

Background: NPC2 is a protein that negatively regulates fibroblast activation.
Results: NPC2-null human fibroblasts display induction of cPLA2, COX-2, and mPGES-1 expression that may contribute to the observed robust and selective activation of PGE2 biosynthesis.
Conclusion: NPC2 may regulate the activated fibroblast inflammatory program through modulation of a cPLA2-dependent biosynthetic pathway.
Significance: NPC2 may represent a novel therapeutic tool for the treatment of inflammatory diseases.
Keywords: Arachidonic Acid, Cell Biology, Inflammation, Myofibroblast, Prostaglandins
Abstract
Activated fibroblasts, also known as myofibroblasts, are mediators of several major human pathologies including proliferative fibrotic disorders, invasive tumor growth, rheumatoid arthritis, and atherosclerosis. We previously identified Niemann-Pick type C2 (NPC2) protein as a negative regulator of fibroblast activation (Csepeggi, C., Jiang, M., Kojima, F., Crofford, L. J., and Frolov, A. (2011) J. Biol. Chem. 286, 2078–2087). Here we report that NPC2-deficiency leads to a dramatic up-regulation of the arachidonic acid (AA) metabolic pathway in human fibroblasts. The major enzymes in this pathway, cPLA2 type IVA, COX-2, and mPGES-1, were dramatically up-regulated at both the transcriptional and translational levels. The specific phenotypic changes resulted in a >10-fold increase in the production and secretion of a key modulator of inflammation and immunity, prostaglandin E2. More importantly, AA metabolome profiling by liquid chromatography/tandem mass-spectrometry revealed the very specific nature of prostaglandin E2 up-regulation as the other analyzed AA metabolites derived from the COX-2, cytochrome P450, 5/15-lipoxygenase, and non-enzymatic oxidative pathways were mostly down-regulated. Blocking activity of cPLA2 efficiently suppressed expression of inflammatory cytokines, IL-1β and IL-6, thereby identifying cPLA2 as an important regulator of the inflammatory program in NPC2-null cells. Altogether, these studies highlight NPC2 as a specific regulator of AA metabolism and inflammation that suggests potential for NPC2 protein or its related signaling in the treatment of inflammatory diseases characterized by the presence of activated fibroblasts.
Introduction
Activated fibroblasts (AFs)3 are increasingly recognized as key effector cells in the development of major human diseases, including cancer (1), tissue fibrosis (2, 3), atherosclerosis (4), and rheumatoid arthritis (5, 6). Originally termed as myofibroblasts due to their expression of α-smooth muscle cell actin, AFs have now been shown to represent a diverse cell population originating from multiple precursors whose functions extend from the governance of connective tissue remodeling (7) to regulation of the innate and adaptive immunity (8, 9). With regard to the latter, AF are known to serve as nonprofessional antigen-presenting cells as well as to modulate immune inflammatory functions by influencing a variety of immune cells through either direct cell-cell contact or by secretion of soluble mediators that include PGE2 (10).
PGE2 production in AF can either be induced by exogenous stimuli such as thrombin (11) and TNF-α (12) or be constitutive in nature as reported for AF from lamina propria (13) and cancer-associated AF (14). In addition to its well known role as a mediator of classical inflammation through its effects on endothelial cells and nociceptive pathways, PGE2 can serve as a potent regulator of innate immunity, suppressing neutrophil and macrophage functions. Indeed, PGE2 has been shown to suppress granulocyte functions (15), inhibit CCL5 expression in activated macrophages (16), reprogram classically activated macrophages (M1) into the alternatively activated macrophages (M2) (17), and skew monocyte-to-macrophage differentiation toward M2 (18).
NPC2 protein was originally identified as a human epididymis 1 (HE1), which represents a major secretory protein of epididymal fluid (HE1) (19) and was later assigned to the second NPC disease locus, NPC2 (20). NPC2 gene encodes ubiquitous, 14–18-kDa protein whose precise physiologic function remains to be elucidated. NPC2 is highly conserved among species and has a strong amino acid sequence homology and striking structural resemblance to the dust mite allergen Der p2 (21) as well as the Toll-like receptor 4 (TLR4) adaptor protein MD-2 (22). Besides its strong presence in epididymal fluid, NPC2 is also found in large quantities in milk, bile, and plasma (21, 23). Because NPC2 is able to bind unesterified cholesterol with a high affinity (24), it was thought to participate, along with NPC1, in regulation of intracellular cholesterol trafficking (25). However, this view has been recently challenged (26, 27). Consistent with this notion, several very important physiologic processes such as hematopoiesis (28), immunity (29), and somatic cell plasticity (30, 31) have been shown to be regulated by NPC2 independently of its cholesterol binding ability.
