

Abstract
Two major β-adrenergic receptor (βAR) subtypes, β1AR and β2AR, are expressed in mammalian heart with β1AR coupling to Gs and β2AR dually coupling to Gs and Gi proteins. In many types of chronic heart failure, myocardial contractile response to both β1AR and β2AR stimulation is severely impaired. The dysfunction of βAR signaling in failing hearts is largely attributable to an increase in Gi signaling, because disruption of the Gi signaling restores myocardial contractile response to β1AR as well as β2AR stimulation. However, the mechanism terminating the β2AR-Gi signaling remains elusive, while it has been shown activation of the Gi signaling is dependent on agonist stimulation and subsequent PKA-mediated phosphorylation of the receptor. Here we demonstrate that regulator of G protein signaling 2 (RGS2) is a primary terminator of the β2AR-Gi signaling. Specifically, prolonged absence of agonist stimulation for 24h impairs the β2AR-Gi signaling, resulting in enhanced β2AR- but not β1AR-mediated contractile response in cultured adult mouse cardiomyocytes. Increased β2AR contractile response is accompanied by a selective upregulation of RGS2 in the absence of alterations in other major cardiac RGS proteins (RGS3-5) or Gs, Gi or βAR subtypes. Administration of a βAR agonist, isoproterenol (ISO, 1.0 nM), prevents RGS2 upregulation and restores the β2AR-Gi signaling in cultured cells. Furthermore, RGS2 ablation, similar to βAR agonist stimulation, sustains the β2AR-Gi signaling in cultured cells, whereas adenoviral overexpression of RGS2 suppresses agonist-activated β2AR-Gi signaling in cardiomyocytes and HEK293 cells. These findings not only define RGS2 as a novel negative regulator of the β2AR-Gi signaling, but also provide a potential novel target for the treatment of chronic heart failure.
Keywords: β2-adrenergic receptor, RGS2, Gi proteins, cardiomyocyte contractility
1. Introduction
Stimulation of β-adrenergic receptor (βAR), a prototypical member of G protein-coupled receptors (GPCR) superfamily, is broadly involved in the regulation of energy metabolism, growth control, and cardiac function [1]. In mammalian myocardium, the closely related βAR subtypes, β1AR and β2AR, activate subtype-specific G protein signaling pathways and elicit distinct actions[2]. While β1AR couples to the classic Gs-adenylyl cyclase (AC)-cAMP-PKA signaling cascade, β2AR activates pertussis toxin (PTX)-sensitive Gi proteins in addition to the Gs pathway, with the Gi signaling negating the Gs-mediated positive inotropic effect in the heart[3-5] and promoting cardiomyocyte survival[6]. However, exaggerated β2AR-Gi signaling is a hallmark of the failing heart of mammalian species, including rat, canine, and human[7-9]. In the failing heart, the enhanced Gi signaling negates β1AR- as well as β2AR-mediated contractile response[10-12], thus contributing to the pathogenesis of heart failure. This perception has been recently corroborated by the fact that selective β2AR-Gs stimulation with fenoterol, bypassing the Gi signaling, markedly improves cardiac structure and function in an ischemic heart failure model[7, 13]. Thus, delineating the mechanism that regulates the β2AR-Gi signaling will not only deepen our understanding of βAR signal transduction but also bear important pathological and therapeutic implications.
The classic view on βAR signaling involves an agonist-induced change in the receptor conformation that causes the activation of the Gs protein, leading the formation of the second messenger, cAMP, which activates PKA and downstream signaling. The termination of this cascade, a process known as desensitization, occurs when G protein-coupled receptor kinases (GRKs) and the second messenger kinase, PKA, phosphorylate the activated receptor and promote the binding of β-arrestins which sterically block the coupling of Gs to the receptor.
In contrast to the βAR-Gs signaling, the β2AR-Gi signaling is enhanced by PKA-mediated phosphorylation of the receptor [14, 15]. The fundamental question is what is the mechanism underlying the termination of the β2AR-Gi signaling. In this regard, it has been shown that upon GPCR activation, GDP is exchanged for GTP on the Gα subunit, resulting dissociation of the Gα from Gβγ subunits and the activation of downstream effortors. The intrinsic GTPase activity of the α subunit of G proteins serves as a molecular clock, turning down GPCR signaling via returning G proteins to the GDP-bond heterotrimeric form. Regulator of G protein signaling (RGS) proteins are GTPase-activating proteins (GAPs) which accelerate GTPase-mediated hydrolysis of GTP to GDP on Gα, thus reconstituting the heterotrimeric G protein complex and terminating G protein signaling[16, 17]. Different RGS proteins have GAP activity towards specific G protein families and, in some cases, even for particular receptors, although the mechanisms for such selectivity remain unknown.
In the heart of mammalian species, major RGS proteins include RGS2-5[18]. In particular, RGS2 can negatively regulate the signaling of Gq-coupled receptors, including α1A-adrenergic receptor, angiotensin II receptor 1A, and interleukin receptor[19, 20]. Deregulation of RGS2 has been implicated in the pathogenesis of cardiac hypertrophy and hypertension[21]. Interestingly, it has been recently reported that RGS2 can physically interact with β2AR[22], in addition to Gq-coupled GPCRs such as M1 muscarinic receptor[23] and α1AAR[24]. However, it is unknown whether RGS2 regulates Gi signaling in general and the β2AR-coupled Gi signaling in particular.
In this study, we seek to determine the potential role of RGS proteins, particularly RGS2 in regulating the β2AR-coupled Gi signaling in a physiologically relevant setting, adult mouse cardiomyocytes. We provide multiple lines of evidence to define RGS2 as a primary terminator of the β2AR-Gi signaling. First, an elevation in RGS2 expression impairs the β2AR-Gi signaling, as manifested by the loss of PTX sensitivity of β2AR contractile response in cultured adult mouse cardiomyocytes and the failure of β2AR stimulation to increase PTX-sensitive ERK1/2 activation in HEK293 cells. In contrast, RGS2 deficiency, similar to βAR agonist stimulation, sustains the Gi signaling in cultured cardiomyocytes even in the prolonged absence of agonist stimulation. These data have revealed a previously unappreciated novel negative regulation of β2AR-activated Gi signaling by RGS2 in mammalian hearts.
