TEM images of β-cells from WT (Fig. 4A,C) and RIP-Cre/β1KO (Fig. 4B,D) mice show morphologically normal insulin granules in both cell preparations, thus demonstrating that ablation of β1 integrin does not negatively affect the cellular machinery responsible for the biosynthesis of these subcellular compartments. Interestingly, despite previous evidence indicating that β1 integrin plays important roles in the assembly and maintenance of basal membranes (Aumailley et al., 2000; Ruoslahti, 1996), we find that β-cells from RIP-Cre/β1KO mice exhibit electron-dense basal membrane structures that are indistinguishable from those observed in WT (Fig. 4D versus 4C, brackets). In addition, microcapillary endothelial cells lining basal membranes adjacent to RIP-Cre/β1KO β-cells exhibit normal fenestrations (Fig. 4C,D, arrowheads), suggesting that ablation of β1 integrin in β-cells has no functional consequences for the morphology of the adjacent vascular endothelium.
Fig. 4.
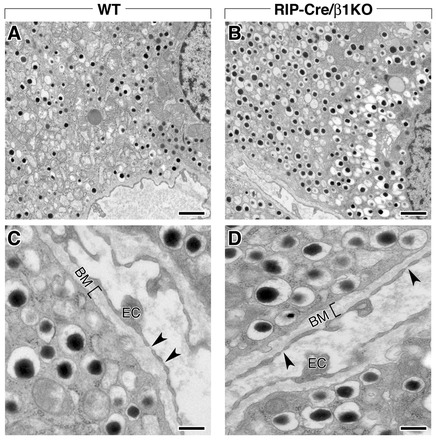
Ultrastructural analysis of WT and RIP-Cre/β1KO pancreatic islets. (A,B) Transmission electron microscopy (TEM) of β-cells in WT (A) and RIP-Cre/β1KO (B) islets showing insulin-containing granules with their typical electron-dense core. (C,D) Higher magnification TEM images of WT (C) and RIP-Cre/β1KO (D) islets identifying the presence of a basal membrane (BM, bracket) at the interface between endothelial cells (EC) and β-cells. Normal endothelial cell fenestrae (C,D, arrowheads) can be identified in both WT and RIP-Cre/β1KO samples. Scale bars: 1 μm in A,B; 0.3 μm in C,D.
In these studies we also observed that β-cells from RIP-Cre/β1KO mice appear to contain an increased number of insulin granules. This observation was validated biochemically by the demonstration that β-cells in RIP-Cre/β1KO mice contain close to 3-fold more insulin than in WT (Fig. 5A).
Fig. 5.
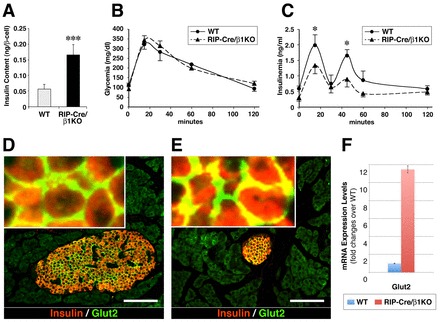
Insulin secretory function of WT and RIP-Cre/β1KO pancreatic islets. (A) Insulin content measured in WT and RIP-Cre/β1KO β-cells (n=4). *P<0.001. (B,C) Blood glucose (B) and plasma insulin (C) quantification during a glucose tolerance test in 4-month-old WT and RIP-Cre/β1KO mice. Values are expressed as mean ± s.e.m. (n=5 animals for each genotype). **P<0.05. (D,E) GLUT2-specific immunoreactivity detected in WT (D) and RIP-Cre/β1KO (E) β-cells. (F) mRNA expression levels of Glut2 measured by qPCR in β-cells isolated from WT and RIP-Cre/β1KO pancreas. Scale bars: 70 μm.
Despite the significant reduction in β-cell numbers (Fig. 3), adult RIP-Cre/β1KO mice did not display abnormal fasting glycaemia, nor altered response in a glucose tolerance test (Fig. 5B). These results are not surprising in consideration of the fact that, in most rodent models, overt diabetes occurs only upon loss of more than 80% of the total pancreatic β-cell number (Bonner-Weir et al., 1983). The only functional difference we observed in RIP-Cre/β1KO mice is a reduced insulin output in response to glucose, although both the first and second phases of insulin release are preserved (Fig. 5C). RIP-Cre/β1KO β-cells also display a normal membrane-targeted expression of the glucose transporter GLUT2 (SLC2A2 - Mouse Genome Informatics) (Fig. 5D,E), and Glut2 expression appears significantly upregulated in RIP-Cre/β1KO β-cells when compared with WT (Fig. 5F). These results indicate that RIP-Cre/β1KO β-cells harbor a functional glucose-sensing machinery that allows for sufficient insulin secretion to maintain normoglycemia. It is possible that, despite the reduced β-cell mass and decreased levels of circulating insulin (Fig. 5C), RIP-Cre/β1KO mice may develop peripheral insulin hypersensitivity as an adaptive counter-regulatory mechanism to maintain normoglycemia.
β1 integrin is required for β-cell expansion
Based on the evidence that β1 integrins may function as positive regulators of cell cycle progression and survival (Novak et al., 1998; Sherr and Roberts, 1995), we postulated that the reduced number of β-cells in RIP-Cre/β1KO mice might result from defective β-cell expansion and/or survival during the developmental and neonatal periods. Indeed, immune-morphometric assessment of pancreata stained for markers of cell proliferation demonstrated that at E17.5 numerous proliferating β-cells can be identified in the pancreas of WT but not RIP-Cre/β1KO mice (Fig. 6A, arrowheads). This analysis revealed a significant reduction of the number of proliferating β-cells, as determined by double immunostaining for insulin and phospho-histone H3 (Fig. 6B), or double staining for insulin and PCNA (Fig. 6C). These results correlated with a reduction of β-cell area already at this relatively early stage of islet cell cluster expansion (Fig. 6D). This phenotype becomes even more striking by P4, when the number of proliferating β-cells increases dramatically in WT but not in RIP-Cre/β1KO mice (Fig. 6E-G), an effect that correlates with a dramatic reduction of the β-cell area (Fig. 6H). Eventually, by 8 weeks of age, although occasional proliferation of β-cells can still be detected in WT mice, BrdU+ (or PCNA+) β-cells become extremely rare in RIP-Cre/β1KO mice (Fig. 6I-K). Accordingly, RIP-Cre/β1KO mice exhibit a β-cell area that is only ∼18% of that measured in WT or in RIP-Cre transgenic mice used as additional controls (Fig. 6L).
