

Abstract
Cell differentiation and proliferation are coordinated during animal development, but the link between them remains uncharacterized. To examine this relationship, we combined single-molecule RNA imaging with time-lapse microscopy to generate high-resolution measurements of transcriptional dynamics in Caenorhabditis elegans embryogenesis. We found that globally slowing the overall development rate of the embryo by altering temperature or by mutation resulted in cell proliferation and transcription slowing, but maintaining, their relative timings, suggesting that cell division may directly control transcription. However, using mutants with specific defects in cell cycle pathways that lead to abnormal lineages, we found that the order between cell divisions and expression onset can switch, showing that expression of developmental regulators is not strictly dependent on cell division. Delaying cell divisions resulted in only slight changes in absolute expression time, suggesting that expression and proliferation are independently entrained to a separate clock-like process. These changes in relative timing can change the number of cells expressing a gene at a given time, suggesting that timing may help determine which cells adopt particular transcriptional patterns. Our results place limits on the types of mechanisms that are used during normal development to ensure that division timing and fate specification occur at appropriate times.
Keywords: Cell division, Gene expression, RNA FISH, Time-lapse, Timing, Transcription
INTRODUCTION
The process of animal embryonic development begins at fertilization and the creation of a single cell zygote. From there, this cell will divide into many more cells, and these cells will eventually acquire different identities, as reflected in their shape, function and position in the animal. Development as such consists of conceptually parallel processes, including cell proliferation, morphogenesis and differentiation. A major goal of developmental biology is to determine where and how these processes may be linked to each other.
The relative timing of cell division and gene expression events is important for development to proceed properly, and has been the subject of considerable attention (Yasuda and Schubiger, 1992; Pritchard and Schubiger, 1996; Lu et al., 2009; Satoh, 1982; Edgar and McGhee, 1988). This is most clearly crucial when a cell divides into daughter cells with different fates. If a perturbation results in a master activator gene for one of those fates expressing before the division, both daughters may adopt the same fate.
In this paper, we combine single-molecule transcript counting and time-lapse imaging-based cell lineaging to quantify transcriptional dynamics in the early Caenorhabditis elegans embryo. Using these measurements, we first examine the extent to which changes in overall metabolic rate affect the relative dynamics of transcription and proliferation during embryogenesis. We find that the timing of both processes scale by the same factor upon changes in temperature or metabolic rate, thus preserving their relative order. Based on these findings, we hypothesized that cell cycle events may directly control the timing of gene expression. However, using mutants that specifically affect cell cycle lengths, we show that the cell division events do not define the time of onset of transcription, which we found can occur before or after particular cell divisions. As the work of others has shown that zygotic transcription does not control the timing of early cell divisions (Powell-Coffman et al., 1996; Edgar et al., 1994), and our results show that the timing of cell division does not determine the timing of expression of the genes examined here, other types of mechanisms must exist to coordinate cell proliferation and gene expression upon perturbations such as temperature shift.
MATERIALS AND METHODS
Worm culture and RNA FISH protocol
C. elegans strains we studied by RNA FISH were N2 wild type, EU548 [div-1(or148)III], GE31 [cib-1(e2300)I], GG19 [div-1(g19)III (originally isolated as emb-16(g19))], GG32 [emb-22(g32)V], GG39 [emb-23(g39)II], GG52 [emb-29(g52)V], HH16 [emb-29(b262)unc-60(m35)V], MQ130 [clk-1(qm30)III], RB1331 [end-3(ok1448)V], WM99 [cdk-1(ne2257)III] and VC271 [end-1(ok558)V]. The elt-7 deletion strain FX840 [elt-7(tm840)V] was obtained from Shohei Mitani (Tokyo Women’s Medical University, Japan). All others were obtained from the Caenorhabditis Genetics Center. For a summary table of mutant strains we studied by RNA FISH and their role in this study, see supplementary material Table S1.
For preparation for RNA FISH experiments, all strains, except for MQ130, were cultured on enriched peptone plates seeded with Na22 strain E. coli for efficient mass culture. MQ130 was grown on NGM agar plates seeded with OP50. Except for MQ130, worm populations were synchronized by releasing eggs with hypochlorite and letting the L1 larvae hatch and undergo growth arrest in M9 solution (at either room temperature or 15°C) before seeding at a density of 6000-10,000 per 9 cm plate. Conditional mutants were grown at 15°C and shifted to 25°C when most worms on the plates were L4, and embryos were harvested the next day. MQ130 were harvested after growth in asynchronous culture for several generations at constant temperature (20°C). To obtain fixed embryos, worms were collected in deionized water, the embryos were released by hypochlorite treatment, washed again in deionized water and transferred to a 4% formaldehyde solution in phosphate-buffered saline (PBS). After 15 minutes, the egg shell and vitelline membrane were permeabilized by freeze cracking in liquid nitrogen for a minute. Once the solution melted, the fixation was continued for 20 minutes on ice, the worms were washed in PBS and transferred and stored in 70% ethanol at 4°C.
RNA FISH staining followed protocols by Raj et al. (Raj et al., 2010; Raj et al., 2008). In some cases, 0.1% Triton X-100 was used in the washes to prevent loss of embryos, though this practice appeared to correlate with poorer RNA FISH signal. We incorporated DAPI staining for identifying embryos and counting cell number during the washes. Samples were mounted in a glucose oxidase 2×SSC buffer between two coverglasses, sealed from the ambient with vacuum grease and imaged on a standard inverted epifluorescence microscope using a 100× 1.4 NA oil immersion objective and a Princeton Instruments PIXIS camera. Z sections with a 0.35 μm spacing were taken for each region. Image analysis was carried out using custom scripts in MATLAB. Briefly, RNA FISH image stacks were filtered with a Laplacian of Gaussian filter optimized to isolate spots corresponding to single RNAs. Local maxima were chosen as candidate spots and fit to a symmetric two-dimensional Laplacian of Gaussian. For any given RNA FISH channel, many embryos showed a cloud of spots that were well separated in size-amplitude space, with sizes appropriate for diffraction-limited spots corresponding to single RNA molecules and a clear threshold between signal and background.
The spots we detect are primarily single molecules of RNA. Arguments for the detection of single RNA molecules follow those presented in previous work (Vargas et al., 2005).
Embryo cell count
The number of nuclei in every embryo were counted manually using the DAPI staining signal. Counts are expected to be accurate to within approximately ±1 before the 60-cell stage, where the relevant changes in our data occur. By overlaying the position of detected RNA molecules with the DAPI images, we were also able to approximately determine the number of cells expressing gut markers in some of the strains.
