

Abstract
We have been studying chaperonins these past twenty years through an initial discovery of an action in protein folding, analysis of structure, and elucidation of mechanism. Some of the highlights of these studies were presented recently upon sharing the honor of the 2013 Herbert Tabor Award with my early collaborator, Ulrich Hartl, at the annual meeting of the American Society for Biochemistry and Molecular Biology in Boston. Here, some of the major findings are recounted, particularly recognizing my collaborators, describing how I met them and how our great times together propelled our thinking and experiments.
Keywords: Protein Folding, Chaperone Chaperonin, Molecular Chaperone, Yeast, Protein Misfolding, Polypeptide
How I Came to Study a Machine That Assists Protein Folding
My pathway to studying a protein folding machine could not have been predicted from the trajectory of my training. I finished a six-year medical program at Brown University in 1975 and then trained in pediatrics at Yale University. During residency, I became fascinated with cell transformation and, in 1978, went off to the Salk Institute to train in tumor virology with Walter Eckhart and Tony Hunter. I participated in early recombinant DNA-mediated expression of tumor virus-transforming proteins and watched Tony discover tyrosine phosphorylation (1). In 1981, with the cloning of human coding sequences becoming increasingly possible, I returned to Yale to pursue genetics training with Leon Rosenberg. In particular, I cloned the cDNA for a nuclear-coded urea cycle enzyme, ornithine transcarbamylase (OTC), which is involved in an X-linked deficiency that results in lethal ammonia intoxication in newborn male infants. It is a devastating clinical situation I indelibly observed during pediatric training. With the cloned cDNA in hand (2), we were able to develop DNA diagnostic approaches to help affected families by providing prenatal diagnosis of the condition, but the cloned cDNA also allowed us to see the cleavable N-terminal mitochondrial targeting sequence in OTC. We soon showed that the targeting sequence contained sufficient information to direct mitochondrial localization because, when we fused it to the cytosolic protein dihydrofolate reductase (DHFR), it now directed DHFR into mitochondria (3). I found these studies of mitochondrial trafficking to be of fundamental interest. When I moved across the hall as an independent investigator in 1984, I wanted to isolate the “machinery” of the mitochondria themselves that was involved with protein import.
Discovery of Chaperonin Action in Mediating Protein Folding
To develop a screen for import machinery, we first tested whether expression of the human OTC cDNA in yeast would lead to its proper targeting, signal cleavage, and maturation into active enzyme in the mitochondrial matrix. Happily, we observed this to be the case (4), allowing us to use OTC as a reporter protein. We produced a bank of temperature-sensitive lethal yeast mutants containing OTC as an inducible reporter protein. The idea was that at the nonpermissive temperature (37 °C), if proteins failed to be imported into mitochondria and no new mitochondria could be produced, cell growth would halt. For the human OTC reporter protein, we could turn on its expression after a shift from 23 °C to 37 °C using a Gal operon promoter (yeast cells lack a mitochondrial OTC enzyme, and we deleted the gene for their cytosolic OTC). The idea was that if OTC protein was made but no enzyme activity was detected in one or more of the temperature-sensitive lethal mutants after temperature shift, this would indicate that mitochondrial import was blocked. This could occur at any number of possible points, e.g. recognition of the newly translated precursor protein at the mitochondrial outer membrane, translocation through the membranes, or maturational cleavage of the N-terminal signal peptide. We began screening our temperature-sensitive lethal mutants and soon identified ones with mutations that affected the cleavage of the signal peptide by a matrix-localized peptidase. Then one night, after a long day of screening, it dawned on Ming Cheng (one of my first graduate students) and me that a very interesting type of mutant might be present in our bank, one that would affect the folding of the newly imported OTC to its native form. After all, there were studies from Gottfried Schatz's group in Basel showing that imported proteins had to be completely unfolded to pass through the mitochondrial membranes (5). Also, there was increasing evidence, much of it coming from Hugh Pelham's group at the MRC Laboratory of Molecular Biology, that there was a class of specialized proteins known as heat shock proteins (induced by thermal exposure of cells) that could adjust the conformation of other proteins, preventing them from aggregating with each other under stress conditions (6). Could there be such a thing as a machine that supported de novo protein folding of imported proteins? Here, in our mitochondrial system, we were in a position to address this question.
Within a week, Ming found such a mutant in our library; in particular, OTC was imported and its signal peptide was cleaved, but there was no detectable OTC enzyme activity (7). We were stunned. People had always believed that mitochondrial proteins spontaneously refolded into their native form after import through the membranes. After all, the work of Christian Anfinsen at the National Institutes of Health had shown that the primary structure of a polypeptide chain contains all of the information required to reach the native state (8), but even Anfinsen had pondered the observation that not all proteins could refold in vitro after dilution from denaturant (9), and many proteins expressed in bacteria failed to properly fold, lodging in inclusion bodies. It had seemed as if something more was needed in some situations providing, for example, kinetic assistance.
We next tested an endogenous yeast mitochondrial matrix protein, the β-subunit of the F1-ATPase. We found that at the nonpermissive temperature, it also failed to reach its destination in the stalk structure that faces the mitochondrial matrix (Fig. 1). The protein was present, but it appeared to be lodged in an insoluble fraction, suggesting that it had aggregated (7). Now, we were really excited but also uncertain how to proceed to further prove that this yeast mutant failed to fold newly imported proteins.
FIGURE 1.
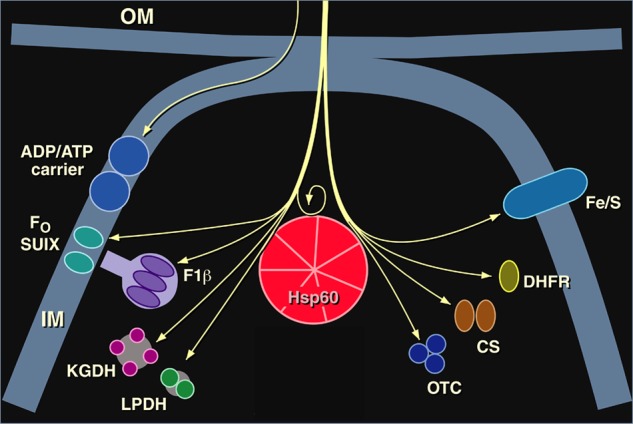
Imported mitochondrial proteins whose folding to the native form in the matrix compartment was deficient in our Hsp60-deficient yeast mutant. Ketoglutarate dehydrogenase (KGDH) and lipoamide dehydrogenase (LPDH) were observed to be affected in Ref. 39. Pre-existent functional Hsp60 was observed to be required for proper folding and subsequent assembly of newly imported Hsp60 in Ref. 40: no new Hsp60 complex could be produced in the mutant yeast after temperature shift. OM, outer membrane; IM, inner membrane; Fo SUIX, subunit 9 of F0-ATPase; F1β, F1-ATPase β-subunit; CS, citrate synthase. This figure was modified from Ref. 45.