Fig. 6.
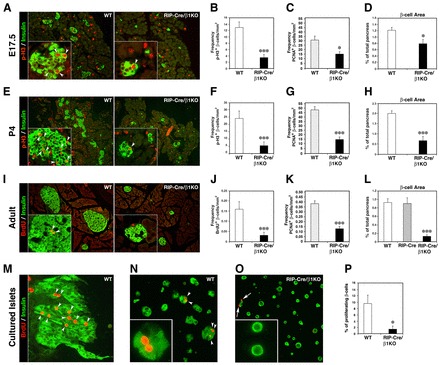
Defective β-cell expansion in RIP-Cre/β1KO mice during pancreas development. (A) Representative images of pancreatic sections from WT and RIP-Cre/β1KO E17.5 embryos immunostained for phospho-histone H3 (p-H3) and insulin. (B-D) Frequency of p-H3+ (B) and PCNA+ (C) β-cells and measurement of β-cell area (D) in WT and RIP-Cre/β1KO E17.5 pancreas. (E) Pancreatic sections of P4 WT and RIP-Cre/β1KO mice immunostained for p-H3 and insulin. (F-H) Frequency of p-H3+ (F) and PCNA+ (G) β-cells and measurement of β-cell area (H) in P4 WT and RIP-Cre/β1KO mice. (I) Adult pancreatic sections from WT and RIP-Cre/β1K mice stained for BrdU and insulin. (J-L) Frequency of BrdU+ (J) and PCNA+ (K) β-cells and measurement of β-cell area (L) in pancreatic sections from adult WT and RIP-Cre/β1K mice. (M-O) Immunostaining for BrdU and insulin in cultures to assess the induction of β-cell proliferation by 804G matrix and HGF in WT whole islets (M), partially dissociated WT (N) or RIP-Cre/β1KO (O) islet cells. Insets in A,E,I,N,O show higher magnification of islet cells. Arrowheads in A,E,I,M,N indicate nuclei of proliferating β-cells. Arrows in O indicate proliferating insulin-negative cells. (P) Quantification of BrdU+ β-cells measured for WT and RIP-Cre/β1KO islet cell preparations. (A-D) n=8 pancreata for each genotype; (E-L) n=5 pancreata for each genotype; (M-P) representative of n=4 independent experiments using both WT and RIP-Cre/β1KO pancreatic islets. Values in graphs represent mean±s.e.m. (B) ***P<0.001, (C,D) *P<0.05, (F-H,J-L) ***P<0.001.
To investigate whether β-cells from RIP-Cre/β1KO mice can be induced to proliferate in vitro, we isolated islets from both WT and RIP-Cre/β1KO mice, and cultured them on 804G extracellular matrix-coated coverslips in the presence of HGF as previously described (Hayek et al., 1995). WT islets established colonies that comprised numerous BrdU-incorporating insulin+ cells over a 3-day culture period (Fig. 5M). By contrast, islets isolated from RIP-Cre/β1KO mice failed to adhere, spread or generate colonies (Fig. 2F) and did not show any significant cell proliferation (not shown). In similar experiments, plating of partially dissociated islets onto the 804G extracellular matrix gave similar results, with numerous BrdU-incorporating β-cells detected in WT (Fig. 6N,P) but not in RIP-Cre/β1KO islet cell preparations (Fig. 6O,P). Hence, ablation of β1 integrin in β-cells severely impacts on their ability to respond to mitogenic stimuli provided by ECM cues and by growth factors such as HGF.
Ablation of β1 integrin affects the expression of genes that regulate cell cycle progression
To investigate the effect of β1 integrin ablation on global gene expression we performed a microarray analysis on FACS-sorted β-cells using the Illumina mouse-6 v2 Expression BeadChip array. As shown in Fig. 7A, these experiments revealed a significant upregulation of genes encoding ECM ligands of β3, β4 or β5 integrin receptors (Vtn, Lamb3, Ntn1, Ntn4), non-integrin receptors such as DDR1 and DDR2 (Col1a1, Col3a1, Col14a1) and genes involved in basement membrane assembly (Col6a1, Col7a1, Col8a1) (van der Rest and Garrone, 1991; Gelse et al., 2003; Vogel et al., 1997). Conversely, genes encoding ECM ligands of β1 integrin receptors (Col4a2) are downregulated. Similarly, genes regulating MAPK/Rho activity (Map4, Mapk8ip2, Pik3cg, Jun, Arhgef10, Fgd2) are downregulated, possibly reflecting reduced integrin activation and utilization due to the ablation of the β1 integrin subunit.
Fig. 7.
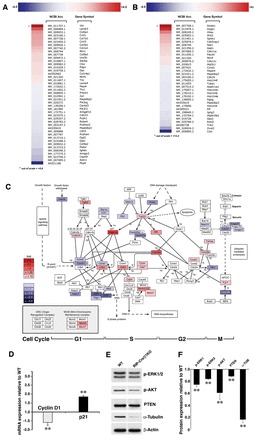
β1 integrin-dependent regulation of global gene expression in RIP-Cre/β1KO sorted β-cells. (A,B) Heat maps of selected genes involved in cell adhesion (A) or the cell cycle (B) that are downregulated or upregulated in RIP-Cre/β1KO sorted β-cells. (C) Altered expression levels of cell cycle genes plotted onto the cell cycle pathway map using GenMAPP software. (D) Changes in expression of the key regulators of cell cycle progression cyclin D1 and Cdkn1a (p21) validated by qPCR. Values are expressed as fold expression ± s.d. relative to WT (WT value=1), and normalized to Gapdh. **P<0.01. (E) Western blot analysis of isolated pancreatic islets probed for phospho-ERK1/2, phospho-PKB, PTEN and α-tubulin. (F) Densitometric quantification of immunoblot bands from E. Values are representative of three independent experiments and are expressed as fold expression ± s.d. relative to WT and normalized to β-actin. **P<0.01.