Automated cell lineage tracing with reporter expression
To determine the embryonic lineage and division timing of the mutant strains we studied, we first crossed mutant hermaphrodites with males containing integrated his-72::HIS-72::GFP (stIs10026 or stIs10064) or pie-1::H2B::mCherry and his-72::HIS-24::mCherry (itIs38; stIs10116) transgenes. clk-1(qm30); itIs38; stIs10116 worms were grown at 20°C for over two generations before embryos were imaged at 20°C. Strains containing the temperature-sensitive alleles div-1(or148), emb16(g19) or emb-29(g52) were grown at 15°C before L4 hermaphrodites were incubated overnight at 25°C and their embryos were imaged the following morning at 25°C. The same procedure was followed for div-1(or148) embryos carrying the transgene hlh-1::HLH-1::GFP (stIs10436). The lineage data for wild-type embryos at 15°C was obtained from worms carrying ujIs113. Lineages of a minimum of three embryos were collected to generate division times used for subsequent analyses.
We acquired confocal images with a Leica TCS SP5 (67 z-planes at 0.5 μm spacing and 1.5-minute time spacing) and generated lineages using StarryNite and AceTree as previously described (Bao et al., 2006; Boyle et al., 2006; Murray et al., 2006; Murray et al., 2008) but by applying a newer segmentation algorithm (Santella et al., 2010) and higher resolution imaging (Richards et al., 2013). Embryos were mounted in a solution of 20 μm beads in egg buffer/methyl cellulose (Bao and Murray, 2011). Images were acquired with 0.5 μm z-spacing every 1.5 minutes for each embryo.
Method for reconstructing RNA time traces
The lineage data constrains the age of each fixed embryo to an interval between the last cell division and the next division based on its cell count. We considered all embryos from a sample within an interval, equally spaced them in time and ordered them as increasing or decreasing, or randomly, based on their expression level and depending on which behavior we assumed for that gene during those particular stages. For example, elt-2 mRNA levels were assumed to be increasing in time within the developmental window we study, but end-3 mRNA levels were assumed first to increase and then to decrease.
Calculation of scaling predictions
We defined a rate of progress of the transcriptional dynamics clock, ρ(t), such that if in wild-type 20°C conditions the RNA dynamics follow the trajectory r(t)=F(t - t0), where t0 is a reference time, then the RNA trajectory under slow conditions is r*(t)=F(τ(t) - τ(t0*)), where dτ(t)/dt=ρ*(t)/ρ(t′); * indicates slow conditions; t′ is the time at which the cell stage in the wild type that is equivalent to that occurring under slow conditions at time t is reached. We defined an embryo-wide rate of the cell cycle clock, k(t), as the average of the inverse of the lengths of the cell cycles in all cells existing at time t. Perfect scaling of RNA dynamics to cell cycle dynamics is defined in this framework as ρ*(t)/ρ(t′)=k*(t)/k(t′). No scaling at all, meaning a total insensitivity of RNA dynamics to the conditions that slow down cell cycling, was taken to mean ρ*(t)=ρ(t′). We parametrized uniform but imperfect scaling either as ρ*(t)/ρ(t′)=(1 - α) + α [k*(t)/k(t′)] (linear parametrization) or ρ*(t)/ρ(t′)=[k*(t)/k(t′)]α (geometric parametrization). When 0≤α<<1, it implies less slowdown of transcription than of cell cycling; when α>1, it implies a greater slowdown in transcription than in cell cycling.
For the calculations, F, was fitted by inspection to the wild-type 20°C elt-2 dynamics as a curve with a wait time during which there is no transcription followed by a straight-line rise in elt-2. t0 is chosen as the time of birth of the E cell, which is the progenitor of the gut. k was calculated using live cell lineage data, averaging at any given moment over all cells with known future division times.
Mutant selection
We selected mutant strains previously reported to have altered cell cycle lengths. We rejected from further analysis strains in which elt-2 mRNA levels were much lower than in wild type [this excludes emb-23(g39) and cib-1(e2300)]. We also did not analyze further a mutant [cdk-1(ne2257)] in which elt-2 mRNA was observed very early and throughout the entire embryo at a very low density. We did not analyze the mutant emb-22(g32) because it had many more than the usual number of cells expressing gut lineage markers.
Genetics of the emb-16 and emb-29 mutations
We prepared genomic DNA from the strains GG52 [emb-29(g52)], HH16 [emb-29(b262)] and GG19 [emb-16(g19)], following a method similar to that of Sarin et al. (Sarin et al., 2010) using a Qiagen Gentra Puregene Kit. Libraries for sequencing were prepared using a Nextera DNA Sample Preparation Kit (Illumina). Eighty-three, 36 and 46 million 100 bp reads were obtained respectively for GG52, GG19 and HH16 on an Illumina HiSeq instrument. We computed homozygous variants with respect to the WS220 (ce10) C. elegans reference using CloudMap (Minevich et al., 2012). We eliminated background variants present in all three strains from further analysis.
For GG19[emb-16(g19)], after removing background mutations found in strains GG52 and HH16, we found three protein-coding mutations within the emb-16 genetic limits (Cassada et al., 1981): dos-1(S81N), M01A8.2 (P292S) and div-1 (P353L). Of these, deletions of dos-1 and M01A8.2 are viable, as determined by availability of unbalanced homozygous deletion strains from the C. elegans Knockout Consortium. Conditional and non-conditional alleles of div-1 cause maternally rescued embryonic lethality and slowed cell divisions, like emb-16(g19). In fact, div-1(or27), a non-conditional embryonic lethal allele of div-1, has a mutation at the same amino acid as strain GG19 (P353H).
We tested whether emb-16(g19) is an allele of div-1 by complementation testing. We found that 70% (±20% s.d.) of embryos derived from g19/div-1(or148) trans-heterozygotes mothers failed to hatch when the mothers were grown at 25°C, whereas all progeny from +/div-1(or148) heterozygous mothers hatch under these conditions. Post embryonic development in the progeny of g19/or148 was also notably slower at the restrictive temperature. This indicates that g19 is an allele of div-1.
To test the molecular identity of emb-29, we looked for genes within the known genetic interval that also carried protein-altering mutations in both GG52 [emb-29(g52)] and HH16 [emb-29(b262)]. Previously, Hecht et al. found that embryos homozygous for either g52 or b262 fail to produce antigens detected by a mitosis-specific antibody at restrictive temperature and suggested that emb-29 encodes a cell division cycle function (Hecht et al., 1987). Consistent with this, we found that only one gene, cdc-25.2, had the same mutation (S239F) in both strains, and neither strain had any other coding mutations within the previously mapped interval (Cassada et al., 1981). A deletion and putative null allele of cdc-25.2 exists and is homozygous sterile (Kim et al., 2010), and exhibits cell proliferation defects during embryonic development primarily in the E lineage (Yhong-Hee Shim, personal communication). By contrast, emb-29(g52) causes a stronger phenotype, 100% embryonic lethality (Denich et al., 1984), at the restrictive temperature. This defect cannot be rescued by a maternal contribution of emb-29(+) (Denich et al., 1984). We performed complementation testing and found that at the restrictive temperature, only ∼21% (93/440) of the progeny of cdc-25.2(ok597)/emb-29(g52) F1s arrested as embryos, suggesting that cdc-25.2(ok597) complements the zygotic lethality of emb-29(g52). However, 90% of L4 animals picked from the resulting plates were sterile (compared with 33% expected if the alleles were on different genes). We conclude that emb-29(g52) is most likely a non-null allele of cdc-25.2, with the embryonic lethality possibly resulting from a gain of function.