We have recently reported that human skin fibroblasts with a natural nonsense mutation in the NPC2 gene were characterized by constitutive receptor tyrosine kinase (RTK)/growth factor receptor (GFR) → ERK 1/2 signaling (30). Constitutive RTK/GFR activation has also been observed in mature human adipocytes after silencing NPC2 gene expression by siRNA (31). This evidence points strongly toward RTK/GFR signaling module as a proximal target for the negative regulation by NPC2.
RTK/GFR → ERK1/2 signaling is one of the major driving forces for PGE2 production where it up-regulates the expression and activity of the components in the cPLA2 → COX-1/2 → mPGES-1 → PGE2 biosynthetic pathway (32–34). Furthermore, sustained ERK1/2 activation is required for maximal PGE2 production (35). We, therefore, reasoned that this pathway should be persistently activated in NPC2-null human fibroblasts that bear the AF phenotype (30).
In the present study we confirmed our hypothesis and demonstrated that NPC2 deficiency results in the up-regulation of cPLA2 → COX-1/2 → mPGES-1 biosynthetic axis at both the transcriptional and translational levels, leading to a robust and specific increase in PGE2 production. Furthermore, inhibiting this biosynthetic pathway in NPC2-deficient cells through suppression of cPLA2 activity resulted in a significant reduction of proinflammatory cytokine expression. Altogether, our studies begin to unveil a novel and important role for NPC2 in regulation of the PGE2 biosynthetic pathway that could modulate inflammation and innate immunity.
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, glutamine, and penicillin/streptomycin were obtained from Cellgro. cPLA2 specific inhibitor was purchased from Calbiochem (catalog #525143).
Cell Lines
Normal skin fibroblasts (CRL-1474) were obtained from ATCC. NPC2-null human dermal fibroblasts were described elsewhere (36, 37).
Stable overexpression of NPC2 gene in NPC2-deficient synovial fibroblasts (SF) isolated from patients with rheumatoid arthritis (RA-83) was achieved using the respective OmicsLink ORF cDNA lentiviral expression clone from GeneCopoeia. The human NPC2 pseudovirus-containing lentiviral expression construct was produced by the Cincinnati Children's Hospital Research Core Facility. RA-83 cells (30) were transduced with the above construct using Magnetofection Protocol from OZ Biosciences. Stable transducers were selected by culturing cells in the medium supplemented with 3.0–3.5 μg of puromycin. The human RA-83 cell line stably overexpressing NPC2 gene was termed RA-83/NPC2.
DNA Microarray Assay
Total RNA was isolated from the normal and NPC2-null confluent cells that were cultured under the normal conditions (36, 37) by using Qiagen RNeasy kit following the manufacturer instructions. Samples were hybridized to the human U133A microarray (Affimetrix) by the Alvin Siteman Cancer Center's Multiplexed Gene Analysis Core at the Washington University in St. Louis. Signal intensity ratios of >2-fold (up-regulated) or <2-fold (down-regulated) in at least two trials were considered regulated. Expression of the genes that were undetectable in the wild type cells and were only present in NPC2-null fibroblasts was termed “induced.”
Real-time RT-PCR
Total RNA from unelicited normal and NPC2-deficient human skin fibroblasts was quantitatively converted into the single-stranded cDNA by using a High Capacity cDNA Archive kit (Applied Biosystems). The particular genes were detected using the respective TaqMan Gene Expression Assays (Applied Biosystems) on the 7300 Real-time RT-PCR system from the same manufacturer by relative quantitation employing β-glucuronidase as the reference gene.