As shown in Fig. 7B, many genes reported to act as positive regulators of cell cycle arrest, such as ERK-dependent tumor suppressors (Dmbt1, Azgp1, Pdcd4), inhibitors of cyclins (Cdkn1a, Cdkn1c), and cyclin dependent kinases (Cdk5rap1), are upregulated in the RIP-Cre/β1KO β-cells. Surprisingly, we also observed that some genes known to foster cell cycle progression (Hhex, Ifrd2, Sphk1) are upregulated in RIP-Cre/β1KO β-cells, possibly as a result of an attempt to overcome the proliferation defect. Among genes known to function as strong positive regulators of cell cycle progression, Ovol2 and Tuba1 are the most significantly downregulated. A further representation of these results is shown in Fig. 7C, where individual genes and their fold changes in expression (normalized to values recorded in WT β-cells) are clustered based on their involvement in the regulation of select phases of the cell cycle, i.e. G1 through M phase. This representation facilitates a visual inspection of both positive and negative regulators of the cell cycle while identifying the effects of β1 integrin deletion on their levels of expression. In this display, Ccnd1, Ccnd2 and Ccne1, which regulate the progression from G1 to S phase, are downregulated, possibly owing to the increased expression of the inhibitor Cdkn1a (p21) (Cozar-Castellano et al., 2006). Significant downregulation of E2f1 and E2f2 is also observed, which in turn is likely to contribute to cell cycle arrest in G1 phase (Wu et al., 2001). As a possible rebound mechanism, genes involved in S- and G2-phase checkpoints, such as Cdc25a, are upregulated, whereas the cyclin inhibitor Gadd45 and components of the APC complex (Anapc5, Anapc7 and Cdc27) are downregulated, possibly as a result of an unsuccessful attempt to push the cell through interphase. Changes in the expression levels of select genes were validated by qPCR. As shown in Fig. 7D, β-cells isolated from RIP-Cre/β1KO mice show significant downregulation of cyclin D1 (Ccnd1), which is known to be required for postnatal β-cell growth (Kushner et al., 2005), and an upregulation of the cyclin inhibitor Cdkn1a, which is reported to negatively regulate cell cycle progression in β-cells (Cozar-Castellano et al., 2006) and in other cell types (Aszodi et al., 2003; Li et al., 2005).
In parallel experiments, western blotting analysis revealed a slight reduction of phosphorylated ERK1/2 (MAPK3/1 - Mouse Genome Informatics), phosphorylated AKT and PTEN (Fig. 7E,F), which are all known to be directly regulated by integrins (Saleem et al., 2009; Velling et al., 2004). In addition, RIP-Cre/β1KO β-cells exhibited a dramatic decrease in α-tubulin levels (Fig. 7E,F), a protein that, in addition to its function in microtubule assembly, has been shown to directly control cell cycle entry and progression by positively regulating ERK activation and cyclin B synthesis (Vée et al., 2001).
DISCUSSION
It is well established that β1 integrin receptors regulate multiple functions, encompassing cell adhesion, migration, differentiation, growth and survival (Giancotti and Ruoslahti, 1999; Liddington and Ginsberg, 2002; Wang et al., 1998). In this study, we provide original evidence for a crucial role of the β1 integrin subunit in the in vivo expansion of pancreatic β-cells following their endocrine specification. Our results indicate that ‘pro-proliferative’ signaling cues transduced by β1 integrins in pancreatic β-cells are uncoupled from ‘pro-differentiative’ signals attributed to these receptors in other cell types, suggesting that alternative integrins, and cell-cell adhesion receptors, are likely to be responsible for mediating the interaction of β-cells with their extracellular microenvironment and with neighboring cells, and for regulating the maintenance of their differentiated endocrine phenotype.
The signaling properties of integrin receptors are dictated primarily by their state of activation resulting from ECM ligand occupancy and/or cis-transactivation through interaction with growth factor receptors harboring tyrosine kinase functions, rather than by their levels of expression (Fujita et al., 2013; Giancotti and Ruoslahti, 1999; Ju and Zhou, 2013; Liddington and Ginsberg, 2002; Margadant et al., 2011; McCall-Culbreath et al., 2008; Mitra et al., 2011; Wang et al., 1998). Nevertheless, it is reasonable to infer that the levels of β1 integrin expression detected in different pancreatic epithelial cell types correlate with cell type-specific proliferative capabilities. Accordingly, ductal cells expressing high levels of β1 integrin exhibit a higher propensity to respond to mitogenic stimuli than islet cells and acinar cells (Beattie et al., 1999; De Lisle and Logsdon, 1990; Githens et al., 1994; Means et al., 2005; Rooman et al., 2000). Conversely, acinar cells, which we find to express low levels of β1 integrin, are unable to undergo significant proliferation unless they enter an acinar-to-ductal transdifferentiation (Fukuda et al., 2012; Kopp et al., 2012; Reichert et al., 2013). Islet cells have been reported to undergo epithelial-to-mesenchymal transition when replicating (Kaido et al., 2010; Montgomery and Yebra, 2011), a phenomenon that has been described in many epithelial cell types and that is associated with significant upregulation of β1 integrin (Lim and Thiery, 2012; Yeh et al., 2010). This provides a possible mechanism by which cells about to enter the cell cycle can enhance their propensity to use this integrin subunit to respond to microenvironmental cues and activate pro-proliferative signaling cascades.
Following their endocrine specification and insulin expression, pancreatic β-cells enter a spatiotemporally regulated expansion that starts to become evident at ∼E17.5, continues to increase during the last week of embryonic life, reaches a plateau during the first 2 weeks of postnatal life (Georgia and Bhushan, 2006; Georgia et al., 2006), and gradually declines thereafter (Dhawan et al., 2009; Teta et al., 2005; Tschen et al., 2009). Based on this well-established timeline of islet cell mass development, and on the established pro-proliferative functions of β1 integrin described in other epithelia, our finding that RIP-Cre/β1KO mice exhibit significantly reduced numbers of β-cells led us to postulate that the ablation of β1 integrin in β-cells might negatively impact on their replication and/or survival. Accordingly, our analysis of RIP-Cre/β1KO pancreas reveals that the frequency of proliferating β-cells is significantly decreased already at E17.5, a defect that becomes even more dramatic during the first week of postnatal life, and in adult mice. This proliferative defect leads to a severe reduction in β-cell numbers in adult RIP-Cre/β1KO pancreas as compared with WT mice. Collectively, these results clearly demonstrate that β1 integrin functions as a crucial positive regulator of β-cell expansion during development and in postnatal life.