2. Materials & Methods
2.1. Isolation, Culture and Adenoviral Infection of Adult Cardiomyocytes Isolated from Wild-type or RGS2 Knockout Mice
Single myocytes were isolated from the hearts of 2-3 month-old Black C57 or RGS2 Knockout mice using a standard enzymatic technique, as described previously[25]. In a subset of experiments, isolated myocytes were cultured and infected with target gene-carrying adenoviral vectors in the presence or absence of isoproterenol (ISO, 10 nM). Adenovirus-mediated gene transfer was implemented by adding adenoviral vectors carrying β-galactosidase (Adv-β-gal, as a control virus) or N-terminally Flag-tagged RGS2 (Adv-RGS2), RGS3 (Adv-RGS3), RGS4 (Adv-RGS4) or RGS5 (Adv-RGS5) (each at a multiplicity of infection, M.O.I., of 100). At the M.O.I. used, almost 100% myocytes were positively infected, as evidenced by β-gal staining[25] or GFP fluorescent signal (Adv-GFP infection, unpublished data). All experiments were performed after cells were cultured for 24 h, unless indicated otherwise.
2.2. Cell Contraction Measurements
Cells were perfused with a HEPES-buffered solution, and electrically paced at 0.5 Hz at 23°C. To inhibit Gi signaling, freshly isolated or cultured myocytes were incubated with PTX (1.5 μg/ml, at 37°C for at least 3 h), prior to contraction measurements. Cell sarcomere length was monitored by an optical tracking method using a photodiode array (Model 1024 SAQ, Reticon) as described previously [25].
2.3. Radioligand-Binding Assay
A βAR-specific ligand [125I]-iodocyanopindolol ([125I]-CYP) binding assay was performed, as described previously[26]. Competition experiments were carried out at 50 pM [125I]-CYP in the presence of a highly selective β2AR antagonist, ICI 118.551, or a highly selective β1AR antagonist, CGP 20712A. Non-specific binding was determined in the presence of 10 μM propranolol and was about 10-30% of total binding of [125I]-CYP (50 pM).
2.4. Western Blotting of Gs and Gi Proteins and RGS proteins
The expression of either Gs or Gi proteins was measured by Western blotting with specific antibodies, as previous described[7]. Endogenous RGS2 protein abundance was assayed by an affinity-purified antibody from GenWay Biotech[26]. Endogenous RGS3, RGS4 and RGS5 protein levels were detected with specific antibodies from Santa Cruz, whereas Flag-tagged RGS2-5 was detected by an antibody specifically reacting with Flag.
2.5 cAMP Measurement
Intracellular cAMP levels were assayed by radioimmunoassay, as previously described [7]. Briefly, cultured mouse cardiomyocytes or HEK 293 cells were treated with the phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine (IBMX, 1 mM), for 30 min at 37°C. The cells then were incubated with zinterol (10 μM, a β2AR agonist) in the presence of CGP27013A (0.3 μM, a β1AR selective antagonist) for 10 min. cAMP levels were assayed with the cAMP assay kit (Parameter TM, R& D Systems, # KGE002B) with a duplicate in each experiment. Protein content was measured using the Piece BCA (Thermo Scientific # 23228).
2.5. Materials
Zinterol was kindly supplied by Bristol-Myers, Evansville, IN. Antibodies recognizing the α-subunits of Gs, Go, Gi1, Gi2, and Gi3, were purchased from Santa-Cruz, USA. The antibody recognizing RGS2 was obtained from GenWay Biotech, while antibodies for other RGS proteins (RGS3, RGS4, RGS5) and secondary antibodies were purchased from Santa Cruz. The antibody reacting with Flag was from Affinity Bioreagents. Pertussis toxin (PTX), isoproterenol (ISO), propranolol and norepinephrine (NE), CGP20712A (CGP), and minimal essential medium (MEM) were purchased from Sigma (St. Louis, MO). ICI 118,551 (ICI) was kindly supplied by ICI Pharmaceutic Group (Wilmington, DE). FBS and Penicillin-Streptomycin were purchased from Gibco (Gaithersburg, MD). Laminin was purchased from Upstate. [125I]-CYP was purchased from NEN Life Science Products, Inc. (Boston, MA).
2.6. Statistics
Data reported are mean ± standard error of mean (SEM). Statistical comparisons were made by student's t test or paired t test when appropriate. Two-factor analysis of variance, ANOVA, was used to analyze the overall drug dose-response. A P value of < 0.05 was considered to be statistically significant.
3. Results
3.1. Prolonged Absence of Agonist Stimulation Desensitizes β2AR-Mediated Gi Signaling and Selectively Elevates RGS2 Expression in Adult Mouse Cardiomyocytes
In adult mouse cardiac myocytes, β2AR stimulation activates both Gs and Gi proteins with the Gi signaling substantially negating the Gs-mediated contractile response[27]. Consistent with the previous notion[27], the contractile response to a selective β2AR agonist, zinterol (10-8 to 10-5 M), was minor and profoundly enhanced by disrupting Gi signaling with PTX in freshly isolated adult mouse ventricular myocytes (Fig. 1A), without altering cell basal contractility (Table A1 of the online-only Data Supplement). Interestingly, prolonged absence of agonist stimulation markedly augmented zinterol-induced contractile responses which could not be further enhanced by PTX treatment in myocytes cultured for 24 h (Fig. 1B). However, the contractile response to β1AR stimulation by norepinephrine (NE, plus an α1AR antagonist prazosin at 10-6M) remained intact in cultured myocytes (Fig. 1C), suggesting that the excitation-contraction machinery and the Gs-AC-cAMP-PKA signaling cascade are unaltered during 24 h cell culture. These results strongly suggest that the β2AR-coupled Gi signaling is severely desensitized, while the Gs pathway is intact during 24h absence of agonist stimulation in cultured rodent cardiomyocytes.
Fig. 1. Prolonged absence of agonist stimulation selectively enhances β2AR-, but not β1AR-, mediated contractile response in cultured adult mouse cardiomyocytes.