Western Blot Analysis
It was performed as previously described (30). Briefly, human fibroblasts were cultured under normal conditions as previously described (36, 37), and both the cytosolic and nuclear protein fractions were isolated from the ∼90%-confluent cells using nuclear/cytosol fractionation kit from BioVision according to the manufacturer's instructions. cPLA2 and phospho-cPLA2, respectively, were probed with the D49A7 rabbit monoclonal antibody (catalog #5249) and rabbit polyclonal antibody (Ser-505, Cat# 2831) from Cell Signaling. COX-2 was detected using rabbit polyclonal antibody from Cell Signaling (catalog #4842). mPGES-1 was probed with the rabbit polyclonal antibody from Cayman (catalog #160140). β-Tubulin was detected using mouse monoclonal antibody from Santa Cruz (catalog #sc-55529) and served as a loading control. That was followed by the incubation with the Alexa Fluor 680 goat anti-mouse (Molecular Probes) and IRDye800CW goat anti-rabbit (Rockland) affinity-purified IgG. Protein bands were identified on the same membrane by simultaneous, dual-color scanning using the LI-COR Odyssey near-infrared imaging system.
LC/MS/MS Analysis of AA Metabolites
It was carried out as previously described in details (38). Briefly, an Agilent 1200 SL liquid chromatography series (Agilent Corp., Palo Alto, CA) with an Agilent Eclipse Plus C18 2.1 × 150-mm, 1.8-μm column was used for the eicosanoid separation. The mobile phase A was water with 0.1% acetic acid, whereas the mobile phase B was composed by acetonitrile/methanol (80/15, v/v) and 0.1% acetic acid. Gradient elution was performed at a flow rate of 250 μl/min. The injection volume was 10 μl, and the samples were kept at 4 °C in the auto sampler. Analytes were detected by negative MRM mode using a 4000 QTrap tandem mass spectrometer (Applied Biosystems) equipped with an electrospray ionization source (Turbo V). Calibration curves were generated by 10-μl injections of seven standards containing each analyte, internal standard I (d4-PGE2, d11–14,15-DiHETrE, d8–5-HETE, and d11–11(12)-EpETrE), and internal standard II (1-cyclohexyl-dodecanoic acid urea) for quantification purpose.
Confocal Immunofluorescence Microscopy
Cells grown under normal conditions on coverslips in 24-well cell tissue culture plate were fixed, permeabilized, and stained with rabbit polyclonal cPLA2 (catalog #sc-438) antibodies from Santa Cruz as previously described (36, 37). That was followed by incubation with the Alexa Fluor 568 goat anti-rabbit antibody (Molecular Probes). Image acquisition was performed on Leica TCS SP5 inverted confocal system.
PGE2 Detection by Enzyme Immunoassay
PGE2 detection in the conditioned medium of SF from RA patient was performed using the PGE2 enzyme immunoassay kit, Monoclonal from Cayman Chemical, following the manufacturer's instructions.
RESULTS
NPC2 Deficiency Confers a Robust and Selective Activation of the PGE2 Biosynthetic Program
We have recently shown that NPC2 deficiency in human fibroblasts resulted in the sustained activation of RTK/GFR → ERK1/2 signaling (30). Given that this signaling pathway provides a driving force for activation of the cPLA2 → COX-1/2 → mPGES-1 → PGE2 biosynthetic axis (Fig. 1) (32–34), we hypothesized that the latter should be significantly stimulated in those cells. To confirm our hypothesis, we first probed by DNA microarray analysis the expression of the genes that regulate prostaglandin biosynthesis and signaling (Fig. 1 and Table 1). These data demonstrated either induced or significantly up-regulated expression of the respective genes. We are using term induced to define expression of the genes that were undetectable in the wild type cells and were only present in NPC2-null fibroblasts. To confirm differential expression of specific genes in human fibroblasts, we performed analysis of the respective mRNA transcripts by real-time RT-PCR (Fig. 2). As in this figure, the data showed a coordinate up-regulation of the genes that regulate PGE2 production, including PLA2G4A (∼60-fold), PTGS2 (∼150-fold), and PTGES (∼1.3-fold) as well as that of PGE2 signaling membrane receptor, PTGER4 (∼3-fold).
FIGURE 1.