A detailed phenotypic analysis of β-cells from RIP-Cre/β1KO mice revealed that, despite defective cell adhesion to fibronectin and collagen type IV, they retain all of the essential hallmarks of endocrine differentiation, normal architectural organization within islet clusters, and glucose-stimulated secretory function. Accordingly, we found that the expression pattern of the transcription factors PDX1, PAX6, NKX6.1 and MAFA appears indistinguishable from that of WT β-cells. Interestingly, the insulin content of β-cells from RIP-Cre/β1KO mice is increased 3-fold, suggesting that maintenance of endocrine differentiation might be regulated by alternative mechanisms of cell-cell and/or cell-matrix interaction. Notably, we found that the expression of the canonical cadherins Cdh1 and Cdh2 (E-cadherin and N-cadherin), which are the primary adhesion receptors responsible for cell-cell aggregation of islet cells (Rouiller et al., 1991; Dahl et al., 1996), is unaffected in β-cells from RIP-Cre/β1KO, suggesting that their pro-differentiative function is preserved in the absence of β1 integrin.
Previous work has proposed that laminins produced by endothelial cells play an important role in regulating insulin gene expression, and that blockade of β1 integrin laminin receptors in β-cells interferes with insulin secretion (Bosco et al., 2000; Nikolova et al., 2006; Parnaud et al., 2006). At variance with these earlier in vitro studies, we find that the genetic ablation of β1 integrin in β-cells in vivo does not affect their insulin production or secretion in response to glucose. These results suggest that, in the absence of β1 integrin, β-cells might activate alternative mechanisms of ECM recognition that support their endocrine phenotype and function. Hence, our findings might be explained by the fact that pancreatic islet cells express not only β1 but also β3, β4 and β5 integrins (Cirulli et al., 2000; Kaido et al., 2004a; Kaido et al., 2004b; Kaido et al., 2006; Kaido et al., 2010; Riopel et al., 2011; Saleem et al., 2009; Yebra et al., 2011; Yebra et al., 2003). These integrins might provide alternative mechanisms of cell-matrix recognition and might contribute/transduce pro-differentiative signaling cues allowing for the development and functional maturation of β-cells. In support of the possible compensatory function of these alternative integrins, there is evidence that β3 and β5 integrins (i.e. αvβ3 and αvβ5) mediate a number of functions ranging from stationary cell adhesion to motility and invasion, as well as cell proliferation or differentiation depending on the cellular context. In addition, it was demonstrated that β1, β3 and β5 integrins share a reciprocal regulatory cross-talk at the transcriptional, translational and post-translational levels, such that significant β3 and/or β5 compensatory functions can be elicited when β1 is either blocked or genetically ablated, resulting in the maintenance or enhancement of cell differentiation programs (Brunetta et al., 2012; Guan et al., 2001; Hirsch et al., 1998; Jeanes et al., 2012; Retta et al., 1998). Further supporting a compensatory function by alternative integrins, the β4 subunit, partnering with α6 to form the α6β4 laminin 5 receptor, has been shown to support a multitude of signaling pathways depending on the cellular context and the complement of other adhesion receptors (Dowling et al., 1996; Giancotti and Ruoslahti, 1999; Giancotti et al., 1992; Niessen et al., 1996; Sonnenberg et al., 1991; van der Neut et al., 1999; van der Neut et al., 1996; Xia et al., 1996; Yebra et al., 2003). Our full-genome Illumina array shows that β-cells from RIP-Cre/β1KO mice exhibit significant upregulation of ECM components, such as vitronectin (Vtn) and laminin 5 β-chain (Lamb3), that serve as ligands for αvβ3 and α6β4, respectively. Among other genes upregulated in β-cells as a result of β1 integrin deletion are those that encode netrin 1 and netrin 4 (Ntn1 and Ntn4), two proteins that share significant homology with laminins and that we have previously reported to be expressed in the developing pancreas in distinct cellular compartments, where they support cell adhesion, migration and endocrine differentiation (Cirulli and Yebra, 2007; Yebra et al., 2011; Yebra et al., 2003). Based on evidence demonstrating that netrins may also contribute to signaling that supports β-cell survival (Yang et al., 2011), it is possible that ablation of β1 integrin might trigger counter-regulatory mechanisms that protect β-cells from apoptosis.
In addition to regulating mechanisms of cell-matrix adhesion (Hynes, 2002; Vuori, 1998), β1 integrins have been shown to play important roles in the assembly and homeostasis of basal membranes (Ruoslahti, 1996), which are thin proteinaceous layers comprising a mesh-like macromolecular organization of ECM components that cells adhere to and use as a signaling platform during development and in postnatal life (Cheng et al., 1997; Martin and Timpl, 1987; Yurchenco and Wadsworth, 2004). Experimental evidence has shown that deletion of β1 integrins negatively impacts on the assembly and function of basal membranes, which in turn may alter cell attachment, cell phenotype and function (Aumailley et al., 2000; Sasaki et al., 1998). Surprisingly, we find that ablation of β1 integrins in β-cells has no effect on the integrity and ultrastructural appearance of basal membranes that are identifiable at the interface between endothelial cells and β-cells. Several concurrent mechanisms may be invoked to explain these findings. For example, β-cells from RIP-Cre/β1KO mice express and upregulate a number of basal membrane components, such as laminin 5 β-chain (Lamb3), netrin 1 and netrin 4 (Ntn1 and Ntn4), and accessory ECM proteins (e.g. Vtn) (Fig. 7). Based on the reported ability of laminins and netrins to self-assemble into basal membrane-like networks (Cheng et al., 1997; Yurchenco and Wadsworth, 2004), it is possible that, in the absence of a β1 integrin-mediated mechanism of basal membrane assembly, the upregulation of the Lamb3 chain and netrins observed in our RIP-Cre/β1KO β-cells might provide compensatory mechanisms to rescue basal membrane architecture. Further supporting this possibility is the observation that RIP-Cre/β1KO β-cells also exhibit increased transcript levels for collagen isoforms Col6a1, Col7a1 and Col8a1, which have previously been shown to be involved in the assembly and architectural integrity of basal membranes (Gelse et al., 2003; van der Rest and Garrone, 1991). Alternatively, it is possible that the assembly of ECM components into functional basal membranes is mediated by alternative integrin subunits expressed by β-cells (e.g. β3, β5 or β4) or by non-integrin ECM receptors such as DDR1 and DDR2 (Vogel et al., 1997), which we find to be significantly upregulated in β-cells from RIP-Cre/β1KO mice. Finally, endothelial cells expressing high levels of integrin receptor β1, in addition to β3, β5 and β4, are also known contributors to basal membrane biosynthesis and assembly, and may assist in mitigating possible defects caused by the deletion of β1 integrin in adjacent β-cells. Collectively, our observations indicate a dynamic adaptation of β-cells to the loss of β1 integrin function to produce basal membranes, and, together with previous work in rodent and human islets (Otonkoski et al., 2008; Virtanen et al., 2008), indicate that the biosynthesis of functional basal membranes within islet clusters is contributed to not only by endothelial cells, as previously proposed (Nikolova et al., 2006), but also by β-cells (Fig. 7).