Amazingly, at this point, the phone rang, and it was Walter Neupert and Ulrich Hartl calling from the University of Munich to ask if we could use a little biochemical help to study our yeast mitochondrial protein import mutants. The answer was unequivocally yes, as soon as possible. I went immediately to Munich and presented a seminar about our mutants. At the end of my talk, I mentioned the mutant that we thought might have a defect in protein folding. Ulrich and Walter were quite surprised to hear about this phenotype. They were a little worried that we might be studying a translocation defect, i.e. the imported protein could be jammed in the translocation channel. This would allow the N terminus to be cleaved in the matrix, but the mature portion of the protein would be lodged in the import site and unable to fold properly (see Fig. 1). This possibility seemed to be easily addressed by isolating mitochondria from the mutant yeast and applying an exogenous protease to an import reaction. I returned home and mailed off the yeast mutant. Two weeks later, Ulrich called excitedly to report that imported proteins were indeed localized within the matrix, fully protected from the exogenously added protease, apparently failing to refold in the matrix. This was both tremendously exciting and comforting. Now, working together, our labs could employ the wealth of anti-mitochondrial protein antibody reagents in the Neupert lab, as well as their biochemical expertise with isolated organelles, to study other imported proteins, including monomeric ones. We soon observed that the monomeric Rieske iron-sulfur protein failed to undergo normal biogenesis (Fig. 1) (7). This lent enormous strength to the contention that we were studying the nascent folding of newly imported proteins in the mitochondrial matrix, as opposed to oligomeric assembly of already folded proteins. In additional studies with Joachim Ostermann, Ulrich showed that newly imported monomeric DHFR, sent into the mitochondria via an attached targeting peptide, became associated with a large complex, occupying a protease-susceptible, apparently non-native state in this complex in the absence of ATP. However, upon addition of ATP, DHFR was released and became native-like in its protease resistance (Fig. 1) (10). These latter observations further indicated that the yeast mutant was affecting folding of imported proteins as opposed to oligomeric assembly.
We rescued the mutant cells with a yeast library and found that the gene that was able to rescue the folding-defective phenotype encoded an abundant mitochondrial matrix protein. This protein, first identified in Tetrahymena thermophila by Richard Hallberg's group at Iowa State University, was induced by ∼2-fold by heat shock and found to be present in a double-ring assembly (11). Hallberg had been studying the yeast homolog, so we called him up and found that our rescuing sequence was a dead match to his. Thus, much to our delight, a double-ring assembly in the mitochondrial matrix, which we collectively dubbed Hsp60 (but actually is essential for cell growth at all temperatures), appeared to mediate protein folding to the native state in that compartment (Fig. 1). There also was the indication from this conclusion that other similar double-ring assemblies were likely to mediate folding of proteins in their cellular compartments as well, because their subunits bore substantial sequence identity to Hsp60. One of these assemblies was GroEL in the bacterial cytoplasm, which had been previously implicated in phage assembly (e.g. Ref. 12), and another was a complex inside chloroplasts, which had been previously implicated in ribulose-bisphosphate carboxylase/oxygenase (Rubisco) assembly (13).
With such an understanding in hand, Ulrich (Fig. 2) and I had enormous fun thinking further about how the double-ring chaperonin machines could assist folding. Our conversations took place everywhere: the Munich airport while we waited for me to catch a plane and tried to deduce whether the two major domains of the subunits in the rings were arranged “ABAB” versus “ABBA”; my kitchen in Connecticut, where we mused about how the central cavity lining the rings might operate; and on the Merritt Parkway in Connecticut, where we conjectured that maybe Hsp70 class proteins could “hand off” nascent unfolded chains to the chaperonin cavity. It was a time of intense ferment.
FIGURE 2.

Left, Ulrich Hartl in 1989, during a visit to his parents' home in the Black Forest. Food, wine, and ideas flowed freely. Middle, Ming Cheng in 1989. Right, Ming's impenetrable bench in the lab in 1989 (upper) and Ming's similarly occupied kitchen counter in Taipei in 2012 (lower).
Perhaps the most fun of all was had by Ming Cheng (Fig. 2), who had miraculously pulled out the Hsp60 mutant from our library. Ming was a young physician from Taiwan who wanted to gain basic molecular biology training and so joined the Yale Genetics Graduate Program. She was, in two words, experimentally fearless. Her bench reflected this attitude, with the most dense collection of yeast reagents I have ever seen (Fig. 2). There seemed to be simply no room in which to carry out an experimental manipulation, but she was deft. During a recent visit to Taiwan, I observed that her kitchen is no different from the laboratory, with every counter space fully occupied (Fig. 2). Regardless, there were many stories concerning Ming's various outside activities. Driver education was one feature. One morning, we learned that she and her car had done battle with a bush next to her garage and defeated it, but Ming ultimately became a very good driver and also a good skier, regularly participating in what were termed “von Horwich” ski trips to Vermont. She has subsequently pursued both research and teaching at the National Yang-Ming Medical College in Taiwan.
Shortly after our mitochondrial experiments, George Lorimer and his colleagues at DuPont reconstituted a chaperonin reaction in vitro using the bacterial homolog GroEL and a “lid”-like co-chaperonin, a seven-member ring assembly called GroES, whose coding sequence is in an operon with GroEL (14). In a first step, a dimeric form of Rubisco, unfolded in denaturant, was diluted into a mixture with GroEL. One Rubisco monomer formed a stoichiometric complex with one GroEL tetradecamer, in which Rubisco was inactive. In contrast, if Rubisco was diluted from denaturant into buffer without any GroEL present, it underwent wholesale aggregation. In a second step, upon addition of the GroES lid and ATP to the Rubisco-GroEL complex, Rubisco became properly folded over a period of a few minutes and subsequently dimerized to its native active form. Ulrich and our collective soon carried out similar reconstitution studies using GroEL-GroES-ATP to refold monomeric rhodanese and DHFR (15).