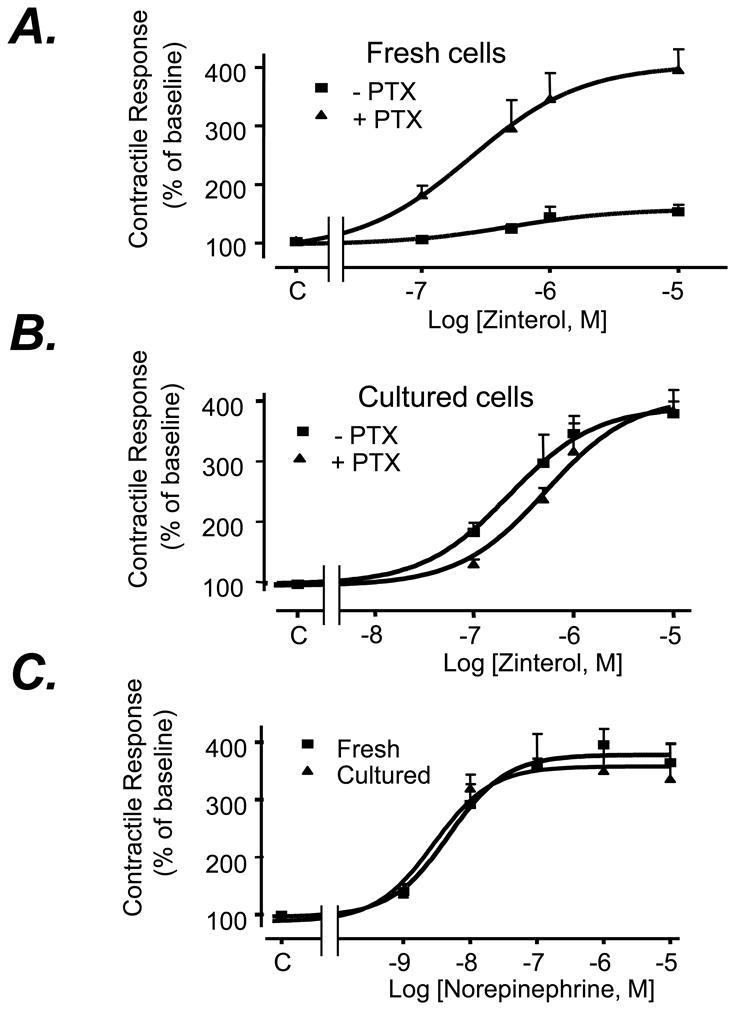
The contractile response of freshly isolated adult mouse ventricular myocytes (Fresh) (Panel A) or those cultured for 24 h (Culture) (Panel B) to a β2AR selective agonist zinterol or a β1AR agonist norepinephrine (NE) in the presence of an αAR antagonist (prazosin, 10-6 M) (Panel C) (n = 8∼14 cells from at least 6 hearts for each data point). Note that PTX markedly enhances β2AR-mediated contractile response in freshly isolated, but not in cultured, cardiomyocytes. There is no significant difference between freshly isolated cells and cultured cells in their basal contraction amplitudes in the presence or absence of PTX treatment (Table 1S, data supplements).
Because many RGS proteins are GAPs for Gαi/o family and negatively regulate Gi/o signaling in in vitro experimental systems[28, 29], we hypothesized that RGS proteins may be involved in the desensitization of the β2AR-Gi signaling. To determine which particular RGS proteins regulate the β2AR-Gi signaling, we examined β2AR-Gi-dependent activation of ERK1/2 MAPK in HEK 293 cells in the presence or absence of adenoviral overexpression of RGS2, RGS3, RGS4 and RGS5. β2AR stimulation with ISO (1.0 μM) increased ERK1/2 phosphorylation level by 3-4 folds in a PTX-sensitive manner in uninfected HEK 293 cells or those infected with a control adenovirus, Adv-β-gal (100 M.O.I..), (Fig. 2 A&B), consistent with previous report[14]. Importantly, adenoviral overexpression of Flag-tagged RGS2 or RGS5 fully abolished the β2AR-Gi-mediated activation of ERK1/2 (Fig. 2A), indicating that both RGS2 and RGS5 negatively regulate the β2AR-mediated Gi signaling. In contrast, overexpression of RGS3 or RGS4 to a greater protein level had no significant effect on β2AR-induced, Gi-dependent ERK1/2 activation (Fig. 2A&B).
Fig. 2. Effects of overexpression of RGS2-5 on the β2AR-Gi-dependent activation of ERK1/2 in HEK293 cells.
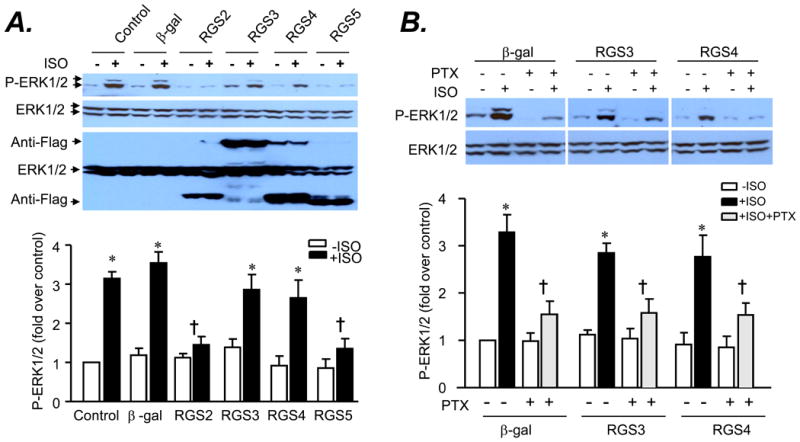
Cells were transferred with an adenovirus (at 10 M.O.I.) expressing β-gal or Flag-tagged RGS2, RGS3, RGS4, and RGS5 for 24 h and subjected to serum starvation overnight. A. Activation of ERK1/2 by β2AR stimulation with isoproterenol (ISO) was inhibited by overexpression of RGS2 or RGS5 but not by overexpression of RGS3 and RGS4. B. Disrupting Gi signaling with PTX treatment suppressed ISO-induced activation of ERK1/2 in HEK293 infected with an adenovirus (at 10 M.O.I.) expressing β-gal or Flag-tagged RGS3 or RGS4. Cells were stimulated with ISO (1 μM) for 5 min with or without PTX (0.5 μg/ml) pretreatment and reactions were stopped by adding 100 μl of cell lysis buffer. Phosphorylation of ERK1/2 was assayed by Western blotting using a site-specific antibody reacting with phosphorylated ERK1/2. * P<0.01 versus respective untreated group; †P<0.01 versus β-gal group in the absence of PTX (n=3-4).
Next, we examined the expression profile of a panel of major cardiac RGS proteins (RGS 2, RGS3, RGS4, and RGS5) at mRNA and protein levels using real-time PCR and Western blotting, respectively, in freshly isolated or cultured cardiomyocytes. To our surprise, among examined RGS proteins, only RGS2 expression was increased at both mRNA (Fig. 3A) and protein levels (Fig. 3 B), in the absence of alterations in other major cardiac RGS proteins (Fig. 3A&B), in cultured cardiomyocytes compared to freshly isolated cells. Thus, RGS2 was selectively upregulated in response to the prolonged absence of agonist stimulation (Fig. 3), although when overexpressed both RGS2 and RGS5 blocked the β2AR-Gi-dependent ERK1/2 activation (Fig. 2A). RGS2 protein level was similarly elevated in cultured adult rat and mouse cardiac myocytes, but basal abundance of RGS2 was relatively lower in mouse myocytes (Fig. 4A&B). Confocal immunostaining revealed that RGS2 was largely located along cell subsurface membranes in both freshly isolated and cultured myocytes (Fig. 4B).
Fig. 3. Expression of RGS2-5 at mRNA and protein levels in freshly isolated or cultured cardiomyocytes.