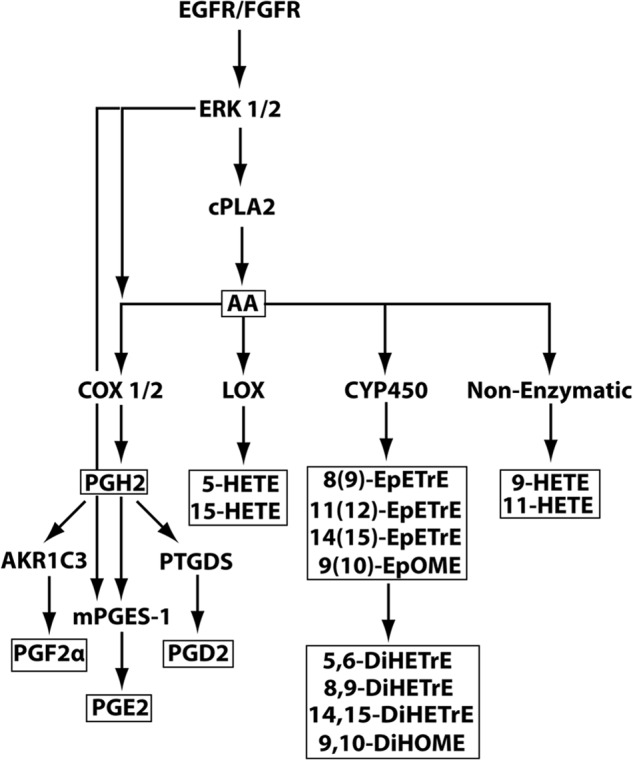
Eicosanoid biosynthetic pathway and its activation by RTK/GFR → ERK1/2 signaling. Four major branches of the AA metabolic pathway are shown. COX-1/2, cyclooxygenase-1/2-dependent branch. COX-1/2 is also known as prostaglandin-endoperoxide synthase (PTGS) 1/2; LOX, lipoxygenase-dependent branch; CYP450, cytochrome P450-dependent branch; Non-enzymatic, free radical only-dependent branch. Unboxed abbreviations depict proteins and biosynthetic pathways. The boxed symbols identify AA and its metabolites that were measured in the current study by LC/MS/MS.
TABLE 1.
NPC2-deficiency confers activation of a genetic program that controls eicosanoid metabolic pathway and signaling; DNA microarray analysis
| Probe set | Gene name | Symbol | Gene expression |
|---|---|---|---|
| Eicosanoid metabolism | |||
| 210145_at | Phospholipase A2, group IVA (cytosolic, calcium-dependent) | PLA2G4A | Induced |
| 205128_x_at | Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase) | PTGS1 (COX-1) | ↑5.3 |
| 204748_at | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | PTGS2 (COX-2) | Induced |
| 210367_s_at | Prostaglandin E synthase | PTGES (mPGES-1) | ↑2.9 |
| 211748_x_at | Prostaglandin D2 synthase 21 kDa (brain) | PTGDS | Induced |
| 209160_at | Aldo-keto reductase family 1, member C3 (3α-hydroxysteroid dehydrogenase, type II) | AKR1C3 | ↑10.5 |
| Eicosanoid receptors | |||
| 206631_at | Prostaglandin E receptor2 (subtype EP2), 53 kDa | PTGER2 (EP2) | ↑4.7 |
| 204897_at | Prostaglandin E receptor4 (subtype EP4) | PTGER4 (EP4) | ↑3.5 |
FIGURE 2.
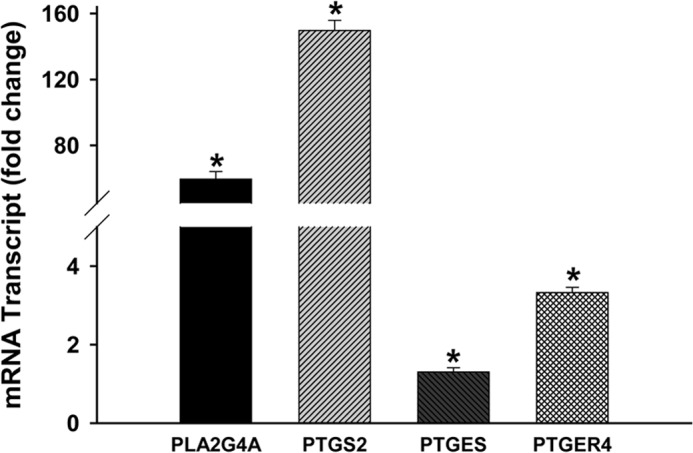
Activation of a genetic program that controls eicosanoid metabolic pathway and signaling in unelicited NPC2-fibroblasts; real-time RT-PCR analysis. Total RNA was isolated from unelicited normal and NPC2-deficient human skin fibroblasts and quantitatively converted into the single-stranded cDNA by using a High Capacity cDNA Archive kit (Applied Biosystems). The particular genes were detected using the respective TaqMan Gene Expression Assays (Applied Biosystems) by relative quantitation employing β-glucuronidase as the reference gene. Data were normalized to their respective controls and are shown as the mean ± S.E. * depicts p ≤ 0.05 as compared with control (n = 3–6).