Structural Analyses
By 1992, there were almost as many models for how GroEL-GroES might be working as there were investigators in the field. We were busy localizing bound protein to the central cavity of the GroEL ring (16, 17), but we were unsure how to proceed to a more general understanding of the reaction mechanism. This was changed by an administrative mission that took me across the Yale campus to chat with Paul Sigler, a newly arrived crystallographer from the University of Chicago (Fig. 3). Paul immediately brushed the administrative matter aside and began to discuss the chaperonins, marveling at their symmetries (two back-to-back 7-fold radially symmetric rings) and enjoining us that we would never figure out how the chaperonins work without an x-ray structure. The collaboration was born at that minute. He brought Zbyszek Otwinowski into the room (Fig. 3), who just shook his head at the computational problem of dealing with an 800-kDa assembly. Zbyszek reminded us that it would take us a few years to crystallize the molecular machine and that the computing capability would be ready by then to deal with it. He was right! Once we produced two data sets, one of the native complex and another with an ethyl mercury derivative, Zbyszek astonishingly solved the structure in a single day in the fall of 1993 using a search program he had devised to sort through the large number of heavy atoms (18). As added testimony to Zbyszek's brilliance and daring, when we initially went to collect native data at the Cornell High Energy Synchrotron Source (CHESS) beamline for the first time in the summer of 1993, he wondered if we could collect a few reflections at very low resolution from which he might be able to phase the structure. We tried moving the detector way back and collecting a reflection or two at 150 Å. Obviously, this did not succeed, but it was surely fun to try.
FIGURE 3.

Left, Andrzej Joachimiak (left), Paul Sigler (middle), and Zbyszek Otwinowski (right) in 1989, inspecting a crystallographic model of the trp repressor-operator complex, a structure they had recently solved. Middle, Kerstin Braig generated the first well diffracting crystals of GroEL in 1993. Right, David Boisvert, here in 1995, provided major impetus to the crystallization work. He worked on a monoclinic crystal form of GroEL and solved the structure of GroEL-ATPγS (work that was carried out with help from Jimin Wang). Note that many members of the Sigler lab, including Dan Gewirth, Greg Van Duyne, and Rashmi Hegde, also contributed to the GroEL structure determination, helping particularly with data collection and refinement. Refinement efforts ultimately relied heavily on the group of Axel Brunger (e.g. Ref. 41).
Andrzej Joachimiak from Paul's lab (Fig. 3) was a wonderful companion and collaborator during the period of generating GroEL protein and testing crystallizations. The two of us would converse about expression and purification of GroEL every day, our collaboration accelerated to white heat by the proximity of our group and the Sigler group next to each other in the newly built Boyer Center for Molecular Medicine at the Yale School of Medicine in late 1991. Ultimately, we produced GroEL to the level of 95% of total bacterial protein. I remember the look on Andrzej's face when I showed him a gel with a massive Coomassie blotch at the position of GroEL. I said, “I have a gram of this stuff, Andrzej, from 2 liters.” “This is amazing,” was his response. It was enormous fun some months later to drive up to CHESS with Andrzej for the first synchrotron data collection. I remember walking down the hill to grab breakfast after an overnight fire drill with putting up image plates, shooting x-rays, and then fetching the plates and putting them into the Fuji scanner at the F1 beamline (this was before charge-coupled device cameras), thinking, “This is really going to change our understanding of this machine.”
Equally good times have subsequently been had visiting Andrzej at the ID19 beamline at the Argonne National Laboratory, trying any and all means to see GroEL-bound polypeptide by crystallography. However, we did not have much success. These were trips made with George Farr, who came as a postdoctoral fellow to my laboratory in the mid-1990s but stayed for fifteen years, enjoying the biochemistry and structural aspects of the chaperonin system. I can only say that as we have migrated toward cell biology and physiology of neurodegeneration, I have missed going to the synchrotron, but I continue to enjoy Andrzej's Christmas cards, loaded with images of newly solved structures.
As for Paul, he was like a father to me for a period of ten years. We had the Chicago scene in common (I grew up there), the Bulls, the Cubs, the Ohio Street pizza places, but we also shared a love of jazz and an early training in medicine. Like me, Paul had been medically trained and had been an intern with my postdoctoral genetics mentor, Leon Rosenberg. There was a memorable account of his taking care of Marilyn Monroe as a patient: no further comment needed. Anyway, he osmotically taught me crystallography, showing all aspects of the process when we were next-door neighbors in the Boyer Center. It was wonderful to hear the stories about the MRC Laboratory of Molecular Biology in the 1960s, where Paul had trained with David Blow, working on chymotrypsin, in the circle of Max Perutz, Sydney Brenner, and many others. Many of these stories were shared in our lunchroom, where Paul, an avid fan of Slim-Fast as the solution to good health, would rant at the messy state of the kitchen as he prepared the next dose of the reagent. I can attest that it remains, even ten years after Paul's passing, a constant challenge to keep the lab kitchen workable. It was a magical time, where ideas flowed freely between our two labs on the mechanism of GroEL-GroES, and we watched the Sigler group reveal the beauties of heterotrimeric G proteins, NF-κB, phospholipases, and other structures. It was inspiring.