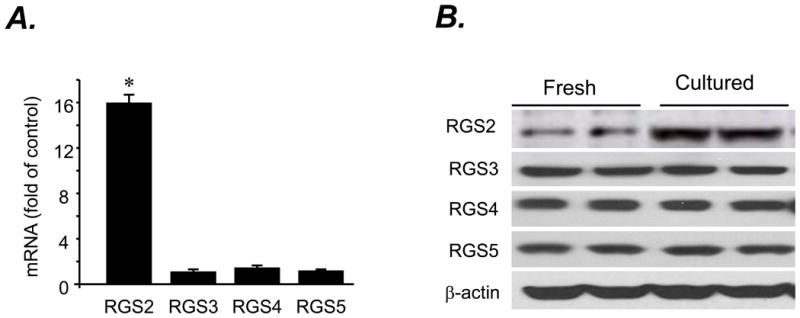
A. mRNA levels of RGS2-5 assayed by real-time PCR (n= 3 independent experiments; * P< 0.01 vs. RGS3, RGS4 and RGS5). B. Representative Western blots probed with an antibody reacting with RGS2, RGS3, RGS4, or RGS5 or β-actin in freshly isolated or cultured cardiomyocytes.
Fig. 4. Expression and intracellular distribution of RGS2 in freshly isolated or cultured adult rat or mouse cardiomyocytes.
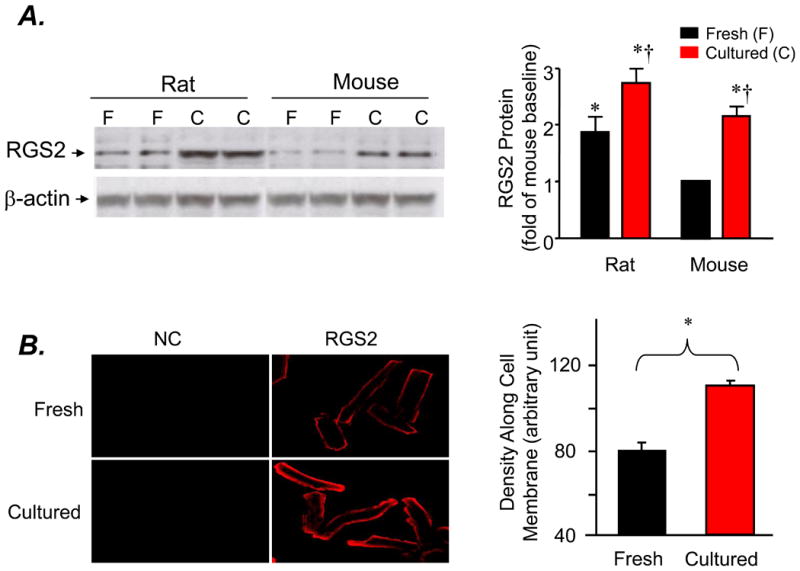
A. A representative Western blot and average data of RGS2 probed with an antibody reacting with RGS2 (* P< 0.05 vs. freshly isolated mouse cardiomyocytes; †P<0.01 vs. respective freshly isolated myocytes; n=6 hearts for each group). B. Typical confocal immunacytochemical imaging and average data of RGS2 in freshly isolated or cultured cardiomyocytes (n= 41 and 43 cells from 6 hearts, respectively; *P<0.001 vs. the value of freshly isolated cells)
3.2. Agonist Stimulation Prevents RGS2 upregulation and Restores β2AR-Gi Signaling in Cultured Cardiomyocytes
Next, we determined whether the selective upregulation of RGS2 can be prevented by adding a βAR agonist during cell culture. Indeed, administration of a βAR agonist, ISO (1.0 nM for 24 h), blocked the upregulation of RGS2 in cultured adult mouse cardiomyocytes (Fig. 5 A&B). Previous studies have reported that activation of PKA with βAR stimulation or forskolin increases rather than decreases the expression of RGS2 in cardiac myocytes and other cell type[30-32] However, in those previous studies, cells were subjected to a short-term (2-3h) instead of 24h treatment of βAR agonists or forskolin. We then compared the expression of RGS2 in cardiomyocytes subjected to either a short-term (2 h) or a prolonged (24 h) treatment with ISO (1 nM) or forskolin (1 μM). Consistent with previous studies, incubation of cells with ISO (1 nM) or forskolin (1 μM) for 2h significantly elevated RGS2 protein abundance by 2-3 folds (Fig. 5 A&B). In sharp contrast, treatment of myocytes from the same hearts with the same agents for 24h reduced RGS2 expression as compared to the baseline of cultured cells in the absence of ISO (Fig. 5), suggesting there is a biphasic regulation of RGS2 expression at protein level by either ISO or forskolin. Together, our data indicate that the upregulation of RGS2 was largely prevented by the presence of the βAR agonist, ISO (1.0 nM for 24 h), during cell culture.
Fig. 5. Biphasic regulation of RGS2 expression by βAR stimulation with ISO (1.0 nM) or adenylate cyclase activation with forskolin (1.0 μM).
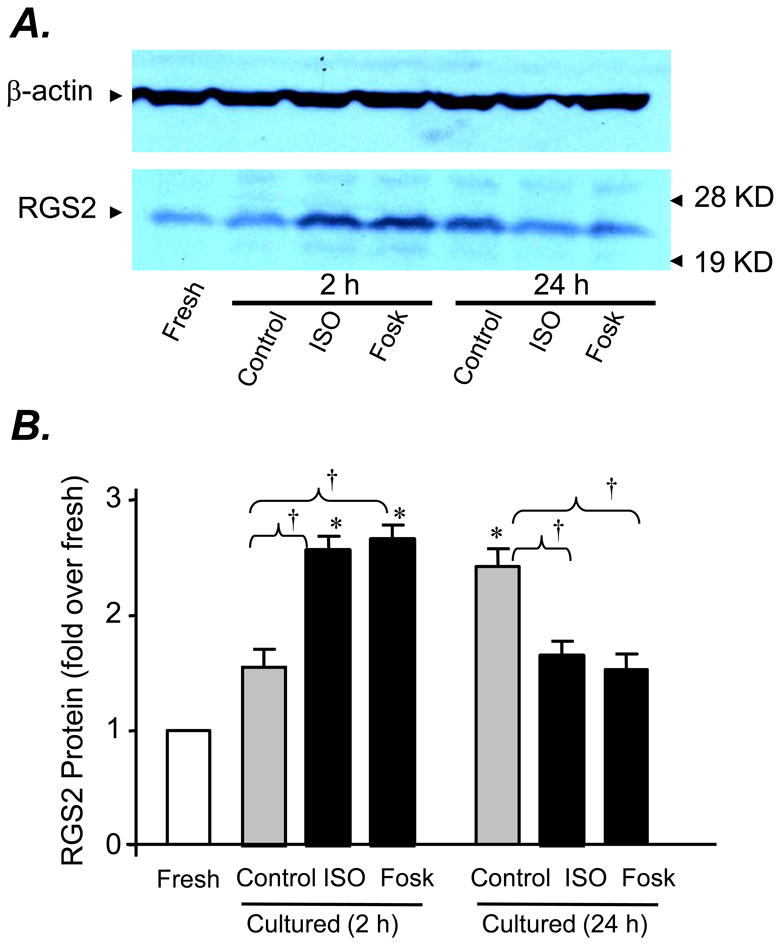
Panels A and B show, respectively, a representative Western blot and the average data probed with an antibody reacting with RGS2 in freshly isolated adult mouse cardiomyocytes or those cultured for 2h or 24 h in the presence or absence of ISO (1.0 nM) or forskolin (1.0 μM). The same membrane was striped and re-probed with anti-β-actin (the top panel). * P<0.01 vs. fresh mouse cardiomyocytes; †P<0.01 vs. as indicated; at least three independent experiments for each group.