RESULTS
Transcription dynamics from single molecule RNA FISH
In order to quantify precisely the relationship between transcription and cell division, we developed a strategy that combined transcript counting in individual fixed C. elegans embryos with time-lapse lineage tracing of live embryos. To illustrate our method, we focused first on the transcription factors end-3, end-1 and elt-2, which form a transcriptional cascade involved in gut specification (Fig. 1A) (Maduro and Rothman, 2002). We measured transcription by performing single molecule RNA FISH (Raj et al., 2008) to count the number of individual end-3, end-1 and elt-2 transcripts in a large collection of mixed-stage fixed embryos (Fig. 1B; supplementary material Fig. S1). We simultaneously counted the number of nuclei in each embryo via the DAPI DNA stain. Using this staging information, one can reconstruct the trajectory of transcription during early embryogenesis (Fig. 1C).
Fig. 1.
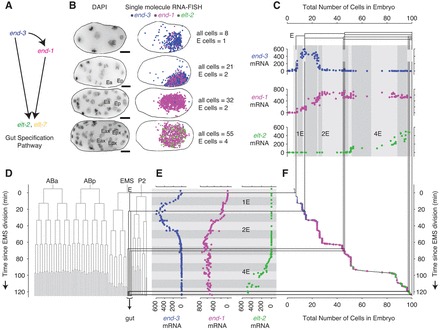
Gene expression dynamics in early gut specification, as determined by single-molecule RNA FISH. (A) A portion of the gene network responsible for specifying the gut. (B) Images of fixed N2 (’wild type’) strain C. elegans embryos grown at 20°C. Scale bars: 10 μm. Left column: maximum merges of DAPI nuclear staining fluorescence images. ‘E’ indicates E cells (as determined by shape, location and gut gene expression). Right column: location of individual transcripts of end-3, end-1 and elt-2 (blue, magenta and green, respectively) obtained from single-molecule RNA FISH staining and subsequent image processing. Filtered RNA FISH fluorescence images are shown in supplementary material Fig. S1. (C) Gene expression trajectories for a sample of N2 embryos grown at 20°C. The total number of end-3, end-1 and elt-2 mRNA in each embryo is plotted against the total number of cells. In C,E, thin black lines correspond to divisions in the E lineage and shaded blocks in the backgrounds correspond to 10 minutes of developmental time. (D) Embryonic lineage of N2 C. elegans based on data from Bao et al. (Bao et al., 2008). The division times were scaled to match the average 20°C division rate observed by Sulston et al. (Sulston et al., 1983). (E) mRNA counts in each embryo as in C but plotted against the estimated age at which the embryos were fixed. (F) Embryo age versus total number of cells in the embryo. The data points from C and E are overlaid with symbol area proportional to the mRNA level.
To determine the temporal dynamics of transcription, we used time lapse microscopy to determine the lineages of embryos under the same conditions (Bao et al., 2006) (see Fig. 1D). We used these data to assign a time window (after the previous division to before the next division) to each embryo that we imaged (Fig. 1F). For example, the 46-cell stage in an N2 embryo grown at 20°C, is the period between 70 and 74 minutes after the EMS cell division. We then assigned an approximate age within each embryo’s respective time window by assuming that RNA transcription within each window follows the same increasing or decreasing trend as between nearby windows and obtained transcriptional trajectories as mRNA versus time (Fig. 1E). The order of transcriptional onset we observe for all the genes we studied agrees well with fluorescent protein reporter data acquired from live embryos (Murray et al., 2012), when taking note of the additional time required for reporter protein synthesis and maturation.
Transcription dynamics and proliferation remain synchronized when the overall developmental rate is changed
We used this measurement approach to look for changes in the degree of synchronization of proliferation and differentiation. We began by searching for such changes upon global slowdown of proliferation, either by temperature drop or a clk-1(qm30) mutational background.
Cell cycles were markedly slower in all wild-type embryos at 15°C and in a fraction of clk-1(qm30) mutant embryos at 20°C (see Fig. 2B; supplementary material Figs S2, S3). Under these conditions, elt-2 expression is also likely to be delayed, although it need not necessarily slow to the same extent as proliferation. If the transcription onset time is slowed down to a different degree from proliferation, the number of cell divisions that occur in the whole embryo between transcriptional events may change. However, if both slow to the same extent, the relative timing with respect to cell division will be preserved, and the number of transcripts observed in embryos of any particular number of cells would be very similar to those obtained from wild-type 20°C embryos (Fig. 2A). Our data closely follow the latter scenario (Fig. 2C), in which transcriptional dynamics are slowed down to a similar degree to cell proliferation.
Fig. 2.
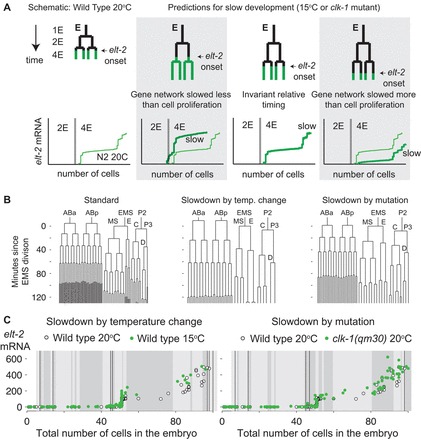
Experimental strategy to study the relative timing of cell proliferation and gene expression. (A) Cell proliferation and the gene expression network can be slowed down by different factors under conditions of overall metabolic slowdown. This will be observable as a difference in the RNA versus total embryo cell count trajectories compared with standard (N2 20°C) development. If gene expression and proliferation are synchronized, the trajectories will be similar. (B) From left to right: wild-type embryonic lineage grown at 20°C (as in Fig. 1D), cell lineage of a representative wild-type embryo grown at 15°C, and cell lineage of a clk-1(qm30) mutant embryo grown at 20°C with a strong slowdown phenotype (embryo 6 from supplementary material Fig. S3). (C) Green circles: gene expression trajectories of elt-2 plotted against total number of cells in embryos of wild-type C. elegans grown at 15°C (left) or clk-1(qm30) mutant worms grown at 20°C (right). White circles: data from N2 worms grown at 20°C (Fig. 1). Thin black vertical lines correspond to divisions in the E lineage and shaded blocks in the backgrounds correspond to 10 minutes of developmental time. For clk-1(qm30), the lineage data are from the same embryo as in B. For 15°C data, we scaled the N2 20°C lineage by a factor of 1.77, consistent with the smallest slowdown observed in any of the embryos we lineaged. The data agree with the ‘invariant timing’ scenario.