Maintenance of cell differentiation is constantly at balance with mechanisms regulating cell proliferation. This is achieved through the interplay between positive and negative regulators of the cell cycle that are controlled by both cell-autonomous and non-autonomous mechanisms of gene expression (Georgia and Bhushan, 2006; Georgia et al., 2006; Miller et al., 2007). Integrins provide an example of multimodular receptors that, depending on their αβ heterodimeric composition, can function as highly specialized transducers of extracellular cues capable of activating outside-in signaling pathways ultimately affecting cellular decisions. Our experiments clearly demonstrate how ablation of β1 integrin in β-cells can dramatically alter their ability to respond to growth stimuli provided by ECMs and growth factors, both in vivo and in vitro. Thus, our full-genome microarray analysis shows that whereas positive regulators of the cell cycle are downregulated, genes that prevent or interfere with cell replication, and thus foster cell differentiation, are upregulated (Fig. 7). The upregulation of genes promoting cell cycle arrest might be explained by mechanisms of derepression invoked as a result of the ablation of β1 integrin, whereas the upregulation of genes supporting cell cycle progression might reflect an attempt to overcome the proliferative defect caused by the loss of β1 integrin signaling. Accordingly, loss of β1 integrin signaling is supported by the observed downregulation of some of the cyclins or cyclin kinases, as well as of MAPK activity, which are known downstream effectors of β1 integrin signaling (Chiang et al., 2011; D’Amico et al., 2000; El Azreq et al., 2012; Saleem et al., 2009; Velling et al., 2004). Also of interest is the downregulation of Ovol2 and Tuba1. Ovol2 downregulation has been shown to interfere with c-MYC and NOTCH1 activity, thus causing cell cycle arrest in G1/G0 phase (Wells et al., 2009). Similarly, blockade of α-tubulin has been reported to inhibit cyclin B accumulation and ERK2 activation, leading to arrest in interphase (Vée et al., 2001). Both ERK and AKT are known to be directly regulated by integrins (Saleem et al., 2009; Velling et al., 2004), and their activity has been shown to regulate cell proliferation and survival (Chambard et al., 2007; Lawlor and Alessi, 2001). Hence, reduced activation of the ERK and AKT pathways may impact negatively on the regulation of cyclin D1 transcription. Based on evidence that c-MYC, NOTCH1 and ERK2 are regulated by β1 integrin activation, our results suggest that Ovol2 and Tuba1 might function as novel effectors of β1 integrin utilization in islet β-cell expansion.
Collectively, our studies demonstrate that, in the absence of β1 integrins, pancreatic β-cells fail to activate the signaling machinery that would otherwise lead to their expansion during development and in postnatal life. Although loss of β1 integrin-mediated outside-in signaling renders β-cells unresponsive to the extracellular cues that would normally elicit their expansion for the establishment of a normal baseline β-cell mass, this defect does not affect the state of differentiation nor the secretory function of this endocrine cell population.
Supplementary Material
Acknowledgments
We thank Dr Roman Sásik at the UCSD BIOGEM Laboratory for expert bioinformatics analysis and Jennifer Lapira for Illumina BeadChip array processing; Makoto S. Chino and Christopher Mitchell for technical assistance; Dr Anthony Montgomery and Dr Mayra Yebra (UCSD, Department of Pediatrics) for insightful discussions; and Dr Robert Florkiewicz (University of Washington, Department of Medicine, Seattle, WA, USA) for advice and comments on the manuscript.
Footnotes
Funding
This work was supported by an American Diabetes Association (ADA) Basic Research Award [1-11-BS-28] and Juvenile Diabetes Research Foundation (JDRF) Research Grants [#1-2005-1084 and #1-2004-13] to V.C.; and by National Institutes of Health grants [RO1 DK55267 to C.J.R., RO1 HL075270 to L.C., P30 DK063491 to G.H.]; G.R.D. was the recipient of a Postdoctoral Fellowship from the JDRF; P.R. was supported by a T32 fellowship [T32 DK007247] from the National Institutes of Health (NIH); and W.Y. by a Pharmacological Sciences Training Grant [5T32GM007750-34] from the NIH and by a scholarship from the Howard Hughes Medical Institute [Med-into-Grad Program #56006778] to the University of Washington. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
G.R.D. performed experiments, analyzed data and contributed to the writing of the manuscript. A.J.J.-C. performed some immunostaining experiments and morphometric analysis. P.R. and W.Y. performed qPCR experiments; G.H. contributed to the analysis of microarray data; C.J.R. contributed to expression profiling analysis of signaling molecules p-ERK1/2, p-AKT, PTEN and α-tubulin. L.C. performed flow cytometry experiments and helped in the analysis of results. V.C. conceived experiments, performed studies of transmission electron microscopy, analyzed data and wrote the manuscript.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.098533/-/DC1
References
- Aszodi A., Hunziker E. B., Brakebusch C., Fässler R. (2003). Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 17, 2465–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley M., Pesch M., Tunggal L., Gaill F., Fässler R. (2000). Altered synthesis of laminin 1 and absence of basement membrane component deposition in (beta)1 integrin-deficient embryoid bodies. J. Cell Sci. 113, 259–268 [DOI] [PubMed] [Google Scholar]
- Beattie G. M., Itkin-Ansari P., Cirulli V., Leibowitz G., Lopez A. D., Bossie S., Mally M. I., Levine F., Hayek A. (1999). Sustained proliferation of PDX-1+ cells derived from human islets. Diabetes 48, 1013–1019 [DOI] [PubMed] [Google Scholar]
- Belkin A. M., Stepp M. A. (2000). Integrins as receptors for laminins. Microsc. Res. Tech. 51, 280–301 [DOI] [PubMed] [Google Scholar]