Next, we investigated whether ISO can restore β2AR-mediated Gi signaling as indexed by the PTX sensitivity of the receptor-induced contractile response in cultured adult mouse cardiomyocytes. Remarkably, ISO (1.0 nM for 24 h) fully prevented the augmentation of β2AR-mediated contractile response, and restored its PTX sensitivity (Fig. 6), implying a full restoration of the receptor-coupled Gi signaling by agonist stimulation. These results suggest that RGS2 negatively regulated the β2AR- Gi signaling, and that agonist -induced β2AR-Gi signaling is likely mediated by suppressing RGS2 expression.
Fig. 6. Agonist stimulation restores β2AR-mediated Gi signaling in cultured myocytes.

Addition of 1.0 nM ISO into the culture medium fully restores the PTX sensitivity of β2AR contractile response. Treatment with ISO or PTX did not alter the baseline contraction (see Table S1, online-only data supplement).
3.3. Overexpression of RGS2 Suppresses Agonist-Restored β2AR-Gi Signaling in Cultured Mouse Cardiomyocytes
To determine whether there is a causative relationship between the upregulation of RGS2 and the impairment of β2AR-activated Gi signaling in cultured myocytes, we overexpressed RGS2 protein by approximately 2-fold over baseline using adenoviral gene transfer (Fig. 7A). The increase in the contraction amplitude induced by the β2AR agonist, zinterol (5×10-7 M, a concentration close to EC50), was clearly augmented in cultured adult mouse cardiomyocytes infected with Adv-RGS2, regardless of the presence of ISO (1.0 nM for 24 h) and the absence of PTX (Fig. 7B). Importantly, overexpression of RGS2 diminished PTX-sensitive component of β2AR-mediated contractile response (Fig. 7B). These results indicate that similar to the situation in HEK293 cells (Fig. 2A), an elevation of RGS2 abundance is sufficient to suppress agonist-induced β2AR-Gi signaling in cardiac myocytes and subsequently augments β2AR-induced contractile response.
Fig. 7. Adenoviral gene transfer-mediated overexpression of RGS2 abolishes agonist-restored β2AR-Gi coupling in cultured adult mouse cardiomyocytes.
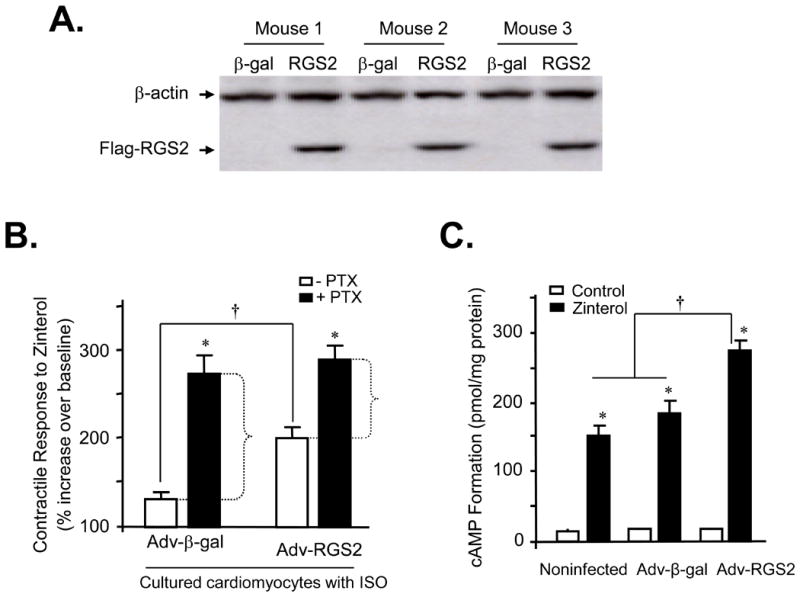
Panel A shows a typical western blot of RGS2 in cultured adult mouse cardiomyocytes infected by Adv-β-gal or Adv-RGS2 (both at m.o.i. 100 for 24 h). Panel B the average contractile response to the β2AR agonist, zinterol (5×10-7 M) in cultured adult mouse cardiomyocytes infected by Adv-β-gal or Adv-RGS2 (both at m.o.i. 100 for 24 h) (*P<0.01 vs. groups without PTX; †P<0.01 as indicated; n = 6-13 cells from 3 mouse hearts). Note that RGS2 overexpression led to augmented β2AR contractile response with reduced PTX-sensitive component, indicating that RSG2 overexpression suppresses ISO-restored β2AR-Gi signaling in cultured myocytes. Panel C Overexpression of RGS2 with adenoviral gene transfer increased β2AR-induced cAMP formation in cultured adult mouse cardiomyocytes infected by Adv-β-gal or Adv-RGS2 (both at m.o.i. 100 for 24 h). Cultured myocytes were stimulated with a β2AR agonist, zinterol (10 μM) in the presence of β1AR selective antagonist, CGP27013A (0.3 μM) and IBMX (1 mM) for 10 min (*P<0.01 vs. respective control for each group; †P<0.01 as indicated; n = 4-6 independent experiments).
3.4 RGS2 Ablation Sustains the b2AR-Gi Signaling in Cultured Cardiomyocytes in the Absence of Agonist Stimulation
To further define the role of RGS2 protein in regulating the β2AR-coupled Gi signaling, we took advantage of a gene-targeted RGS2 knockout (KO) mouse model[33]. Western blotting confirmed the absence of RGS2 protein in myocytes isolated from RGS2 KO mice (Fig. 8A). The most important difference between RGS2 KO and WT groups is that β2AR-mediated contractile response is sensitive to PTX treatment in cultured RGS2 KO cardiomyocytes but not in cultured WT cells (Fig. 8B), indicating that RGS2 ablation is able to retain the β2AR-Gi signaling even in the prolonged absence of βAR agonist stimulation. Thus, RGS2 deficiency leads to constitutive β2AR-Gi signaling which is independent of agonist stimulation, whereas upregulation of RGS2 suppresses the β2AR-mediated Gi signaling and subsequently augments β2AR-induced contractile response.
