
Figure 1.
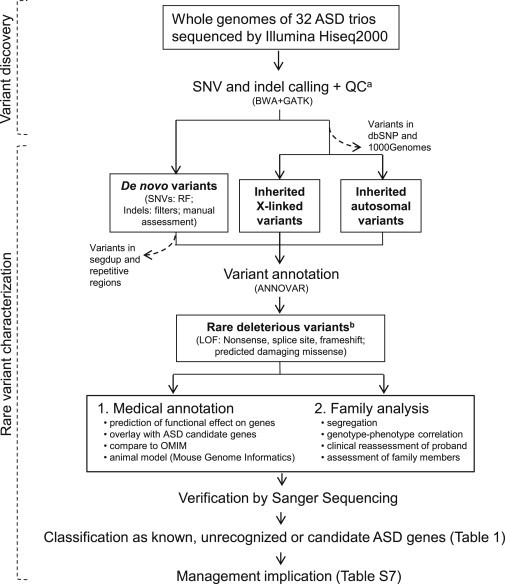
Detection and Classification of Medically Relevant Genetic Variants
Rare SNV and indel variants were assessed as putative etiologic factors in ASD. Samples were also run on high-resolution microarray for CNV calling as indicated in the Material and Methods. Dashed arrows indicate variants not included in downstream analyses. In addition to applying random forests (RF) algorithms and filtering methods, we used Sanger sequencing to perform a manual assessment of potential de novo variants annotated by GATK.
aLow-quality variants were eliminated with GATK’s filters.
bRare variants were defined as those not found in dbSNP or the 1000 Genomes Project. Each gene bearing rare deleterious variants was classified as (1) a "known" (also called "linked") ASD gene if it was previously identified to be involved in ASD, according to lists developed by the Autism Genome Project Consortium,3,7 (2) an "unrecognized" ASD gene if it was not previously recognized to carry a loss-of-function mutation (nonsense, splice site, or frameshift) and if it showed an ASD-related phenotype, as reported in OMIM and/or the MGI mouse knockout database, or (3) a "candidate" ASD gene if it was affected by a de novo deleterious variant. In the case of rare inherited X-linked deleterious variants found in males, the same gene also needed to be implicated in other sequencing studies. Abbreviations are as follows: GATK, Genome Analysis Toolkit; BWA, Burrows-Wheeler Aligner; and segdup, segmental duplication