Next, we examined protein expression levels of cPLA2, COX-2, and mPGES-1 by Western blotting of the cytoplasmic and nuclear fractions, recognizing that membrane-associated and soluble proteins are present in the respective fractions (Fig. 3). Under unstimulated culture conditions, cPLA2 and mPGES-1 expression was induced strongly in NPC2-null cells but was undetectable in normal fibroblasts. More importantly, cPLA2 in NPC2-null cells was present in its activated form as shown by its phosphorylation on Ser-505 (Fig. 3). COX-2, on the other hand, displayed a different expression pattern. In NPC2-null cells, COX-2 was present in both the cytoplasmic and nuclear fractions, whereas in normal fibroblasts COX-2 was undetectable in the cytoplasmic fraction and displayed similar expression levels to NPC2-null cells in the nuclear fraction (Fig. 3).
FIGURE 3.

Activation of eicosanoid metabolic program in NPC2-null fibroblasts. Protein expression was detected by Western blotting in the cytoplasmic and nuclear protein fractions isolated from unelicited WT and NPC2-null fibroblasts. The respective proteins were labeled with the specific sets of primary/fluorescently tagged secondary antibodies and were identified on the same membrane by simultaneous dual-color scanning using the high resolution LI-COR Odyssey near-infrared imaging system. The phospho-cPLA2 detection was performed after stripping and re-probing a membrane that was originally stained with the cPLA2 antibodies. Quantitative analysis of COX-2 in nuclear protein fractions from control and NPC2-null cells revealed no statistically significant difference in the relative protein expression levels with the respective mean ± S.E. of 1.22 ± 0.26 (p = 0.4, n = 3).
Altogether, the above data provide strong evidence for constitutive activation of the PGE2 biosynthetic program in NPC2-null fibroblasts. To verify this conclusion, we performed lipidomic profiling of AA metabolites secreted from NPC2-null and normal human fibroblasts by LC/MS/MS. The analysis revealed distinct and detectable levels of prostaglandins, leukotrienes, and thromboxanes as well as mono- and dihydroxy-fatty acids and epoxy-fatty acids (Fig. 4A) with PGE2 being a dominant prostaglandin AA metabolite (Fig. 4B). The specific AA metabolites were further identified and quantified (Fig. 5). As predicted from the transcriptional and translational up-regulation of cPLA2 → COX-1/2 → mPGES-1 biosynthetic axis in NPC2-null fibroblasts, these cells display a robust (∼10-fold) up-regulation of PGE2 production. Unexpectedly, this robust PGE2 up-regulation appeared to be highly specific as the other AA metabolites were either slightly up-regulated (PGD2 and PGF2α) or mostly down-regulated (Fig. 5). The very modest, in contrast to PGE2, up-regulation of PGD2 and PGF2α production in the setting of the significant up-regulation of their respective enzymes, PTGDS and AKR1C3 (Table 1), points toward preferential cPLA2 → COX-2 → mPGES-1 metabolic coupling in NPC2-null cells (Figs. 1 and 5).
FIGURE 4.

Quantitative analysis of selected arachidonic acid metabolites by LC/MS/MS. Lipids were extracted from the conditioned media of near confluent (∼90%), unelicited normal, and NPC2-null cells and analyzed by liquid chromatography. A, shown is a complete LC chromatogram. B, shown is a prostaglandin LC chromatogram.
FIGURE 5.