Fig. 8. RGS2 ablation retains the β2AR-Gi signaling in cultured mouse cardiomyocytes in the absence of agonist stimulation.
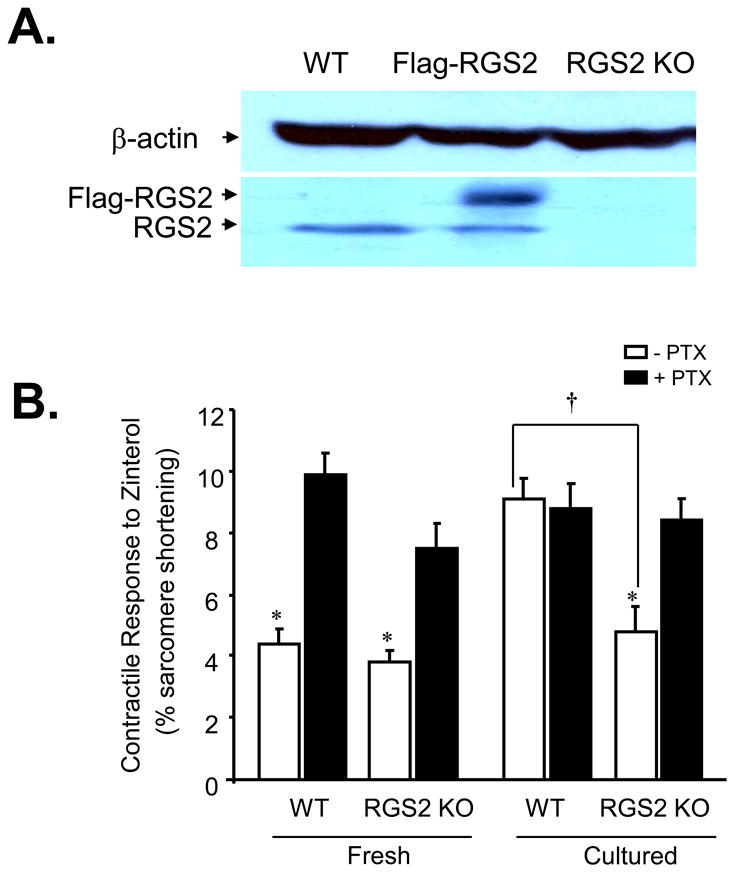
The cell contractile response to zinterol (5×10-7 M, a dose close to the EC50) in freshly isolated or cultured myocytes from WT or RGS2 KO mice (n = 10∼16 cells from 8 hearts for each data point; *, P<0.01 vs. all groups with PTX and cultured WT without PTX; †P< 0.05 as indicated). Note that in cultured cardiomyocytes, 24h-culture impaired the β2AR-Gi signaling in WT but not RGS2-deficient myocytes.
3.5. Neither the Expression Level of Gs and Gi Proteins Nor the Expression and Ligand Binding Properties of β1AR and β2AR Are Altered in Cultured Cardiomyocytes
In principle, prolonged absence of agonist stimulation-caused desensitization of the β2AR-Gi signaling could be attributable to a downregulation of Gi proteins, in addition to upregulation of RGS2. To test this possibility, we first examined the protein level of both Gs and PTX-sensitive G proteins (Gαo, Gαi1, Gαi2, Gαi3) in freshly isolated or cultured cells, and found no significant difference between the two groups (Fig. A1 of the online-only Data Supplement).
Table A2 (online-only Data Supplement) summarizes β1AR and β2AR densities and ligand binding properties assayed with a β1AR or a β2AR selective antagonist, CGP20712A or ICI 118,551, respectively. The density of total βARs, the β1AR and β2AR subpopulations, and their ratio in freshly isolated cardiomyocytes were similar to those of cultured cells. In addition, there was no significant difference between the two groups in β1AR or β2AR binding properties for a radioligand, [125I]-CYP. Thus, the failure of β2AR to activate Gi signaling in cultured myocytes is not caused by alterations in the expression of G proteins or β2AR density or ligand binding properties.
4. Discussion
4.1. RGS2 Constitutively Inhibits β2AR-mediated Gi Signaling in Cardiac Myocytes
Multiple RGS proteins, including RGS2-5, are expressed in the heart of mammalian species[18, 19]. In the present study, we have defined RGS2 as a powerful endogenous terminator of β2AR-mediated Gi signaling in the physiologically relevant setting, adult mouse cardiomyocytes. Upregulation of RGS2 caused by the lack of βAR agonist stimulation suppresses the β2AR-Gi signaling in cultured cardiomyocytes, as evidenced by the lack of PTX sensitivity of β2AR contractile response. Similarly, overexpression of RGS2 with adenoviral gene transfer markedly diminished agonist-restored PTX sensitivity of β2AR contractile response in cultured cardiomyocytes, and fully blocked β2AR-Gi-dependent activation of ERK1/2 in HEK293 cells. In contrast, RGS2 ablation is able to sustain the β2AR-Gi signaling even in the prolonged absence of βAR agonist stimulation in cells cultured for 24 h. To our knowledge, this is a previously unappreciated intrinsic cellular mechanism responsible for the termination of β2AR-activated Gi signaling in a physiologically relevant system, adult mouse cardiomyocytes.
Since RGS2 constitutively binds to β2AR[22], it is reasonable to assume that the β2AR-coupled Gi signaling is constitutively suppressed by RGS2 under physiological conditions, leading to apparent Gs-predominant β2AR signaling in cardiac myocytes of most mammalian species except mouse[2, 27]. The high efficiency of β2AR-mediated Gi signaling in mouse cardiomyocytes is likely attributed to the relatively low basal level of cardiac RGS2 expression in this particular species (Fig. 4A). While these present findings seem to be contradicting the previous notion that RGS2 selectively regulates the signaling of Gq but not Gi/o proteins in cultured cardiac myocytes or in the heart[19] and in vitro experimental systems[34-36], emerging evidence suggests that RGS2 regulates Gi/o-, but not Gq-, mediated signaling pathway in hippocampal neurons[37] and in cardiomyocytes derived from embryonic stem cells[38].