Kerstin Braig was the true artist through all of this (Fig. 3). She produced the first well diffracting crystals of GroEL. She was a visiting student from Berlin, supposedly for one year, but she stayed for five years. Her first study visualized a gold particle-labeled substrate polypeptide in the GroEL cavity by scanning transmission electron microscopy at the Brookhaven National Laboratory (16), in collaboration with Joe Wall and Jim Hainfeld. She then took on the crystallization and structure determination of GroEL. In her outside life, Kerstin required a strong artistic involvement, such as sculpture classes, and many other outside activities, probably to maintain her sanity during the difficult task of crystallization trials, which were a manual undertaking at that point of history. Perhaps her boldest exploit was a weekend jaunt to Mongolia. She spent one day flying and two days on the ground enjoying the local scene and then flew back to have a look at her new crystals. Indeed, when she pulled the initial well diffracting ammonium sulfate crystal, it emerged from a literal wall of trays stacked to massive heights side-by-side at room temperature on shelves above her bench. Kerstin was helped considerably by another student, David Boisvert, who also brought a lot of personality to the lab, as well as experience in crystallization. David had assisted in crystallization studies of reverse transcriptase in Tom Steitz's laboratory at Yale before matriculating in the graduate school (Fig. 3). David brought with him a wealth of expertise and ultimately crystallized GroEL in complex with adenosine 5′-O-(3-thiotriphosphate) (ATPγS) (19).
These wonderful people and many others, including Axel Brunger, contributed as a team to the crystallization and refinement efforts. With a structure of GroEL, we saw a central cavity that is 45 Å in diameter (Fig. 4, left). This cavity is blocked at the waistline of the cylinder by disordered C-terminal tails of each subunit (25 residues each, amounting to ∼20 kDa per ring), visible as collective masses by EM but not visible by crystallography. Thus, GroEL contains a central cavity at either end of the cylinder, large enough to house a polypeptide of 30–40 kDa. Of course, in the absence of a bound GroES, larger proteins can be both bound in the cavity and partly present in the bulk solution, bound as if a champagne cork (20). Mutational studies soon revealed that the polypeptide-binding site is present on the cavity surface of the terminal apical domains of the subunits, presenting hydrophobic residues to the solvent (Fig. 4, right, bottom ring) (21). Non-native proteins, presenting their own hydrophobic surfaces, which would be buried in the interior in the native state, are bound on this surface. This binding prevents them from forming associations that would lead to aggregation. Later studies, both genetic and EM, have also shown that such binding is multivalent in character, with a polypeptide bound to three or four consecutive apical domains (22, 23). The polypeptide occupies an unstructured state while bound (24); indeed, fluorescence studies have indicated that a loosely folded but kinetically trapped state can be “pulled apart” upon binding to the apical domains (25).
FIGURE 4.

Cutaway images of models of GroEL alone (left) and complexed with nucleotide and GroES (middle and right). The left and middle images are Cα traces of the machine, with one of the seven subunits of GroEL colored green and the subunits of GroES colored white. In the image of GroEL alone (left), the equatorial domains are visible, forming the waistline of the cylinder, making slender covalent connections via the intermediate domains to the terminal apical domains, at either end of the cylinder. Note that the central cavity is blocked at the equatorial level of both rings by the collective of disordered C-terminal tails. The hydrophobic polypeptide-binding surface at the inside aspect of the apical domains of an open ring is colored yellow in the space filling model (right, bottom ring). When nucleotide and GroES bind, the intermediate and apical domains undergo rigid body movements that involve a downward rotation of the intermediate domain that locks the nucleotide into the bound ring (middle, red and blue sphere) and involves overall elevation and clockwise twisting movements of the apical domains that remove their hydrophobic surfaces from facing the cavity, ejecting the initially bound polypeptide into it, whereupon folding commences. Note that the ejected polypeptide resides in a hydrophilic chamber, denoted by blue in the cavity of the top ring (right). This figure is modified from Ref. 33.
The apical domains of GroEL are connected to a “base” composed of α-helical equatorial domains that make tight contacts both going around a ring and across the ring-ring interface (Fig. 4). Each of these domains exhibits a pocket that could house an ATP molecule (Fig. 4, middle, top ring). David Boisvert soon co-crystallized GroEL-ATPγS, observing the stereochemistry of nucleotide binding in the pocket (19).
Mechanism Studies and Structures of GroES-bound GroEL
How does folding ensue in the GroEL system? Early EM studies had shown that the lid-shaped GroES co-chaperonin can bind to GroEL in the presence of ATP to form asymmetric complexes (Fig. 4, middle and right) (26), but does the polypeptide fold inside these complexes or only outside in solution? When we looked at the native structure of one of our favorite substrate proteins, rhodanese, even in its native state, it clashed with the GroEL apical domains when imposed on the unliganded GroEL x-ray model. It seemed that folding might have to occur in solution, but our EM collaborator, Helen Saibil at Birkbeck College in London, changed our thinking about that about a month later with beautiful new images in both negative stain and cryo-EM (27). Helen observed that when GroES bound to GroEL, it opened up the apical domains of the chaperonin ring to which it bound, producing an enlarged cavity underneath bound GroES (Fig. 4, middle). These images stunned us and reversed our thinking.
Here, I would like to recognize Helen more fully (Fig. 5). A bedrock collaboration with her commenced in 1991. We were both participating in a Royal Society meeting in London, and after a long day's session, we began to chat about how things could be working. She had already published an image of an asymmetric GroEL-GroES complex (26), and we had just presented the gold-labeled DHFR positioned in the central “hole” of GroEL (16). We enjoyed the discussion and each other's company greatly. It was really a beginning of an effort that got stronger and stronger, aimed at capturing states in solution, many of which could not be accessed in crystals. Most recently, this has provided a trajectory of ATP-directed movements of the subunits of a GroEL ring (28). Helen's outside interests, such as visiting all corners of the world, have taken on a fascination of their own. The arrival of a photo by E-mail from Ghana of the skeleton of a giant tortoise in the museum in Accra could only be a product of Helen's (Fig. 5), and I surely do not know anyone else who would take a vacation in Uzbekistan and attend, as a passerby, a local wedding there!
FIGURE 5.

Upper row, left to right, Helen Saibil, our long-time EM collaborator, pictured at home in London in her backyard reading area on a surprisingly sunny day; image of a tortoise skeleton (not a lab member) from one of Helen's travels to Ghana; Jonathan Weissman, circa 1995; Wayne Fenton, timeless; and Krystyna Furtak, who has been with me from day one and produced approximately one-hundred GroEL mutant constructs. Lower row, left to right: Hays Rye, who fluorescently labeled the reaction components and watched them come and go in real time; Zhaohui Xu, who crystallized GroEL-GroES-ADP; Eric Bertelsen, unafraid to design and produce the molecules enabling NMR approaches to the machinery; and Kurt Wüthrich, our long-time NMR collaborator and originator of TROSY techniques for studying large proteins.