- Bombardelli L., Carpenter E. S., Wu A. P., Alston N., DelGiorno K. E., Crawford H. C. (2010). Pancreas-specific ablation of beta1 integrin induces tissue degeneration by disrupting acinar cell polarity. Gastroenterology 138, 2531-2540, 2540.e1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S., Trent D. F., Weir G. C. (1983). Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J. Clin. Invest. 71, 1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D., Meda P., Halban P. A., Rouiller D. G. (2000). Importance of cell-matrix interactions in rat islet beta-cell secretion in vitro: role of alpha6beta1 integrin. Diabetes 49, 233–243 [DOI] [PubMed] [Google Scholar]
- Brunetta I., Casalotti S. O., Hart I. R., Forge A., Reynolds L. E. (2012). β3-integrin is required for differentiation in OC-2 cells derived from mammalian embryonic inner ear. BMC Cell Biol. 13, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambard J. C., Lefloch R., Pouysségur J., Lenormand P. (2007). ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 [DOI] [PubMed] [Google Scholar]
- Cheng Y. S., Champliaud M. F., Burgeson R. E., Marinkovich M. P., Yurchenco P. D. (1997). Self-assembly of laminin isoforms. J. Biol. Chem. 272, 31525–31532 [DOI] [PubMed] [Google Scholar]
- Chiang J. K., Sung M. L., Yu H. R., Chang H. I., Kuo H. C., Tsai T. C., Yen C. K., Chen C. N. (2011). Homocysteine induces smooth muscle cell proliferation through differential regulation of cyclins A and D1 expression. J. Cell. Physiol. 226, 1017–1026 [DOI] [PubMed] [Google Scholar]
- Cirulli V., Yebra M. (2007). Netrins: beyond the brain. Nat. Rev. Mol. Cell Biol. 8, 296–306 [DOI] [PubMed] [Google Scholar]
- Cirulli V., Beattie G. M., Klier G., Ellisman M., Ricordi C., Quaranta V., Frasier F., Ishii J. K., Hayek A., Salomon D. R. (2000). Expression and function of alpha(v)beta(3) and alpha(v)beta(5) integrins in the developing pancreas: roles in the adhesion and migration of putative endocrine progenitor cells. J. Cell Biol. 150, 1445–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozar-Castellano I., Weinstock M., Haught M., Velázquez-Garcia S., Sipula D., Stewart A. F. (2006). Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 55, 70–77 [PubMed] [Google Scholar]
- D’Amico M., Hulit J., Amanatullah D. F., Zafonte B. T., Albanese C., Bouzahzah B., Fu M., Augenlicht L. H., Donehower L. A., Takemaru K., et al. (2000). The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem. 275, 32649–32657 [DOI] [PubMed] [Google Scholar]
- Dahl U., Sjødin A., Semb H. (1996). Cadherins regulate aggregation of pancreatic beta-cells in vivo. Development 122, 2895–2902 [DOI] [PubMed] [Google Scholar]
- De Lisle R. C., Logsdon C. D. (1990). Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 51, 64–75 [PubMed] [Google Scholar]
- Dhawan S., Tschen S. I., Bhushan A. (2009). Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 23, 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J., Yu Q. C., Fuchs E. (1996). Beta4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J. Cell Biol. 134, 559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Azreq M. A., Naci D., Aoudjit F. (2012). Collagen/β1 integrin signaling up-regulates the ABCC1/MRP-1 transporter in an ERK/MAPK-dependent manner. Mol. Biol. Cell 23, 3473–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M., Takada Y. K., Takada Y. (2013). Insulin-like growth factor (IGF) signaling requires αvβ3-IGF1-IGF type 1 receptor (IGF1R) ternary complex formation in anchorage independence, and the complex formation does not require IGF1R and Src activation. J. Biol. Chem. 288, 3059–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A., Morris J. P., 4th, Hebrok M. (2012). Bmi1 is required for regeneration of the exocrine pancreas in mice. Gastroenterology 143, 821-831 e1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelse K., Pöschl E., Aigner T. (2003). Collagen - structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 55, 1531–1546 [DOI] [PubMed] [Google Scholar]
- Georgia S., Bhushan A. (2006). p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes 55, 2950–2956 [DOI] [PubMed] [Google Scholar]
- Georgia S., Soliz R., Li M., Zhang P., Bhushan A. (2006). p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev. Biol. 298, 22–31 [DOI] [PubMed] [Google Scholar]
- Giancotti F. G., Ruoslahti E. (1999). Integrin signaling. Science 285, 1028–1033 [DOI] [PubMed] [Google Scholar]
- Giancotti F. G., Stepp M. A., Suzuki S., Engvall E., Ruoslahti E. (1992). Proteolytic processing of endogenous and recombinant beta 4 integrin subunit. J. Cell Biol. 118, 951–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Githens S., Schexnayder J. A., Moses R. L., Denning G. M., Smith J. J., Frazier M. L. (1994). Mouse pancreatic acinar/ductular tissue gives rise to epithelial cultures that are morphologically, biochemically, and functionally indistinguishable from interlobular duct cell cultures. In Vitro Cell. Dev. Biol. Anim. 30A, 622–635 [DOI] [PubMed] [Google Scholar]
- Guan K., Czyz J., Fürst D. O., Wobus A. M. (2001). Expression and cellular distribution of alpha(v)integrins in beta(1)integrin-deficient embryonic stem cell-derived cardiac cells. J. Mol. Cell. Cardiol. 33, 521–532 [DOI] [PubMed] [Google Scholar]
- Hayek A., Beattie G. M., Cirulli V., Lopez A. D., Ricordi C., Rubin J. S. (1995). Growth factor/matrix-induced proliferation of human adult beta-cells. Diabetes 44, 1458–1460 [DOI] [PubMed] [Google Scholar]
- Herrera P. L. (2000). Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127, 2317–2322 [DOI] [PubMed] [Google Scholar]
- Hirsch E., Lohikangas L., Gullberg D., Johansson S., Fässler R. (1998). Mouse myoblasts can fuse and form a normal sarcomere in the absence of beta1 integrin expression. J. Cell Sci. 111, 2397–2409 [DOI] [PubMed] [Google Scholar]
- Howe A., Aplin A. E., Alahari S. K., Juliano R. L. (1998). Integrin signaling and cell growth control. Curr. Opin. Cell Biol. 10, 220–231 [DOI] [PubMed] [Google Scholar]
- Hynes R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- Jeanes A. I., Wang P., Moreno-Layseca P., Paul N., Cheung J., Tsang R., Akhtar N., Foster F. M., Brennan K., Streuli C. H. (2012). Specific β-containing integrins exert differential control on proliferation and two-dimensional collective cell migration in mammary epithelial cells. J. Biol. Chem. 287, 24103–24112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju L., Zhou C. (2013). Association of integrin beta1 and c-MET in mediating EGFR TKI gefitinib resistance in non-small cell lung cancer. Cancer Cell Int. 13, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaido T., Perez B., Yebra M., Hill J., Cirulli V., Hayek A., Montgomery A. M. (2004a). Alphav-integrin utilization in human beta-cell adhesion, spreading, and motility. J. Biol. Chem. 279, 17731–17737 [DOI] [PubMed] [Google Scholar]