In addition to its GAP activity, RGS2 has been shown to inhibit adenylyl cyclase (AC) activity by directly binding to Gαs (without displaying GAP activity for this Gα subunit)[19] or AC[39]. But we have demonstrated that the upregulation of RGS2 in cultured cardiomyocytes has no detectable effect on β1AR-Gs-AC-mediated contractile response (Fig. 1C), suggesting RGS2 does not directly regulate AC activity in rodent cardiac myocytes, consistent with our previous notion[19].
4.2. Dynamic Regulation of RGS2 Expression by G Protein-mediated Signaling
Recent studies have shown that mRNA and protein levels of RGS2, but not RGS3-5, are increased in response to enhanced signaling of Gq and Gs (βAR or forskolin stimulation for 3 h) in adult rat ventricular cardiomyocytes[19] and osteoblasts[31, 39]. Interestingly, we have demonstrated, for the first time, that there is a biphasic regulation of RGS2 expression by enhanced Gs signaling induced by ISO or AC activation with forskolin. Short-term (2 h) treatment of cells with ISO or forskolin increases RGS2 protein level by 2-fold, whereas prolonged treatment (24 h) with the same stimuli significantly reduces RGS2 expression relative to untreated cultured myocytes (Fig. 5). Although the underlying mechanism of the biphasic regulation of RGS2 expression by G protein-mediated signaling remains to be explored, it might explain the opposing regulation of RGS2 expression by acute and chronic augmentation of Gq signaling. For instance, acute activation of Gq induced by α1AR stimulation increases RGS2 expression[19], whereas sustained enhancement of Gq signaling, as is the case in cardiac hypertrophy models, selectively suppresses RGS2 expression[40]. Taken together, the present and previous findings suggest that RGS2 is dynamically regulated by GPCR-mediated signaling, highlighting important physiological and pathological relevance of the fine tune of this particular RGS protein.
4.3. Receptor Activation-Dependent β2AR-Gi signaling Is Attributable to Downregulation of RGS2
In this study, we have shown that prolonged absence of agonist stimulation causes the loss of β2AR-activated Gi signaling, resulting in enhanced β2AR contractile response in cultured cardiomyocytes. Chronic agonist stimulation (ISO 1.0 nM included in the culture medium) rescues the Gi signaling. Consistent with the perception that phosphorylation of the receptor by PKA is required for the Gi signaling[14], these present data indicates that the β2AR-Gi signaling is dependent on the receptor activation. Most importantly, the present study has revealed, for the first time, that the switch of the receptor signaling from Gs to Gi pathway is mediated, at least in part, by βAR-induced downregulation of RGS2 protein (Figs. 5&6).
4.4. Potential Clinical Implications of Cardiac RGS2 Deregulation
The selective upregulation of RGS2 and the resultant augmentation of β2AR contractile response induced by the lack of βAR stimulation in cultured myocytes are intriguing given the fact that β-blocker therapy can resensitize βAR-mediated contractile support and improve cardiac function in patients with congestive heart failure. The beneficial effects of β-blockers might be mediated, in part, by increasing RGS2 expression. In contrast, adrenergic overdriving, as is the case in hypertension and cardiac hypertrophy in different animal models, is accompanied by a selective downregulation of RGS2[40-42]. Under those pathological circumstances, the reduction in RGS2 expression is expected to enhance Gq- and Gi-mediated signaling. In this regard, recent studies have shown that RGS2 gene silencing blocks α1AR-induced cardiac myocyte hypertrophy [19], and that pressure overload by trans-aortic constriction results in enhanced Gq signaling, exacerbated cardiac hypertrophy, heart failure and premature death in RGS2-deficient mice as compared to wild type counterparts[33], implying that RGS2 may play a central role in protecting the heart against stress-induced maladaptive remodeling.
In addition to the well-established Gq signaling, the current study has demonstrated that the lack of this RGS protein promotes β2AR-mediated Gi signaling, whereas overexpression of RGS2 suppresses the Gi signaling in cardiomyocytes. In the failing heart, both catecholamine levels and Gi expression are elevated, the Gi signaling is exaggerated[7-9], leading to defects of both β1AR- and β2AR-mediated positive inotropic effects[10-12]. The downregulation of RGS2 likely contributes to the enhancement of the β2AR-coupled Gi signaling, which, in turn, may accelerate the development of hypertension and cardiac hypertrophy, eventually resulting in heart failure.
5. Conclusion
In the present study, we have demonstrated, for the first time, that RGS2 confers a powerful negative regulation of β2AR-activated Gi signaling in mammalian cardiac myocytes. Deregulation or malfunction of RGS2 may constitute a pathogenic element for the development of heart failure in addition to its known role in the pathogenesis of hypertension. Thus, the current findings not only define RGS2 as a novel cellular mechanism responsible for the termination of β2AR-mediated Gi signaling, but also bear important pathogenic and therapeutic implications.
Appendix A
Supplementary data associated with this article can be found in the online version.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Institute on Aging (RP Xiao, W Zhu, S Tsang, AYH Woo, D Yang, X Wang, X Zeng, EG Lakatta) and, in part, by NIH extramural grants to DA Kass, U Mende, and KJ Blumer.
Abbreviations
- AC
adenylyl cyclase
- Adv
adenovirus
- GPCR
G protein-coupled receptor
- βAR
β-adrenergic receptor
- WT
wild type
- ISO
isoproterenol
- CGP
CGP20712A
- ICI
ICI 118,551
- Tpeak
the time from stimulation to peak shortening
- T50
the time from the peak to 50% relaxation
- [125I]-CYP
[125I]- iodocyanopindolol
- m.o.i
multiplicity of infection
- PKA
protein kinase A
- PKI
a peptide inhibitor of PKA
- RGS
regulator of G-protein signaling
- PTX
pertussis toxin
- GAP
GTPase-activating protein
- ERK
Extracellular signal-regulated kinases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None.