A key mechanistic experiment followed Helen's observations of a cavity underneath bound GroES. Jonathan Weissman carried out an order-of-addition proteolysis experiment that showed that a polypeptide, bound first to a ring that then became bound by GroES (i.e. in cis), was protected from exogenously added protease. However, a polypeptide bound to the open GroEL ring opposite the one bound by GroES (i.e. in trans) was fully degraded upon addition of protease (29). The topologies were further supported by hit-and-run radioiodine photocross-linking studies. I should say that Jonathan was one of the most enjoyable collaborators to come into the lab as a postdoctoral fellow (Fig. 5). We had a more-or-less continuous dialogue every day concerning the chaperonin mechanism and had wonderful times together. Often, we would be chatting over his terrarium, housing one or more pet chameleons, which were more finicky eaters than any children I have encountered. They required a steady diet of crickets, which had to be correctly aged, fed, and placed at the right place in the tank to be consumed. Jonathan hovered over both the chaperonins and these creatures, probably more successfully with the former.
One of our greatest discussions, which really possessed the whole lab, was whether polypeptides could leave GroEL during its reaction cycle in a non-native state. Was the reaction committed or not? Jonathan felt that it was not. Wayne Fenton, commented on below, felt that it might be. I was neutral. I just wanted to add a “trap” version of GroEL to an ongoing reaction and see what it did to it. If polypeptides were leaving in a non-native state, then the trap would halt the further production of the native state. Indeed, we already had such GroEL mutants from a structure-function analysis that Wayne and I had been conducting. These mutants could bind non-native protein but not release it, even in the presence of ATP and GroES (21). Jonathan carried out the experiment, and we observed immediate cessation of folding. In chromatography studies, we observed the rapid transfer of the polypeptide to the trap molecules (30). Thus, the chaperonin reaction cycle is all-or-none (Fig. 6): the polypeptide either reaches the native state during a round at the machine or is discharged in a non-native state that has to be rebound for another trial for folding to the native form.
FIGURE 6.
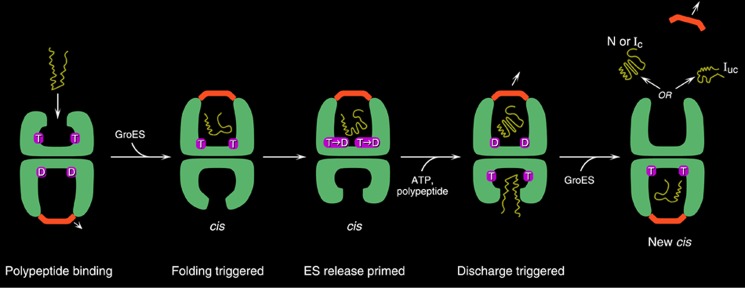
GroEL-GroES reaction cycle. An open ring becomes rapidly occupied with ATP in the seven equatorial sites, with binding within a ring occurring cooperatively and between rings with negative cooperativity (42). This dictates an inherent asymmetry to the machine, as GroES can bind only to ATP-mobilized apical domains. Following rapid ATP binding, the polypeptide arrives, coming on at about one-tenth the rate of ATP over several hundred milliseconds (first panel) (43). Subsequently, GroES collides with the ATP-mobilized apical domains, forming a ternary collision complex in which both polypeptide and GroES are simultaneously bound to the apical domains, preventing any chance of polypeptide escape. Next, large and forceful rigid body elevation and twisting of the apical domains lead to release of the polypeptide into the encapsulated chamber, where the chain commences folding (second panel). This is the longest step of the reaction cycle, ∼10 s, following which ATP hydrolysis (third panel) weakens the affinity for GroES, and subsequent binding of ATP in the opposite ring sends an allosteric signal that ejects GroES, polypeptide, and hydrolyzed ADP from what had been the folding-active ring (fourth panel) (44). At the same time, the opposite ring is now set up to become the folding-active ring (fifth panel). T, ATP; D, ADP; N, natively folded protein; Ic, intermediate committed to the native state; Iuc, intermediate not committed to the native state. This figure is from Ref. 45.
In relation to these studies and many more, including a direct next test of whether “cis” complexes were productive compared with trans ones, my longest collaboration, extending more than thirty years, is with Wayne Fenton, a marvelous and indefatigable investigator, whose Santa Claus appearance is matched by a personality that resembles that icon if he were accessible (Fig. 5). Wayne was a product of the enzymological powerhouse that was Brandeis University's biochemistry program in the 1960s, a product from Bob Abeles's laboratory. I first met Wayne in Leon Rosenberg's lab at Yale when I joined to work on cloning the cDNA for OTC. It was Wayne's unenviable task to try to teach me to write more coherently. Tony Hunter and Walter Eckhart had already given good effort to teaching me how to think and perform experimentally and had done what they could with my writing skills. Wayne had to deal with the final step of maturation. Wayne was willing to mark up the whole manuscript, so there was a sea of red ink containing corrections and comments that all needed to be addressed. After draft after draft, the amount of red would slowly abate, and there would be agreement on what could be understandable. Wayne has been a member of my group since the mid-1990s, and to this day, I still find red marks on virtually everything I draft. Outside of constantly harassing him with new ideas and relying on him to organize and pilot many of the agreed-upon experiments, we have a long history of sailing together. Wayne is the master racer. These were cruising trips, however, down in the Chesapeake with our urea cycle colleague, Saul Brusilow. I will never forget the pluck of sailing across the bay to Rock Hall on an incredibly blustery Saturday night in late October, the wind blasting us, none of us harnessed, but all thrilling at our speed and lack of clear direction (we could not see the markers while trying to find the route into the rocky harbor). The crabs and beer were great, but the ride back across at midnight was a little beyond my feeling of safety; the wind and choppy waters had further picked up, and we could barely hear each other. There were also lighter times on the bay, when we enjoyed soaking up the sun, anchored on the Wye or Sassafras River, as we discussed how the GroEL reaction cycle might work. One spring, Wayne announced that he was making a trip to the Azores on a 50 footer with some of his yacht club friends. I shuddered. I warned him that a harness was a must; there were still too many scientific questions to solve.