We quantitatively analyzed the degree of synchrony between transcription and proliferation for the case of elt-2 transcription. We predicted the shape of the RNA versus cell stage trajectories for scenarios in which transcriptional dynamics are not slowed down at all under slow conditions (no scaling) and in which they are slowed to scale perfectly or imperfectly with cell cycle.
For 15°C embryos, we found that cell cycles were lengthened by a factor of 1.9 (±0.05 embryo-to-embryo s.d.) relative to 20°C growth, and this slowdown varied little with respect to developmental stage (supplementary material Fig. S2). Even using a conservative slowdown factor of 1.77 [the same found by Wong et al. (Wong et al., 1995) for overall development at 15°C], we found that our data agree with transcription dynamics scaling with cell cycle dynamics to within 10% (supplementary material Figs S4, S5). The clk-1(qm30) strain exhibited dramatic levels of variability in cell cycle slowdown from embryo to embryo, also observed by Wong et al. (Wong et al., 1995), and the slowdown is less pronounced in the earliest cell divisions (supplementary material Fig. S3). However, our time-lapse data indicated that 30% of clk-1(qm30) embryos exhibited a strong enough slowdown to result in readily appreciable differences in our RNA versus cell stage trajectories if cell proliferation and gene expression were not synchronized (supplementary material Fig. S5). The tight spread of the elt-2 RNA versus number of nuclei trajectories in our ensemble clk-1(qm30) RNA FISH data around the predicted curve for perfect synchrony (Fig. 2C; supplementary material Fig. S5) argues that transcriptional dynamics and cell proliferation remain synchronized in the entire range of clk-1(qm30) embryos.
We also tested for synchronization in a panel of genes expressed in early gut progenitors. In addition to end-1, end-3 and elt-2, we analyzed in Fig. 3 pgp-2, a gene involved in the production of gut granules (Schroeder et al., 2007), elt-7, another member of the gut-specification transcriptional network (Sommermann et al., 2010), and nhr-79 (Murray et al., 2012). For each of these genes, we found the RNA versus cell count trajectories are again very similar for N2 worms grown at 20°C (standard), N2 worms grown at 15°C (slow) and clk-1(qm30) mutants grown at 20°C (also slow) (Fig. 3). Incidentally, we found that elt-7 expresses much earlier than elt-2, even though genetic studies have pointed to an approximate redundancy in function between elt-2 and elt-7 (Sommermann et al., 2010). We also found that the enzyme pgp-2 involved in gut granule production begins expressing well before the master regulator elt-2, which may be due to the action of elt-7 or the end gut transcription factors.
Fig. 3.
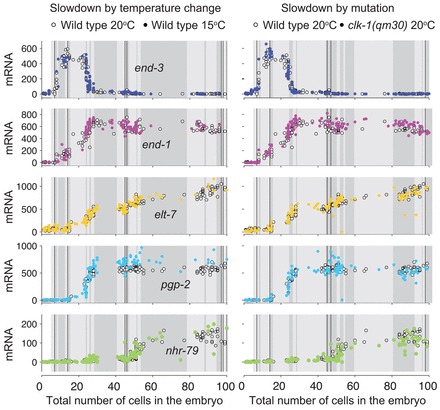
Expression and proliferation are synchronized in the gut specification pathway. Total embryo transcript number versus total cell count trajectories for gut-specific genes end-1, end-3, pgp-2, elt-7 and nhr-79. White symbols are for N2 worms grown at 20°C; colored circles correspond to N2 embryos grown at 15°C (left panels) or clk-1(qm30) embryos grown at 20°C (right panels). Thin black vertical lines correspond to divisions in the E lineage and shaded blocks in the backgrounds correspond to 10 minutes of developmental time. These data agree with the ‘invariant timing’ scenario from Fig. 2.
To check whether synchronization is evident in other lineages, we also measured the transcriptional dynamics of hlh-1 and unc-120. These genes are regulators of body wall muscle (Fukushige et al., 2006), which is produced in a complex lineage pattern that does not include the E lineage (Fig. 4A,B) (Krause, 1995). Nevertheless, we again found invariant timing with respect to cell proliferation (Fig. 4C,D). These results show that the invariant relative timing of transcription dynamics and proliferation is likely a universal phenomenon in the early transcriptional cascades of the C. elegans embryo. We further note that not just the onset time, but also the rate of transcription and the transcript number plateaus are nearly identical under all three conditions, suggesting that many aspects of transcriptional dynamics are preserved under these perturbations.
Fig. 4.
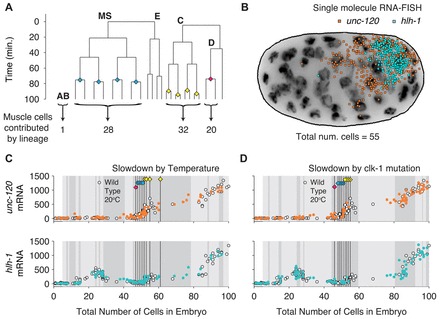
Expression and proliferation are synchronized in the body wall muscle-specification pathway. (A) Part of the N2 20°C lineage diagram from Fig. 1. Body wall muscle arises from the MS (28 cells), C (32 cells) and D (20 cells) lineages, and a single cell from the AB lineage (Krause, 1995). Diamond symbols mark the timing of the 1D, 4MS and 4C divisions. (B) Positions of transcripts of the muscle master regulators unc-120 and hlh-1 overlaid on a grayscale DAPI nuclear stain maximum projection for a 55-cell stage wild-type embryo grown at 20°C. C. elegans embryos are about 50 μm long. (C,D) Total embryo transcript number versus total cell count trajectories for hlh-1 and unc-120. White symbols are for N2 worms grown at 20°C; colored circles correspond to N2 embryos grown at 15°C (C) or clk-1(qm30) embryos grown at 20°C (D). The early hlh-1 expression transient peaking at the 26-cell stage (in the MS lineage) has no known function (Krause, 1995). The black vertical lines show the location of the 1D, 4MS and 4C divisions, and the background shading blocks correspond to 10 minutes of developmental time each. The lineage data used to represent wild type at 15°C and clk-1(qm30) are the same as in Fig. 2.
The timing of elt-2, unc-120 and hlh-1 transcription is not locked to the cell cycle
The data from temperature and mutation (metabolic)-slowed embryos suggested that cell cycle dependence of transcription could be a potential mechanism for synchronization. As seen in Fig. 1C, the onset time of transcription of many genes occurs slightly after cell division events in the expressing lineages. Similarly, we found that the body wall muscle transcription factors hlh-1 and unc-120, like elt-2 in the gut, also accumulate beginning at roughly the 50-cell stage, as seen in Fig. 4B,C, and that their onset time is coincident with the round of divisions between the 4MS,4C,1D and 8MS,8C,2D stages of embryo development. This suggested that activation of these genes could be linked to progression through the cell cycle.