- Kaido T., Yebra M., Cirulli V., Montgomery A. M. (2004b). Regulation of human beta-cell adhesion, motility, and insulin secretion by collagen IV and its receptor alpha1beta1. J. Biol. Chem. 279, 53762–53769 [DOI] [PubMed] [Google Scholar]
- Kaido T., Yebra M., Cirulli V., Rhodes C., Diaferia G., Montgomery A. M. (2006). Impact of defined matrix interactions on insulin production by cultured human beta-cells: effect on insulin content, secretion, and gene transcription. Diabetes 55, 2723–2729 [DOI] [PubMed] [Google Scholar]
- Kaido T. J., Yebra M., Kaneto H., Cirulli V., Hayek A., Montgomery A. M. (2010). Impact of integrin-matrix interaction and signaling on insulin gene expression and the mesenchymal transition of human beta-cells. J. Cell. Physiol. 224, 101–111 [DOI] [PubMed] [Google Scholar]
- Kopp J. L., von Figura G., Mayes E., Liu F. F., Dubois C. L., Morris J. P., IV, Pan F. C., Akiyama H., Wright C. V., Jensen K., et al. (2012). Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner J. A., Ciemerych M. A., Sicinska E., Wartschow L. M., Teta M., Long S. Y., Sicinski P., White M. F. (2005). Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol. Cell. Biol. 25, 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor M. A., Alessi D. R. (2001). PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 114, 2903–2910 [DOI] [PubMed] [Google Scholar]
- Li N., Zhang Y., Naylor M. J., Schatzmann F., Maurer F., Wintermantel T., Schuetz G., Mueller U., Streuli C. H., Hynes N. E. (2005). Beta1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. EMBO J. 24, 1942–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddington R. C., Ginsberg M. H. (2002). Integrin activation takes shape. J. Cell Biol. 158, 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J., Thiery J. P. (2012). Epithelial-mesenchymal transitions: insights from development. Development 139, 3471–3486 [DOI] [PubMed] [Google Scholar]
- Margadant C., Monsuur H. N., Norman J. C., Sonnenberg A. (2011). Mechanisms of integrin activation and trafficking. Curr. Opin. Cell Biol. 23, 607–614 [DOI] [PubMed] [Google Scholar]
- Martin G. R., Timpl R. (1987). Laminin and other basement membrane components. Annu. Rev. Cell Biol. 3, 57–85 [DOI] [PubMed] [Google Scholar]
- McCall-Culbreath K. D., Li Z., Zutter M. M. (2008). Crosstalk between the alpha2beta1 integrin and c-met/HGF-R regulates innate immunity. Blood 111, 3562–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means A. L., Meszoely I. M., Suzuki K., Miyamoto Y., Rustgi A. K., Coffey R. J., Jr, Wright C. V., Stoffers D. A., Leach S. D. (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132, 3767–3776 [DOI] [PubMed] [Google Scholar]
- Miller J. P., Yeh N., Vidal A., Koff A. (2007). Interweaving the cell cycle machinery with cell differentiation. Cell Cycle 6, 2932–2938 [DOI] [PubMed] [Google Scholar]
- Mitra A. K., Sawada K., Tiwari P., Mui K., Gwin K., Lengyel E. (2011). Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 30, 1566–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., Katz B. Z., Lafrenie R. M., Yamada K. M. (1998). Fibronectin and integrins in cell adhesion, signaling, and morphogenesis. Ann. New York Acad. Sci. 857, 119–129 [DOI] [PubMed] [Google Scholar]
- Montgomery A. M., Yebra M. (2011). The epithelial-to-mesenchymal transition of human pancreatic β-cells: inductive mechanisms and implications for the cell-based therapy of type I diabetes. Curr. Diabetes Rev. 7, 346–355 [DOI] [PubMed] [Google Scholar]
- Niessen C. M., van der Raaij-Helmer M. H., Hulsman E. H., van der Neut R., Jonkman M. F., Sonnenberg A. (1996). Deficiency of the integrin beta 4 subunit in junctional epidermolysis bullosa with pyloric atresia: consequences for hemidesmosome formation and adhesion properties. J. Cell Sci. 109, 1695–1706 [DOI] [PubMed] [Google Scholar]
- Nikolova G., Jabs N., Konstantinova I., Domogatskaya A., Tryggvason K., Sorokin L., Fässler R., Gu G., Gerber H. P., Ferrara N., et al. (2006). The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev. Cell 10, 397–405 [DOI] [PubMed] [Google Scholar]
- Novak A., Hsu S. C., Leung-Hagesteijn C., Radeva G., Papkoff J., Montesano R., Roskelley C., Grosschedl R., Dedhar S. (1998). Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc. Natl. Acad. Sci. USA 95, 4374–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L., Unger R. H. (1975). Functional subdivision of islets of Langerhans and possible role of D cells. Lancet 2, 1243–1244 [DOI] [PubMed] [Google Scholar]
- Otonkoski T., Banerjee M., Korsgren O., Thornell L. E., Virtanen I. (2008). Unique basement membrane structure of human pancreatic islets: implications for beta-cell growth and differentiation. Diabetes Obes. Metab. 10 Suppl. 4, 119–127 [DOI] [PubMed] [Google Scholar]
- Pan F. C., Wright C. (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn. 240, 530–565 [DOI] [PubMed] [Google Scholar]
- Parnaud G., Hammar E., Rouiller D. G., Armanet M., Halban P. A., Bosco D. (2006). Blockade of beta1 integrin-laminin-5 interaction affects spreading and insulin secretion of rat beta-cells attached on extracellular matrix. Diabetes 55, 1413–1420 [DOI] [PubMed] [Google Scholar]
- Raghavan S., Bauer C., Mundschau G., Li Q., Fuchs E. (2000). Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J. Cell Biol. 150, 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert M., Takano S., von Burstin J., Kim S. B., Lee J. S., Ihida-Stansbury K., Hahn C., Heeg S., Schneider G., Rhim A. D., et al. (2013). The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes Dev. 27, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta S. F., Balzac F., Ferraris P., Belkin A. M., Fässler R., Humphries M. J., De Leo G., Silengo L., Tarone G. (1998). beta1-integrin cytoplasmic subdomains involved in dominant negative function. Mol. Biol. Cell 9, 715–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riopel M., Krishnamurthy M., Li J., Liu S., Leask A., Wang R. (2011). Conditional β1-integrin-deficient mice display impaired pancreatic β cell function. J. Pathol. 224, 45–55 [DOI] [PubMed] [Google Scholar]