We knew that polypeptides could be housed underneath GroES, but we did not know whether productive folding could occur in such a topology. Wayne prepared cis and trans complexes containing OTC as substrate protein and discharged them under single-turnover conditions (employing a GroES trap). He observed that cis complexes were productive, but trans complexes were not (29). Then Corinne Hohl, a visiting student, and Jonathan carried out an experiment to determine whether folding could go all the way to the native form in a cis cavity. To do this, they formed a stable cis folding chamber. This was accomplished using the knowledge from George Lorimer's studies that ATP in the ring opposite GroES triggers GroES release (31). We decided to simply take that ring away, producing a single-ring version of GroEL called SR1. Rhodanese was bound to SR1, and ATP and GroES were then added. Remarkably, rhodanese regained its activity with the same kinetics as in a wild-type reaction. More strikingly, the 400-kDa complex inside of which it was folding could be isolated at various time points and assayed for rhodanese activity; the regain of activity exactly paralleled that of the unfractionated reaction (30). Thus, polypeptide folding could proceed all the way to the native state in the cis folding chamber.
Here, I wish to digress to describe what is probably my second longest laboratory relationship. Krystyna Furtak joined me on my first day as an independent investigator in 1983 (Fig. 5). Krystyna had been working with Bernard Weinstein at Columbia University, but luckily for me, she was living out in Connecticut and found the commute into New York City and parking to be too much. We looked across the bench at each other that first day and then got right to work characterizing mitochondrial targeting peptides and testing OTC import in yeast. We have now lived thirty years together in the lab, watching each other's kids hatch and grow into mature adults (despite us). We have also been proud lab parents as we have watched our lab teams mature and turn over. Krystyna has also made hundreds of constructs. Few would have dared to undertake some of these constructs, such as a 7-fold tandemized GroEL coding sequence (22).
It was critical, once we understood the topology of productive folding, to “watch” polypeptide to see what was required to trigger folding. For these studies, we wanted to use a fluorescently labeled polypeptide substrate and monitor its behavior. Hays Rye, who was the principal driver of much of our fluorescence work, including building the equipment used for countless kinetic studies, had elected to join the group to study just these questions (Fig. 5). He probably suffered the worst eccentricity of our group right at the outset of his postdoctoral stint. He was ready to move from Berkeley to New Haven to commence work, but we called him up and told him, “No, meet us in Nashville for a week, because we're planning to collect data with Joe Beechem's group there. Don't bother to come to New Haven yet.” Hays landed in a round-the-clock data collection period at Vanderbilt University with Matt Goldberg, Jonathan, Wayne, Joe and his student Jason, and me continuously thinking of experiments and collecting data, sleeping only occasionally. We would have dinner at 1 a.m. and just go back to work. I am sure that Hays concluded that we were completely crazy. In that one trip, we saw that ATP was absolutely required, along with GroES, to mediate polypeptide release into the cis folding chamber, with ADP being unable to substitute (the study was done using fluorescence anisotropy measurements with the fluorophore placed on two different polypeptide substrates) (32). We also measured rotational correlation times of refolded GFP inside the cis folding chamber. We had taken over not only Joe's lab in the process, but all of the chromatography equipment on that floor of the biochemistry department. We more or less shifted the department's resources to chaperonin research for that week.
Then came the next crucial understanding of what a folding chamber looked like. Initial cryo-EM studies from Helen's group indicated that the ring that binds GroES undergoes large changes, but the nature of the movements and of the cis cavity lining and nucleotide pocket was not known. Although GroEL-GroES-nucleotide complexes could easily be made and crystallized, freezing them and retaining diffraction were terrible problems. Zhaohui Xu (Fig. 5), a postdoctoral fellow in Paul's group, took a year to develop a “swish” technique that involved subsequent freezing directly in the stream of cold gaseous nitrogen to produce well diffracting crystals and an x-ray structure of a domed asymmetric GroEL-GroES complex that became a new object of fascination (Fig. 4, middle and right). Remarkably, the changes that occurred were rigid body movements of the top two domains of GroEL, the apical and intermediate domains (33). The apical domains had undergone elevation and twisting movements that removed their hydrophobic binding sites from facing the cavity, replacing the lining with a hydrophilic, very electrostatic surface, composed of a small excess of acidic over basic residues. Meanwhile, the intermediate domain had rotated down into the nucleotide pocket to proffer an aspartate that could serve as a base for activating a water to mediate attack on the γ-phosphate. This was observed in an ADP-AlF3 x-ray structure that was piloted by Charu Chaudhry in a collaboration of my group and Paul's and Axel Brunger's groups (34).
Finally, last, but far from least, were efforts to attack the GroEL-GroES system at the dynamics level using NMR techniques. Despite the huge size of GroEL for NMR studies, we found a fearless collaborator in Kurt Wüthrich (Fig. 5). Our collaboration started at a meeting in Belgium, to which I arrived late but just in time to hear a plenary talk from Kurt on using solution NMR techniques to look at large molecules. I was able to catch him on his way out the door to his next destination, and we agreed in that moment that my group would 15N label and perdeuterate GroEL to enable an inspection by transverse relaxation-optimized spectroscopy (TROSY) techniques at ETH Zürich. Eric Bertelsen, a postdoctoral fellow in the group (Fig. 5), prepared the sample and flew over to Zürich with it several weeks later. He was met at the airport by Kurt, and the first spectra were collected the same day. Needless to say, TROSY techniques provided a new way to look at the machinery. First, we were able to “see” selectively labeled GroES, including its mobile loops, occupying new conformational states while in complex with GroEL, which were visible by chemical shift changes (35). Next, we directly examined isotopically labeled polypeptide while bound to an open GroEL ring, observing that the NMR-visible portions of bound polypeptides lacked any ordered secondary or tertiary structure (24). Then by amide hydrogen-deuterium exchange/NMR studies, we examined the folding trajectory taken by DHFR inside the cis chamber, observing it to be identical to the trajectory in free solution (36). This supported the idea that the folding chamber is a passive compartment in which folding occurs without the chance of aggregation and proceeds according to the Anfinsen principle that the primary sequence dictates folding to the native state (37, 38). Thus, folding can be occurring as if at infinite dilution, despite the nearby walls with which polypeptide can make rotational or translational collisions.