We sought to assess the role of the cell cycle in transcriptional timing by analyzing mutants that delay cell division times. For studying timing changes in the E lineage, we analyzed an emb-29(g52) temperature-sensitive mutant in which the 2E→4E embryonic division is delayed relative to others at the restrictive temperature (Denich et al., 1984). We found by whole-genome sequencing and complementation testing that emb-29(g52) is likely a complex allele of cdc-25.2, a gene encoding cdc-25 phosphatase (a core component of the cell cycle) and is known to affect embryonic cell proliferation selectively in the E lineage (Yhong-Hee Shim, personal communication).
Normally, transcription of elt-2 begins shortly after the 2E→4E round of divisions. If this division were required for the elt-2 transcriptional onset, then the delayed divisions in emb-29(g52) would also cause a delay in the onset time of transcription, as depicted schematically in Fig. 5A. We found by live-embryo cell lineage tracing that the 2E cell cycle of emb-29(g52) embryos is indeed longer than in wild-type embryos grown at the same temperature by 10±1.8 minutes, and, because non-E lineages are not affected, the 2E→4E division occurs at the 50-instead of the 44-cell stage (see Fig. 5D and supplementary material Table S2). In wild-type embryos, we found that elt-2 transcription began around the 51-cell stage, whereas in the emb-29(g52) mutants, we saw elt-2 mRNA as early as the 44-cell stage, which is well before the 2E→4E division in these embryos. Fig. 5C and supplementary material Figs S6, S7 show that elt-2 transcriptional onset begins when there are only two E cells in emb-29(g52) mutants, but does not begin until there are four E cells in wild type. Thus, the elt-2 transcription onset time is not dependent on the occurrence of the 2E→4E divisions.
Fig. 5.
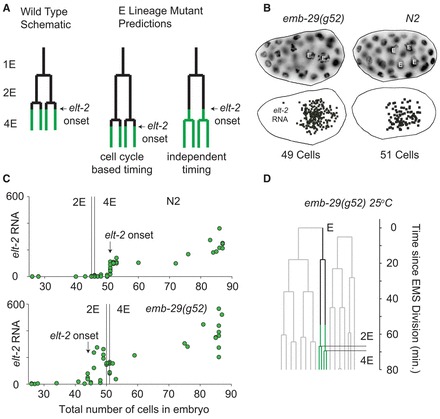
The onset of elt-2 expression is not timed by the 2E→4E divisions. (A) In the gut, elt-2 begins to be expressed soon after the 2E→4E divisions. In the emb-29(g52) mutant, this division is delayed. If gene expression is tied to the cell division, the expression onset will also move, but if it is independent of cell division, expression may begin before the division that normally precedes it. (B) An example of an emb-29(g52) embryo expressing elt-2 with only two E cells. (Top) An individual DAPI nuclear stain image at the E cell plane. (Bottom) Positions of individual elt-2 transcripts determined by single-molecule RNA FISH. (C) elt-2 expression versus total number of cells in the embryo for wild type at 20°C (top) and emb-29(g52) embryos at the restrictive temperature 25°C (bottom). The position of the 2E→4E divisions for each strain are overlaid as black vertical lines. In the emb-29 mutant, elt-2 begins to be expressed well before these divisions. (D) Lineage diagrams for a representative emb-29(g52) embryo grown at 25°C. The beginning of the green lineage coloring indicates the onset of elt-2 expression (see supplementary material Fig. S6).
We also tested the hypothesis of a direct link between elt-2 transcription and E lineage cell cycle progression in a null mutant of end-3, which is known to have a specific effect on the duration of the 2E cell cycles (Boeck et al., 2011). Consistent with our conclusion from studying the emb-29(g52) background, we found that the onset of elt-2 transcription did not shift concomitantly with the earlier 2E→4E division, but rather remained at the same absolute time during development (supplementary material Fig. S8). However, end-3 is an important activator of gut transcription factors, and so we cannot rule out in this case that the delay in onset of elt-2 transcription with respect to the 2E→4E division is partially due to absence of functional end-3.
To test whether transcription depended on cell division in the muscle lineage, we studied the transcriptional dynamics of unc-120 and hlh-1 in two temperature-sensitive mutants of the B subunit of the DNA polymerase α-primase complex, encoded by div-1. Mutants of div-1 have delayed cell cycles in all lineages except the E lineage, because of delays in completing DNA replication (Encalada et al., 2000) but we expect them not to affect gene expression dynamics directly. If the transcriptional onset time of unc-120 and hlh-1 is tied to the 4MS, 4C, 1D→8MS, 8C, 2D round of cell divisions (Fig. 4C,D), then unc-120 and hlh-1 transcripts will only start accumulating after the slowed divisions have occurred. However, if the transcriptional onsets are instead more closely tied to elapsed developmental time, unc-120 and hlh-1 may begin to accumulate in mutant embryos with many fewer cells than in wild type.
We studied two temperature-sensitive alleles of div-1, div-1(or148) (Encalada et al., 2000) and a mutation first identified as emb-16(g19) (Cassada et al., 1981; Denich et al., 1984), which we showed by whole-genome sequencing and complementation testing is also an allele of div-1. When div-1(or148) and div-1(g19) strains are grown at the restrictive temperature (25°C), embryonic cell cycles proceed at 60-70% of the wild-type rate until eventual embryonic arrest around the 200-cell stage [see Encalada et al. (Encalada et al., 2000), Denich et al. (Denich et al., 1984) and Fig. 6C-E]. As shown in Fig. 6; supplementary material Figs S17, S18, this delay occurs equivalently in most lineages (including those that produce muscle) at the restrictive temperature.
Fig. 6.
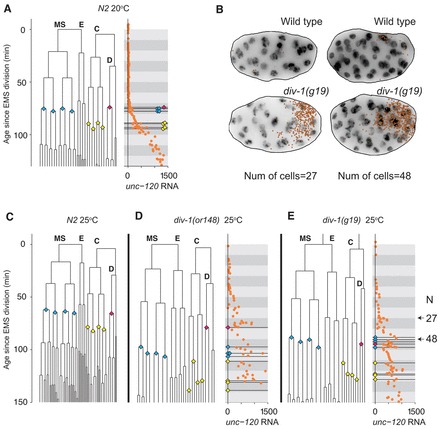
The onset of unc-120 expression is not timed by the 4MS, 4C, 1D→8MS, 8C, 2D divisions. (A) Gene expression trajectory of unc-120 in N2 animals at 20°C, noting the round of divisions that leads from 4MS,4C,1D to 8MS,8C,2D with the same symbols used as in Fig. 4A. Lineage data are the same as in Fig. 1. (B) Positions of unc-120 transcripts overlaid on a DAPI nuclear stain maximum projection in embryos with the same number of cells from wild-type and a div-1(g19) strain with slowed divisions. unc-120 expression onset occurs at a much earlier cell stage in the mutant. The mutant expression pattern is approximately correctly localized (see supplementary material Fig. S15). (C) Lineage of a wild-type embryo at 25°C. (D) (Left) Lineage of a representative div-1(or148) embryo. (Right) Gene expression trajectories for unc-120 in a div-1(or148) strain plotted against the assigned embryo age based on the representative lineage on the left. The marked divisions correspond to the same set of divisions as in A. (E) Same as C for the div-1(g19) strain. In both div-1 mutants, unc-120 rises well over 200 transcripts per embryo before the noted divisions, whereas in wild type the unc-120 expression onset occurs during this block of divisions. This suggests that unc-120 expression onset does not require the cell divisions that normally occur concurrently.