References
- 1.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 2.Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, et al. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–7. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol. 1995;47:322–9. [PubMed] [Google Scholar]
- 4.Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, et al. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 5.Kilts JD, Gerhardt MA, Richardson MD, Sreeram G, Mackensen GB, Grocott HP, et al. Beta(2)-adrenergic and several other G protein-coupled receptors in human atrial membranes activate both G(s) and G(i) Circ Res. 2000;87:705–9. doi: 10.1161/01.res.87.8.705. [DOI] [PubMed] [Google Scholar]
- 6.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A. 2001;98:1607–12. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, et al. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)--but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–9. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
- 8.Kiuchi K, Shannon RP, Komamura K, Cohen DJ, Bianchi C, Homcy CJ, et al. Myocardial beta-adrenergic receptor function during the development of pacing-induced heart failure. J Clin Invest. 1993;91:907–14. doi: 10.1172/JCI116312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohm M, Eschenhagen T, Gierschik P, Larisch K, Lensche H, Mende U, et al. Radioimmunochemical quantification of Gi alpha in right and left ventricles from patients with ischaemic and dilated cardiomyopathy and predominant left ventricular failure. J Mol Cell Cardiol. 1994;26:133–49. doi: 10.1006/jmcc.1994.1017. [DOI] [PubMed] [Google Scholar]
- 10.Sato M, Gong H, Terracciano CM, Ranu H, Harding SE. Loss of beta-adrenoceptor response in myocytes overexpressing the Na+/Ca(2+)-exchanger. J Mol Cell Cardiol. 2004;36:43–8. doi: 10.1016/j.yjmcc.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 11.He JQ, Balijepalli RC, Haworth RA, Kamp TJ. Crosstalk of beta-adrenergic receptor subtypes through Gi blunts beta-adrenergic stimulation of L-type Ca2+ channels in canine heart failure. Circ Res. 2005;97:566–73. doi: 10.1161/01.RES.0000181160.31851.05. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W, Zeng X, Zheng M, Xiao RP. The enigma of beta2-adrenergic receptor Gi signaling in the heart: the good, the bad, and the ugly. Circ Res. 2005;97:507–9. doi: 10.1161/01.RES.0000184615.56822.bd. [DOI] [PubMed] [Google Scholar]
- 13.Ahmet I, Krawczyk M, Zhu W, Woo AY, Morrell C, Poosala S, et al. Cardioprotective and survival benefits of long-term combined therapy with beta2 adrenoreceptor (AR) agonist and beta1 AR blocker in dilated cardiomyopathy postmyocardial infarction. J Pharmacol Exp Ther. 2008;325:491–9. doi: 10.1124/jpet.107.135335. [DOI] [PubMed] [Google Scholar]
- 14.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 15.Tepe NM, Liggett SB. Functional receptor coupling to Gi is a mechanism of agonist-promoted desensitization of the beta2-adrenergic receptor. J Recept Signal Transduct Res. 2000;20:75–85. doi: 10.3109/10799890009150038. [DOI] [PubMed] [Google Scholar]
- 16.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 17.Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–59. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 18.Riddle EL, Schwartzman RA, Bond M, Insel PA. Multi-tasking RGS proteins in the heart: the next therapeutic target? Circ Res. 2005;96:401–11. doi: 10.1161/01.RES.0000158287.49872.4e. [DOI] [PubMed] [Google Scholar]
- 19.Hao J, Michalek C, Zhang W, Zhu M, Xu X, Mende U. Regulation of cardiomyocyte signaling by RGS proteins: differential selectivity towards G proteins and susceptibility to regulation. J Mol Cell Cardiol. 2006;41:51–61. doi: 10.1016/j.yjmcc.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Zou MX, Roy AA, Zhao Q, Kirshenbaum LA, Karmazyn M, Chidiac P. RGS2 is upregulated by and attenuates the hypertrophic effect of alpha1-adrenergic activation in cultured ventricular myocytes. Cell Signal. 2006;18:1655–63. doi: 10.1016/j.cellsig.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Wieland T, Lutz S, Chidiac P. Regulators of G protein signalling: a spotlight on emerging functions in the cardiovascular system. Curr Opin Pharmacol. 2007;7:201–7. doi: 10.1016/j.coph.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Roy AA, Lemberg KE, Chidiac P. Recruitment of RGS2 and RGS4 to the plasma membrane by G proteins and receptors reflects functional interactions. Mol Pharmacol. 2003;64:587–93. doi: 10.1124/mol.64.3.587. [DOI] [PubMed] [Google Scholar]
- 23.Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, et al. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem. 2004;279:21248–56. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- 24.Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR. Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem. 2005;280:27289–95. doi: 10.1074/jbc.M502365200. [DOI] [PubMed] [Google Scholar]
- 25.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, et al. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–36. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 26.Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, et al. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation. 2009;119:1231–40. doi: 10.1161/CIRCULATIONAHA.108.774752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in cardiac beta(2)-adrenergic signal transduction. Circ Res. 1999;85:1092–100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 28.Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86:445–52. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 29.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature. 1996;383:172–5. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 30.Grant SL, Lassegue B, Griendling KK, Ushio-Fukai M, Lyons PR, Alexander RW. Specific regulation of RGS2 messenger RNA by angiotensin II in cultured vascular smooth muscle cells. Mol Pharmacol. 2000;57:460–7. doi: 10.1124/mol.57.3.460. [DOI] [PubMed] [Google Scholar]
- 31.Miles RR, Sluka JP, Santerre RF, Hale LV, Bloem L, Boguslawski G, et al. Dynamic regulation of RGS2 in bone: potential new insights into parathyroid hormone signaling mechanisms. Endocrinology. 2000;141:28–36. doi: 10.1210/endo.141.1.7229. [DOI] [PubMed] [Google Scholar]
- 32.Tsingotjidou A, Nervina JM, Pham L, Bezouglaia O, Tetradis S. Parathyroid hormone induces RGS-2 expression by a cyclic adenosine 3′,5′-monophosphate-mediated pathway in primary neonatal murine osteoblasts. Bone. 2002;30:677–84. doi: 10.1016/s8756-3282(02)00698-1. [DOI] [PubMed] [Google Scholar]
- 33.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T, et al. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest. 2009;119:408–20. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci U S A. 1997;94:14389–93. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, et al. G protein selectivity is a determinant of RGS2 function. J Biol Chem. 1999;274:34253–9. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- 36.Doupnik CA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 1997;94:10461–6. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han J, Mark MD, Li X, Xie M, Waka S, Rettig J, et al. RGS2 determines short-term synaptic plasticity in hippocampal neurons by regulating Gi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron. 2006;51:575–86. doi: 10.1016/j.neuron.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Fu Y, Huang X, Zhong H, Mortensen RM, D'Alecy LG, Neubig RR. Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ Res. 2006;98:659–66. doi: 10.1161/01.RES.0000207497.50477.60. [DOI] [PubMed] [Google Scholar]
- 39.Ko JK, Choi KH, Kim IS, Jung EK, Park DH. Inducible RGS2 is a cross-talk regulator for parathyroid hormone signaling in rat osteoblast-like UMR106 cells. Biochem Biophys Res Commun. 2001;287:1025–33. doi: 10.1006/bbrc.2001.5692. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, et al. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J Biol Chem. 2006;281:5811–20. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Adams LD, Pabon LM, Mahoney WM, Jr, Beaudry D, Gunaje J, et al. RGS5, RGS4, and RGS2 expression and aortic contractibility are dynamically co-regulated during aortic banding-induced hypertrophy. J Mol Cell Cardiol. 2008;44:539–50. doi: 10.1016/j.yjmcc.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 42.Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J Clin Invest. 2003;111:445–52. doi: 10.1172/JCI15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.