I would like to thank Richard Lerner in particular for enabling the NMR studies. He enabled my group to maintain a small lab at The Scripps Research Institute, where Kurt was already partly localizing his own group. With the two groups working physically together, things could really click with the biochemistry of preparing the labeled samples ready to be handed off to the spectroscopy carried out by masters. I reveled in spending time at the 800-MHz machine with Reto Horst, observing the spectra and thinking in real time about how the system might be working. Perhaps most remarkable was our ability later on to collect spectra from GroEL itself, e.g. observing >300 15N-1H-correlated resonances, well dispersed, and to assign most of these backbone amides of the machine using triple labeling. It was particularly fun to get together to brainstorm at Scripps. Kurt would insist that we review the data and discuss them in a relaxed atmosphere of the Faculty Club, replete with good wine. This surely catalyzed new thinking and studies.
In sum, over the years, the willingness, personalities, and even foibles of our team members and collaborators have been just as much fun as the science of working on the chaperonin system. I am very grateful for the richness of these relationships. I note that none of this could have been possible without the support and encouragement of the Howard Hughes Medical Institute, which supported even our wildest efforts to gain new understanding of the chaperonin system. I am forever grateful. I am also grateful to Walter Eckhart, Tony Hunter, and Leon Rosenberg, who trained me. They not only invested time and energy to directly guide me in carrying out solid informative experiments, but were themselves terrific examples of how one conducts great science.
REFERENCES
- 1. Eckhart W., Hutchinson M. A., Hunter T. (1979) An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18, 925–933 [DOI] [PubMed] [Google Scholar]
- 2. Horwich A. L., Fenton W. A., Williams K. R., Kalousek F., Kraus J. P., Doolittle R. F., Konigsberg W., Rosenberg L. E. (1984) Structure and expression of a cDNA for the nuclear coded precursor of human mitochondrial ornithine transcarbamylase. Science 224, 1068–1074 [DOI] [PubMed] [Google Scholar]
- 3. Horwich A. L., Kalousek F., Mellman I., Rosenberg L. E. (1985) A leader peptide is sufficient to direct mitochondrial import of a chimeric protein. EMBO J. 4, 1129–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng M. Y., Pollock R. A., Hendrick J. P., Horwich A. L. (1987) Import and processing of human ornithine transcarbamoylase precursor by mitochondria from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 84, 4063–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eilers M., Schatz G. (1986) Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322, 228–232 [DOI] [PubMed] [Google Scholar]
- 6. Pelham H. R. B. (1986) Speculations on the functions of the major heat-shock and glucose-regulated proteins. Cell 46, 959–961 [DOI] [PubMed] [Google Scholar]
- 7. Cheng M. Y., Hartl F.-U., Martin J., Pollock R. A., Kalousek F., Neupert W., Hallberg E. M., Hallberg R. L., Horwich A. L. (1989) Mitochondrial heat shock protein HSP60 is essential for assembly of proteins imported into yeast mitochondria. Nature 337, 620–625 [DOI] [PubMed] [Google Scholar]
- 8. Anfinsen C. B. (1973) Principles that govern the folding of protein chains. Science 181, 223–230 [DOI] [PubMed] [Google Scholar]
- 9. Epstein C. J., Goldberger R. F., Anfinsen C. B. (1963) The genetic control of tertiary protein structure: studies with model systems. Cold Spring Harb. Symp. Quant. Biol. 28, 439–449 [Google Scholar]
- 10. Ostermann J., Horwich A. L., Neupert W., Hartl F.-U. (1989) Protein folding in mitochondria requires complex formation with HSP60 and ATP hydrolysis. Nature 341, 125–130 [DOI] [PubMed] [Google Scholar]
- 11. McMullin T. W., Hallberg R. L. (1988) A highly evolutionarily conserved mitochondrial protein is structurally related to the protein encoded by the Escherichia coli groEL gene. Mol. Cell. Biol. 8, 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Georgopoulos C. P., Hendrix R. W., Kaiser A. D., Wood W. B. (1972) Role of the host cell in bacteriophage morphogenesis: effects of a bacterial mutation on T4 head assembly. Nat. New Biol. 239, 38–41 [DOI] [PubMed] [Google Scholar]
- 13. Barraclough R., Ellis R. J. (1980) Protein synthesis in chloroplasts. IX. Assembly of newly-synthesized large subunit into ribulose bisphosphate carboxylase in isolated intact pea chloroplasts. Biochim. Biophys. Acta 608, 19–31 [DOI] [PubMed] [Google Scholar]
- 14. Goloubinoff P., Christeller J. T., Gatenby A. A., Lorimer G. H. (1989) Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfolded state depends on two chaperonin proteins and MgATP. Nature 342, 884–889 [DOI] [PubMed] [Google Scholar]
- 15. Martin J., Langer T., Boteva R., Schramel A., Horwich A. L., Hartl F.-U. (1991) Chaperonin-mediated protein folding occurs at the surface of GroEL via a molten globule-like intermediate. Nature 352, 36–42 [DOI] [PubMed] [Google Scholar]
- 16. Braig K., Simon M., Furuya F., Hainfeld J. F., Horwich A. L. (1993) A polypeptide bound to the chaperonin GroEL is localized within a central cavity. Proc. Natl. Acad. Sci. U.S.A. 90, 3978–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langer T., Pfeifer G., Martin J., Baumeister W., Hartl F.-U. (1992) Chaperonin-mediated protein folding: GroES binds to one end of the GroEL cylinder, which accommodates the protein substrate within its central cavity. EMBO J. 11, 4757–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braig K., Otwinowski Z., Hegde R., Boisvert D. C., Joachimiak A., Horwich A. L., Sigler P. B. (1994) Crystal structure of GroEL at 2.8 Å. Nature 371, 578–586 [DOI] [PubMed] [Google Scholar]
