
FIGURE 2.
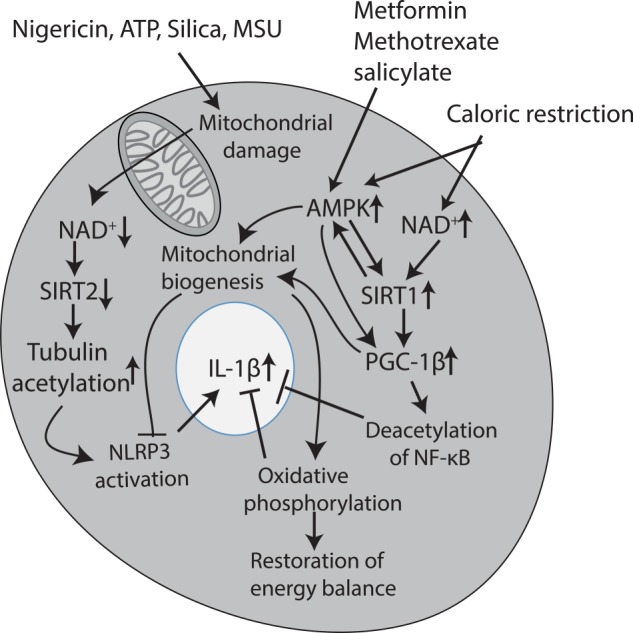
NAD+, AMPK, and SIRT1 as key inflammatory regulators. Agents such as nigericin, ATP, and uric acid crystals lead to mitochondrial damage, which in turn leads to a decrease in NAD+. This limits SIRT2 activity, allowing acetylation of tubulin to persist, which in turn enhances NLRP3 activity by promoting the trafficking of NLRP3 to the mitochondria, enhancing its activation. Because these agents are all NLRP3 activators, this could create a positive feedback loop sustaining NLRP3 activation, leading to IL-1β production. Caloric restriction has the opposite effect and leads to an increase in NAD+. This leads to the activation of SIRT1, a deacetylase that will promote mitochondrial biogenesis and function via PGC-1β and inhibit NF-κB via deacetylation of the p65 subunit. Both of these events will have a net anti-inflammatory effect, with the promotion of mitochondrial function, also inhibiting NLRP3 activation. The increase in oxidative phosphorylation (which appears to be anti-inflammatory probably via an increase in NAD+) will restore the energy balance. Caloric restriction will also activate AMPK, which can also promote mitochondrial biogenesis. There is evidence for cross-talk between SIRT1 and AMPK. Finally, several anti-inflammatory agents, notably methotrexate and salicylate, have been shown to activate AMPK, as has the anti-T2D agent metformin. Metformin may mediate its effects in T2D via this process because limiting inflammation in this way could restore insulin sensitivity. MSU, monosodium urate.