Inconsistent with a model where transcriptional onsets are strictly tied to certain cell cycles, we found that unc-120 transcription starts well before the 4MS, 4C, 1D→8MS, 8C, 2D divisions in both div-1 mutant backgrounds at the restrictive temperature (Fig. 6; supplementary material Figs S9, S10). Similarly, the onset time of hlh-1 also occurs when fewer cells are present, achieving strong transcription just before or early on during the 4MS, 4C, 1D → 8MS, 8C, 2D divisions instead of well after (supplementary material Figs S11, S12). Our data from these two mutants show that the transcription onset times are not determined by waiting for a particular cell division. Although we note that there is considerable embryo-to-embryo variability in lineage for the div-1 strains, the 4MS, 4C, 1D→8MS, 8C, 2D divisions in every div-1 embryo we lineaged are delayed well beyond the transcriptional onset of unc-120, as measured by RNA FISH (supplementary material Figs S17, S18). Therefore, our main conclusion is insensitive to this variability. We also measured elt-2 transcription in these mutants, but the results were inconclusive because the E lineage is not delayed in these mutants and its divisions occur at approximately the same time as in wild type (see supplementary material Figs S13, S14).
Mislocalization of important fate-determining factors, such as PIE-1, is known to occur during the blastomere specification stages in div-1 mutants (Encalada et al., 2000), and it is therefore possible that this opens alternative pathways for early activation of unc-120 and hlh-1 in the mutant that are not relevant in wild-type animals. However, we argue, based on the spatial transcription patterns, that the early unc-120 and hlh-1 transcription is not caused by such dysregulation by another pathway. Our RNA FISH results show that unc-120 and hlh-1 transcription in the div-1(g19) strain is correctly localized to the region of the embryo that eventually gives rise to the body wall muscle in wild type and is similarly colocalized with RNA for the upstream regulator pal-1 in wild type and mutants [see Fig. 5B, supplementary material Fig. S15, and Fukushige and Krause (Fukushige and Krause, 2005) for a discussion of the pal-1 dependence of C and D lineage muscle]. In the div-1(or148) strain, we often found a slightly different spatial pattern of unc-120 and hlh-1 transcription from wild type, but they too overlap strongly with the also altered pal-1 transcription domains, suggesting that the wild-type gene networks are still responsible for activating these muscle regulators in the mutant.
The effects of the relative acceleration of the unc-120 and hlh-1 transcription onset with respect to cell proliferation are apparent in the evolution of the spatial pattern of transcription. In wild-type embryos, unc-120 transcription begins in a few (usually two) cells at around the 48-cell stage (Fig. 6B) but spreads to more than 5 by the 55-cell stage (Fig. 4B). In div-1 mutants, this program begins at a time when the embryo has about half as many cells, and unc-120 transcription has already spread to its full range by the 48-cell stage (Fig. 6B). These results suggest that premature transcriptional onset can increase the number of expressing cells in an embryo with the same total number of cells, indicating that there are not necessarily other gene regulatory mechanisms in place to control the exact number of cells expressing a particular gene.
If the cell cycle is sufficiently slowed such that transcription of a gene that is differentially expressed between sister lineages precedes the division, we expect transcription may also occur in the sister lineages. Clonal commitment to the body wall muscle fate occurs in the D lineage from its birth, in the C lineage at the 4C stage, and in the MS lineage approximately at the 16MS stage (Sulston et al., 1983). In the C lineage, muscle versus epidermal clonal commitment at the 4C stage is thought to occur by the activity of POP-1 in response to an anterior-posterior Wnt signaling gradient (Sugioka et al., 2011; Yanai et al., 2008). Our data shows substantial unc-120 mRNA accumulation in div-1 mutants before even the 2C→4C (Fig. 6D,E), suggesting a possible aberration in muscle specification arising due to mistiming of expression and cell divisions. To test this, we examined hlh-1 expression in div-1(or148) mutants by time lapse imaging of a hlh-1 reporter. In div-1(or148) mutants, the decision to express hlh-1 reporters frequently occurred early, such that expression spread into anterior sister cells of those normally expressing hlh-1 reporters (supplementary material Fig. S16). This indicates that alterations of cell cycle lengths can lead not only to defects in expression timing but also to defects in expression pattern for major developmental regulators.
DISCUSSION
In this paper, we have observed that the onset of transcription of genes often occurs in close temporal proximity to cell division events, and the relative order of these events is maintained upon global perturbations such as changes in metabolic rate by temperature or mutation. The hypothesis that aspects of the cell cycle regulate the timing of transcriptional onset is attractive because it could explain the robustness of development across these conditions. We tested that hypothesis by measuring transcription in mutants in which the relevant cell division occurs after their normal time. Our results show that the timing of transcriptional onset is unaffected by changes in the timing of cell-cycle progression.
The converse issue - whether transcription affects cell-cycle - is equally important. However, multiple studies (Powell-Coffman et al., 1996; Edgar et al., 1994) have quantitatively demonstrated that during the early embryogenesis of C. elegans (up to the 100-cell stage), maternal components are sufficient for most aspects of cell cycle timing. Even a complete block of transcription does not block the progression of the cell cycle, and the rate of cell divisions is unchanged to within 6% (Powell-Coffman et al., 1996). The one change that does occur is a faster rate of cell divisions in the E lineage, probably because end-3 and other E lineage fate specification regulators induce expression of cell cycle inhibitors (Powell-Coffman et al., 1996; Edgar et al., 1994; Hebeisen and Roy, 2008; Bao et al., 2008; Boeck et al., 2011), but notably this is the opposite of the cell-cycle delay phenotype observed in the mutants studied here.
During animal development, in general, both cases where gene expression timing depends on cell cycle timing and is independent of cell cycles have been found. A considerable body of work has dealt with the timing of the first transcriptional events in an embryo, referred to as a transition from maternal to zygotic expression (Yasuda and Schubiger, 1992; Pritchard and Schubiger, 1996; Lu et al., 2009). However, in ascidians and nematodes like C. elegans, zygotic expression begins only a few divisions after fertilization. RNA FISH and microarray studies have shown that zygotic transcription in C. elegans starts at the latest at the four-cell stage (Seydoux and Fire, 1994; Baugh et al., 2003; Raj et al., 2010). Within the developmental time window of our present study, zygotic transcription has been established for more than one cell cycle.