- 19. Boisvert D. C., Wang J., Otwinowski Z., Horwich A. L., Sigler P. B. (1996) The 2.4 Å crystal structure of the bacterial chaperonin GroEL complexes with ATPγS. Nat. Struct. Biol. 3, 170–177 [DOI] [PubMed] [Google Scholar]
- 20. Thiyagarajan P., Henderson S. J., Joachimiak A. (1996) Solution structures of GroEL and its complex with rhodanese from small-angle neutron scattering. Structure 4, 79–88 [DOI] [PubMed] [Google Scholar]
- 21. Fenton W. A., Kashi Y., Furtak K., Horwich A. L. (1994) Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371, 614–619 [DOI] [PubMed] [Google Scholar]
- 22. Farr G. W., Furtak K., Rowland M. B., Ranson N. A., Saibil H. R., Kirchhausen T., Horwich A. L. (2000) Multivalent binding of non-native substrate proteins by the chaperonin GroEL. Cell 100, 561–573 [DOI] [PubMed] [Google Scholar]
- 23. Elad N., Farr G. W., Clare D. K., Orlova E. V., Horwich A. L., Saibil H. R. (2007) Topologies of a substrate protein bound to the chaperonin GroEL. Mol. Cell 26, 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horst R., Bertelsen E. B., Fiaux J., Wider G., Horwich A. L., Wüthrich K. (2005) Direct NMR observation of a substrate protein bound to the chaperonin GroEL. Proc. Natl. Acad. Sci. U.S.A. 102, 12748–12753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin Z., Rye H. S. (2004) Expansion and compression of a protein folding intermediate by GroEL. Mol. Cell 16, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saibil H., Dong Z., Wood S., auf der Mauer A. (1991) Binding of chaperonins. Nature 353, 25–26 [DOI] [PubMed] [Google Scholar]
- 27. Saibil H. R., Zheng D., Roseman A. M., Hunter A. S., Watson G. M., Chen S., auf der Mauer A., O'Hara B. P., Wood S. P., Mann N. H., Barnett L. K., Ellis R. J. (1993) ATP induces large quaternary rearrangements in a cage-like chaperonin structure. Curr. Biol. 3, 265–273 [DOI] [PubMed] [Google Scholar]
- 28. Clare D. K., Vasishtan D., Stagg S., Quispe J., Farr G. W., Topf M., Horwich A. L., Saibil H. R. (2012) ATP-triggered conformational changes delineate substrate-binding and -folding mechanics of the GroEL chaperonin. Cell 149, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weissman J. S., Hohl C. M., Kovalenko O., Kashi Y., Chen S., Braig K., Saibil H. R., Fenton W. A., Horwich A. L. (1995) Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell 83, 577–587 [DOI] [PubMed] [Google Scholar]
- 30. Weissman J. S., Kashi Y., Fenton W. A., Horwich A. L. (1994) GroEL-mediated protein folding proceeds by multiple rounds of release and rebinding of non-native forms. Cell 78, 693–702 [DOI] [PubMed] [Google Scholar]
- 31. Todd M. J., Viitanen P. V., Lorimer G. H. (1994) Dynamics of the chaperonin ATPase cycle: implications for facilitated protein folding. Science 265, 659–666 [DOI] [PubMed] [Google Scholar]
- 32. Weissman J. S., Rye H. S., Fenton W. A., Beechem J. M., Horwich A. L. (1996) Characterization of the active intermediate of a GroEL-GroES-mediated protein folding reaction. Cell 84, 481–490 [DOI] [PubMed] [Google Scholar]
- 33. Xu Z., Horwich A. L., Sigler P. B. (1997) The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature 388, 741–750 [DOI] [PubMed] [Google Scholar]
- 34. Chaudhry C., Farr G. W, Todd M. J., Rye H. S., Brunger A. T., Adams P. D., Horwich A. L., Sigler P. B. (2003) Role of the γ-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure, and energetics. EMBO J. 22, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fiaux J., Bertelsen E. B., Horwich A. L., Wüthrich K. (2002) NMR analysis of a 900 kDa GroEL/GroES chaperonin complex. Nature 418, 207–211 [DOI] [PubMed] [Google Scholar]
- 36. Horst R., Fenton W. A., Englander S. W., Wüthrich K., Horwich A. L. (2007) Folding trajectories of human dihydrofolate reductase inside the GroEL-GroES chaperonin cavity and free in solution. Proc. Natl. Acad. Sci. U.S.A. 104, 20788–20792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Apetri A. C., Horwich A. L. (2008) Chaperonin chamber accelerates protein folding through passive action of preventing aggregation. Proc. Natl. Acad. Sci. U.S.A. 105, 17351–17355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tyagi N. K., Fenton W. A., Deniz A. A., Horwich A. L. (2011) Double mutant MBP refolds at same rate in free solution as inside the GroEL/GroES chaperonin chamber when aggregation in free solution is prevented. FEBS Lett. 585, 1969–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Glick B. S., Brandt A., Cunningham K., Müller S., Hallberg R. L., Schatz G. (1992) Cytochromes c1 and b2 are sorted to the intermembrane space of yeast mitochondria by a stop-transfer mechanism. Cell 69, 809–822 [DOI] [PubMed] [Google Scholar]
- 40. Cheng M. Y., Hartl F.-U., Horwich A. L. (1990) Hsp60, the mitochondrial chaperonin, is required for its own assembly. Nature 348, 455–458 [DOI] [PubMed] [Google Scholar]
- 41. Braig K., Adams P. D., Brunger A. T. (1995) Conformational variability in the refined structure of the chaperonin GroEL at 2.8 Å resolution. Nature Struct. Biol. 2, 1083–1094 [DOI] [PubMed] [Google Scholar]
- 42. Yifrach O., Horovitz A. (1995) Nested cooperativity in the ATPase activity in the oligomeric chaperonin GroEL. Biochemistry 34, 5303–5308 [DOI] [PubMed] [Google Scholar]
- 43. Cliff M. J., Limpkin C., Cameron A., Burston S. G., Clarke A. R. (2006) Elucidation of steps in the capture of a protein substrate for efficient encapsulation by GroE. J. Biol. Chem. 281, 21266–21275 [DOI] [PubMed] [Google Scholar]
- 44. Rye H. S., Burston S. G., Fenton W. A., Beechem J. M., Xu Z., Sigler P. B., Horwich A. L. (1997) Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature 388, 792–798 [DOI] [PubMed] [Google Scholar]
- 45. Horwich A. L., Farr G. W., Fenton W. A. (2006) GroEL-GroES-mediated protein folding. Chem. Rev. 106, 1917–1930 [DOI] [PubMed] [Google Scholar]