- Rooman I., Heremans Y., Heimberg H., Bouwens L. (2000). Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia 43, 907–914 [DOI] [PubMed] [Google Scholar]
- Rouiller D. G., Cirulli V., Halban P. A. (1991). Uvomorulin mediates calcium-dependent aggregation of islet cells, whereas calcium-independent cell adhesion molecules distinguish between islet cell types. Dev. Biol. 148, 233–242 [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. (1996). Integrin signaling and matrix assembly. Tumour Biol. 17, 117–124 [DOI] [PubMed] [Google Scholar]
- Saleem S., Li J., Yee S. P., Fellows G. F., Goodyer C. G., Wang R. (2009). beta1 integrin/FAK/ERK signalling pathway is essential for human fetal islet cell differentiation and survival. J. Pathol. 219, 182–192 [DOI] [PubMed] [Google Scholar]
- Sasaki T., Forsberg E., Bloch W., Addicks K., Fässler R., Timpl R. (1998). Deficiency of beta 1 integrins in teratoma interferes with basement membrane assembly and laminin-1 expression. Exp. Cell Res. 238, 70–81 [DOI] [PubMed] [Google Scholar]
- Schoenwaelder S. M., Burridge K. (1999). Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11, 274–286 [DOI] [PubMed] [Google Scholar]
- Sherr C. J., Roberts J. M. (1995). Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149–1163 [DOI] [PubMed] [Google Scholar]
- Sonnenberg A., Calafat J., Janssen H., Daams H., van der Raaij-Helmer L. M., Falcioni R., Kennel S. J., Aplin J. D., Baker J., Loizidou M., et al. (1991). Integrin alpha 6/beta 4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J. Cell Biol. 113, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- Teta M., Long S. Y., Wartschow L. M., Rankin M. M., Kushner J. A. (2005). Very slow turnover of beta-cells in aged adult mice. Diabetes 54, 2557–2567 [DOI] [PubMed] [Google Scholar]
- Tschen S. I., Dhawan S., Gurlo T., Bhushan A. (2009). Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 58, 1312–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Neut R., Krimpenfort P., Calafat J., Niessen C. M., Sonnenberg A. (1996). Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat. Genet. 13, 366–369 [DOI] [PubMed] [Google Scholar]
- van der Neut R., Cachaço A. S., Thorsteinsdóttir S., Janssen H., Prins D., Bulthuis J., van der Valk M., Calafat J., Sonnenberg A. (1999). Partial rescue of epithelial phenotype in integrin beta4 null mice by a keratin-5 promoter driven human integrin beta4 transgene. J. Cell Sci. 112, 3911–3922 [DOI] [PubMed] [Google Scholar]
- van der Rest M., Garrone R. (1991). Collagen family of proteins. FASEB J. 5, 2814–2823 [PubMed] [Google Scholar]
- Vée S., Lafanechère L., Fisher D., Wehland J., Job D., Picard A. (2001). Evidence for a role of the (alpha)-tubulin C terminus in the regulation of cyclin B synthesis in developing oocytes. J. Cell Sci. 114, 887–898 [DOI] [PubMed] [Google Scholar]
- Velling T., Nilsson S., Stefansson A., Johansson S. (2004). beta1-Integrins induce phosphorylation of Akt on serine 473 independently of focal adhesion kinase and Src family kinases. EMBO Rep. 5, 901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen I., Banerjee M., Palgi J., Korsgren O., Lukinius A., Thornell L. E., Kikkawa Y., Sekiguchi K., Hukkanen M., Konttinen Y. T., et al. (2008). Blood vessels of human islets of Langerhans are surrounded by a double basement membrane. Diabetologia 51, 1181–1191 [DOI] [PubMed] [Google Scholar]
- Vogel W., Gish G. D., Alves F., Pawson T. (1997). The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 [DOI] [PubMed] [Google Scholar]
- Vuori K. (1998). Integrin signaling: tyrosine phosphorylation events in focal adhesions. J. Membr. Biol. 165, 191–199 [DOI] [PubMed] [Google Scholar]
- Wang F., Weaver V. M., Petersen O. W., Larabell C. A., Dedhar S., Briand P., Lupu R., Bissell M. J. (1998). Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 95, 14821–14826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J., Lee B., Cai A. Q., Karapetyan A., Lee W. J., Rugg E., Sinha S., Nie Q., Dai X. (2009). Ovol2 suppresses cell cycling and terminal differentiation of keratinocytes by directly repressing c-Myc and Notch1. J. Biol. Chem. 284, 29125–29135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Timmers C., Maiti B., Saavedra H. I., Sang L., Chong G. T., Nuckolls F., Giangrande P., Wright F. A., Field S. J., et al. (2001). The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414, 457–462 [DOI] [PubMed] [Google Scholar]
- Xia Y., Gil S. G., Carter W. G. (1996). Anchorage mediated by integrin alpha6beta4 to laminin 5 (epiligrin) regulates tyrosine phosphorylation of a membrane-associated 80-kD protein. J. Cell Biol. 132, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. H., Szabat M., Bragagnini C., Kott K., Helgason C. D., Hoffman B. G., Johnson J. D. (2011). Paracrine signalling loops in adult human and mouse pancreatic islets: netrins modulate beta cell apoptosis signalling via dependence receptors. Diabetologia 54, 828–842 [DOI] [PubMed] [Google Scholar]
- Yebra M., Montgomery A. M., Diaferia G. R., Kaido T., Silletti S., Perez B., Just M. L., Hildbrand S., Hurford R., Florkiewicz E., et al. (2003). Recognition of the neural chemoattractant Netrin-1 by integrins alpha6beta4 and alpha3beta1 regulates epithelial cell adhesion and migration. Dev. Cell 5, 695–707 [DOI] [PubMed] [Google Scholar]
- Yebra M., Diaferia G. R., Montgomery A. M., Kaido T., Brunken W. J., Koch M., Hardiman G., Crisa L., Cirulli V. (2011). Endothelium-derived Netrin-4 supports pancreatic epithelial cell adhesion and differentiation through integrins α2β1 and α3β1. PLoS ONE 6, e22750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh Y. C., Wei W. C., Wang Y. K., Lin S. C., Sung J. M., Tang M. J. (2010). Transforming growth factor-beta1 induces Smad3-dependent beta1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am. J. Pathol. 177, 1743–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco P. D., Wadsworth W. G. (2004). Assembly and tissue functions of early embryonic laminins and netrins. Curr. Opin. Cell Biol. 16, 572–579 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.