For genes transcribed well after the onset of zygotic transcription, a dependence on cell cycle may arise due to a requirement for a particular cell-cycle protein at the promoter of a gene (Cosma et al., 1999; Cosma et al., 2001). Alternatively, it is possible that the very process of cell division, which involves DNA replication and thus stripping of the vast majority of transcription factors from their binding sites, could profoundly influence transcriptional regulation, a notion often referred to as the ‘quantal cell cycle’ hypothesis (Egli et al., 2008). Cascades of the sort we have looked at in C. elegans have been studied in the neural lineages of Drosophila melanogaster (Fichelson et al., 2005). Examples have been found where progress along a cascade of gene expression that occurs in parallel with cell proliferation is dependent on cell cycle and examples have also been reported of independence from the cell cycle (Grosskortenhaus et al., 2005; Weigmann and Lehner, 1995; Cui and Doe, 1995). Early work in ascidian embryos (for a review, see Satoh, 1982) has shown examples analogous to the ones we contemplated in which progress through the cell cycle is necessary for differentiation even in cell lineages that have been specified. Intriguingly, the example of cytoplasmic cycles in ascidians (Satoh, 1982) suggests that there may be biochemical cycles that continue to operate in the absence of DNA replication and cytokinesis.
The findings presented here on transcription factor timing fit cohesively with Edgar and McGhee’s beautiful study on the timing of terminal differentiation markers (Edgar and McGhee, 1988). Edgar and McGhee showed that the expression of markers like the gut esterase GES-1, gut granules and muscular paramyosin proceeded normally even when they blocked cell cycle progression in the entire embryo well before the expression of these genes. Whereas their experiments studied whole embryo cell cycle arrest, we also found that transcription of elt-2 onset is also insensitive to a specific delay of just the E cell lineage (Fig. 5B).
Our report has focused on a handful of developmentally important genes and we have focused our measurements on the developmental window beginning after specification of the early blastomeres. It is of course possible that our findings do not hold for all genes, and that there are other genes, possibly earlier in development, whose transcriptional timing does depend on particular cell-cycle events. In fact, Edgar and McGhee found that gut esterase is produced only if their DNA synthesis block was applied after at least a few minutes had transpired after the birth of the E cell, suggesting a coupling of gene expression and cell cycle in these earlier stages of development (Edgar and McGhee, 1988). Our findings show that such mechanisms are not in place in later development for important genes such as elt-2 and hlh-1.
This apparent independence of cell cycle and transcriptional onsets leaves unanswered the question of how cell cycles and transcriptional dynamics remain finely synchronized in the face of large developmental rate perturbations due to temperature or the clk-1(qm30) mutant background. One possibility is that a coupling mechanism exists that can correct for small mismatches in gene expression and proliferation rates, such as via silencing of transcription during and shortly after mitosis, but cannot preserve synchrony in the face of the major cell cycle delay defects in the emb-29 and div-1 mutants we studied. Another is that the rates in the biochemical networks of both cell proliferation and transcriptional timing are independently fine-tuned in such a way that they happen to always remain proportional to each other. Although we cannot formally exclude this possibility, we consider it unlikely because we observed in both temperature-based and mutation-based metabolic slowdown. Last, it is possible that a third ‘clock’ network independently controls both transcription and cell-cycle progression, passively synchronizing the two processes. For example, a small set of rate-limiting metabolic reactions crucial for both cell cycling and transcription could serve such a purpose.
Supplementary Material
Acknowledgments
We thank the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs, the C. elegans Gene KO Consortium and the Japan National BioResource Project for strains. We are grateful to Christopher Fang-Yen for use of equipment. We thank the University of Pennsylvania worm meeting group, especially Meera Sundaram, and members of the Raj and Murray labs for valuable commentary and criticism. We are also grateful for the suggestions made by the anonymous referees during the manuscript review process.
Footnotes
Funding
G.N. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. A.R. acknowledges financial support from a Burroughs-Wellcome Fund Career Award at the Scientific Interface (www.bwfund.org) and a NIH (www.nih.gov) New Innovator Award [1DP2OD008514]. J.I.M. was funded by the Penn Genome Frontiers Institute under a grant with the Pennsylvania Department of Health, which disclaims responsibility for any analyses, interpretations or conclusions. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
G.N., T.W., J.I.M. and A.R. designed research and wrote the manuscript. G.N. and T.W. carried out experiments and analyzed data.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.098012/-/DC1
References
- Bao Z., Murray J. I. (2011). Mounting caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harb. Protoc. doi:10.1101/pdb.prot065599 [DOI] [PubMed]
- Bao Z., Murray J. I., Boyle T., Ooi S. L., Sandel M. J., Waterston R. H. (2006). Automated cell lineage tracing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 103, 2707–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z., Zhao Z., Boyle T. J., Murray J. I., Waterston R. H. (2008). Control of cell cycle timing during C. elegans embryogenesis. Dev. Biol. 318, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh L. R., Hill A. A., Slonim D. K., Brown E. L., Hunter C. P. (2003). Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development 130, 889–900 [DOI] [PubMed] [Google Scholar]
- Boeck M. E., Boyle T., Bao Z., Murray J., Mericle B., Waterston R. (2011). Specific roles for the GATA transcription factors end-1 and end-3 during C. elegans E-lineage development. Dev. Biol. 358, 345–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle T. J., Bao Z., Murray J. I., Araya C. L., Waterston R. H. (2006). AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics 7, 275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassada R., Isnenghi E., Culotti M., von Ehrenstein G. (1981). Genetic analysis of temperature-sensitive embryogenesis mutants in Caenorhabditis elegans. Dev. Biol. 84, 193–205 [DOI] [PubMed] [Google Scholar]
- Cosma M. P., Tanaka T., Nasmyth K. (1999). Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell 97, 299–311 [DOI] [PubMed] [Google Scholar]
- Cosma M. P., Panizza S., Nasmyth K. (2001). Cdk1 triggers association of RNA polymerase to cell cycle promoters only after recruitment of the mediator by SBF. Mol. Cell 7, 1213–1220 [DOI] [PubMed] [Google Scholar]
- Cui X., Doe C. Q. (1995). The role of the cell cycle and cytokinesis in regulating neuroblast sublineage gene expression in the Drosophila CNS. Development 121, 3233–3243 [DOI] [PubMed] [Google Scholar]
- Denich K. T. R., Schierenberg E., Isnenghi E., Cassada R. (1984). Cell-lineage and developmental defects of temperature-sensitive embryonic arrest mutants of the nematode Caenorhabditis elegans. Rouxs Arch. Dev. Biol. 193, 164–179 [DOI] [PubMed] [Google Scholar]
- Edgar L. G., McGhee J. D. (1988). DNA synthesis and the control of embryonic gene expression in C. elegans. Cell 53, 589–599 [DOI] [PubMed] [Google Scholar]
- Edgar L. G., Wolf N., Wood W. B. (1994). Early transcription in Caenorhabditis elegans embryos. Development 120, 443–451 [DOI] [PubMed] [Google Scholar]
- Egli D., Birkhoff G., Eggan K. (2008). Mediators of reprogramming: transcription factors and transitions through mitosis. Nat. Rev. Mol. Cell Biol. 9, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encalada S. E., Martin P. R., Phillips J. B., Lyczak R., Hamill D. R., Swan K. A., Bowerman B. (2000). DNA replication defects delay cell division and disrupt cell polarity in early Caenorhabditis elegans embryos. Dev. Biol. 228, 225–238 [DOI] [PubMed] [Google Scholar]
- Fichelson P., Audibert A., Simon F., Gho M. (2005). Cell cycle and cell-fate determination in Drosophila neural cell lineages. Trends Genet. 21, 413–420 [DOI] [PubMed] [Google Scholar]
- Fukushige T., Krause M. (2005). The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development 132, 1795–1805 [DOI] [PubMed] [Google Scholar]
- Fukushige T., Brodigan T. M., Schriefer L. A., Waterston R. H., Krause M. (2006). Defining the transcriptional redundancy of early bodywall muscle development in C. elegans: evidence for a unified theory of animal muscle development. Genes Dev. 20, 3395–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskortenhaus R., Pearson B. J., Marusich A., Doe C. Q. (2005). Regulation of temporal identity transitions in Drosophila neuroblasts. Dev. Cell 8, 193–202 [DOI] [PubMed] [Google Scholar]
- Hebeisen M., Roy R. (2008). CDC-25.1 stability is regulated by distinct domains to restrict cell division during embryogenesis in C. elegans. Development 135, 1259–1269 [DOI] [PubMed] [Google Scholar]
- Hecht R. M., Berg-Zabelshansky M., Rao P. N., Davis F. M. (1987). Conditional absence of mitosis-specific antigens in a temperature-sensitive embryonic-arrest mutant of Caenorhabditis elegans. J. Cell Sci. 87, 305–314 [DOI] [PubMed] [Google Scholar]
- Kim J., Kawasaki I., Shim Y. H. (2010). cdc-25.2, a C. elegans ortholog of cdc25, is required to promote oocyte maturation. J. Cell Sci. 123, 993–1000 [DOI] [PubMed] [Google Scholar]
- Krause M. (1995). MyoD and myogenesis in C. elegans. Bioessays 17, 219–228 [DOI] [PubMed] [Google Scholar]
- Lu X., Li J. M., Elemento O., Tavazoie S., Wieschaus E. F. (2009). Coupling of zygotic transcription to mitotic control at the Drosophila mid-blastula transition. Development 136, 2101–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M. F., Rothman J. H. (2002). Making worm guts: the gene regulatory network of the Caenorhabditis elegans endoderm. Dev. Biol. 246, 68–85 [DOI] [PubMed] [Google Scholar]
- Minevich G., Park D. S., Blankenberg D., Poole R. J., Hobert O. (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. I., Bao Z., Boyle T. J., Waterston R. H. (2006). The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nat. Protoc. 1, 1468–1476 [DOI] [PubMed] [Google Scholar]
- Murray J. I., Bao Z., Boyle T. J., Boeck M. E., Mericle B. L., Nicholas T. J., Zhao Z., Sandel M. J., Waterston R. H. (2008). Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat. Methods 5, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. I., Boyle T. J., Preston E., Vafeados D., Mericle B., Weisdepp P., Zhao Z., Bao Z., Boeck M. E., Waterston R. H. (2012). Multidimensional regulation of gene expression in the C. elegans embryo. Genome Res. 22, 1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Coffman J. A., Knight J., Wood W. B. (1996). Onset of C. elegans gastrulation is blocked by inhibition of embryonic transcription with an RNA polymerase antisense RNA. Dev. Biol. 178, 472–483 [DOI] [PubMed] [Google Scholar]
- Pritchard D. K., Schubiger G. (1996). Activation of transcription in Drosophila embryos is a gradual process mediated by the nucleocytoplasmic ratio. Genes Dev. 10, 1131–1142 [DOI] [PubMed] [Google Scholar]
- Raj A., van den Bogaard P., Rifkin S. A., van Oudenaarden A., Tyagi S. (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., Rifkin S. A., Andersen E., van Oudenaarden A. (2010). Variability in gene expression underlies incomplete penetrance. Nature 463, 913–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J. L., Zacharias A. L., Walton T., Burdick J. T., Murray J. I. (2013). A quantitative model of normal Caenorhabditis elegans embryogenesis and its disruption after stress. Dev. Biol. 374, 12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A., Du Z., Nowotschin S., Hadjantonakis A.-K., Bao Z. (2010). A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics 11, 580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S., Bertrand V., Bigelow H., Boyanov A., Doitsidou M., Poole R. J., Narula S., Hobert O. (2010). Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics 185, 417–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh N. (1982). Timing mechanisms in early embryonic development. Differentiation 22, 156–163 [DOI] [PubMed] [Google Scholar]
- Schroeder L. K., Kremer S., Kramer M. J., Currie E., Kwan E., Watts J. L., Lawrenson A. L., Hermann G. J. (2007). Function of the Caenorhabditis elegans ABC transporter PGP-2 in the biogenesis of a lysosome-related fat storage organelle. Mol. Biol. Cell 18, 995–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydoux G., Fire A. (1994). Soma-germline asymmetry in the distributions of embryonic RNAs in Caenorhabditis elegans. Development 120, 2823–2834 [DOI] [PubMed] [Google Scholar]
- Sommermann E. M., Strohmaier K. R., Maduro M. F., Rothman J. H. (2010). Endoderm development in Caenorhabditis elegans: the synergistic action of ELT-2 and -7 mediates the specification→differentiation transition. Dev. Biol. 347, 154–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka K., Mizumoto K., Sawa H. (2011). Wnt regulates spindle asymmetry to generate asymmetric nuclear β-catenin in C. elegans. Cell 146, 942–954 [DOI] [PubMed] [Google Scholar]
- Sulston J. E., Schierenberg E., White J. G., Thomson J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 [DOI] [PubMed] [Google Scholar]
- Vargas D. Y., Raj A., Marras S. A. E., Kramer F. R., Tyagi S. (2005). Mechanism of mRNA transport in the nucleus. Proc. Natl. Acad. Sci. USA 102, 17008–17013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann K., Lehner C. F. (1995). Cell fate specification by even-skipped expression in the Drosophila nervous system is coupled to cell cycle progression. Development 121, 3713–3721 [DOI] [PubMed] [Google Scholar]
- Wong A., Boutis P., Hekimi S. (1995). Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139, 1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I., Baugh L. R., Smith J. J., Roehrig C., Shen-Orr S. S., Claggett J. M., Hill A. A., Slonim D. K., Hunter C. P. (2008). Pairing of competitive and topologically distinct regulatory modules enhances patterned gene expression. Mol. Syst. Biol. 4, 163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda G. K., Schubiger G. (1992). Temporal regulation in the early embryo: is MBT too good to be true? Trends Genet. 8, 124–127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.