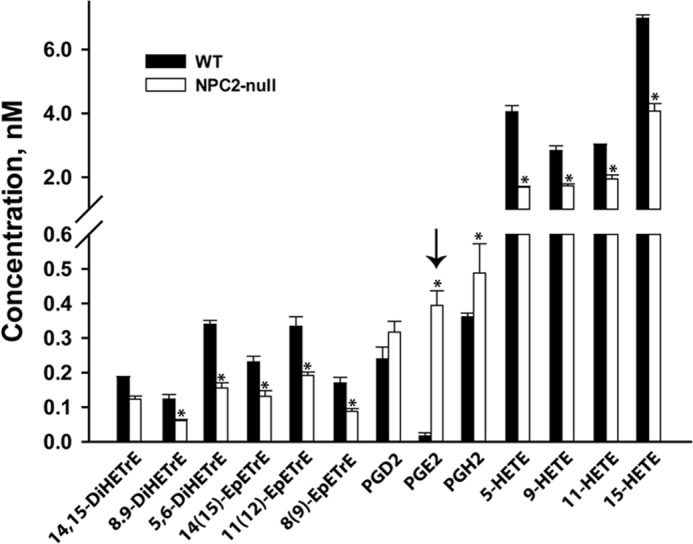
Strong and selective up-regulation of PGE2 production in NPC2-null fibroblasts as measured by LC/MS/MS. Lipid analytes isolated from the conditioned media of near confluent (∼90%), unelicited normal, and NPC2-null cells were detected by tandem mass spectrometry. Data shown are the mean ± S.E. * depicts p ≤ 0.05 as compared with control (n = 4). The arrow points toward strongly and selectively up-regulated PGE2 lipid analyte.
Robust and Selective Up-regulation of PGE2 Biosynthesis in NPC2-null Cells Is Not Controlled by cPLA2 Translocation
It has been previously established that cPLA2 activity is tightly regulated at both the molecular and subcellular levels. The former is more general and includes cPLA2 phosphorylation by ERK1/2 (39) in most of the cell types, whereas the latter is more cell type-specific and includes translocation of the phosphorylated cPLA2 from the cytosol to the perinuclear region (40, 41) and/or nucleus (42) in the immune and endothelial cells. To address a potential cellular mechanism that controls a strong and selective up-regulation of PGE2 production in NPC2-null human fibroblasts, we examined the intracellular localization of cPLA2 in those cells by confocal fluorescence microscopy (Fig. 6). In unelicited normal skin fibroblasts, cPLA2 was virtually undetectable (Fig. 6A), whereas in NPC2-null cells cPLA2 displayed a reticular/punctate cytoplasmic staining pattern (Fig. 6B) that was reported earlier for both unstimulated and stimulated fibroblasts (43, 44). These fluorescence microscopy data correlated well with those of Western blot analysis (Fig. 3) that demonstrated no detectable cPLA2 protein expression in normal fibroblasts along with the exclusive cPLA2 presence in the cytoplasmic fraction of NPC2-null fibroblasts. These findings are consistent with NPC2 regulating cPLA2 activation in human skin fibroblasts at the predominantly molecular level.
FIGURE 6.

Intracellular localization of cPLA2 in unelicited normal (A) and NPC2-null (B) skin fibroblasts as probed by confocal fluorescence microscopy. Cells grown under normal conditions on coverslips in 24-well cell tissue culture plate were fixed, permeabilized, and stained with rabbit polyclonal cPLA2 (catalog #sc-438) antibodies from Santa Cruz. That was followed by the incubation with the Alexa Fluor 568 goat anti-rabbit antibody (Molecular Probes). Image acquisition was performed on Leica TCS SP5 inverted confocal system. The images were taken under the same conditions, and their intensity and contrast were similarly adjusted upward using Adobe Photoshop CS4 Extended software. Arrows point toward individual cells in the confocal section.
cPLA2 Regulates Inflammatory Program in NPC2-null Cells
We have previously shown that NPC2 deficiency in human fibroblasts confers persistent expression and secretion of proinflammatory cytokines, such as IL-1β and IL-6, that was consistent with sustained RTK/GFR → ERK1/2 and NF-κB activation in those cells (30). To further delineate the respective molecular regulatory steps downstream from RTK/GFR → ERK1/2 activation and taking into account the constitutive nature of cPLA2 activation as evidenced by its sustained phosphorylation (Fig. 3), we probed the role of cPLA2 in the induction of IL-1β and IL-6 gene expression by suppressing cPLA2 activity using the cell-permeable, potent, and specific cPLA2 inhibitor CB-525143 (1,2,4-trisubstituted pyrrolidine derivative) (Fig. 7). As shown in this figure, this inhibitor in a dose-dependent manner was able to suppress efficiently both IL-1β and IL-6 mRNA expression, thereby identifying cPLA2 as an important regulator of the inflammatory program in NPC2-null cells.
FIGURE 7.
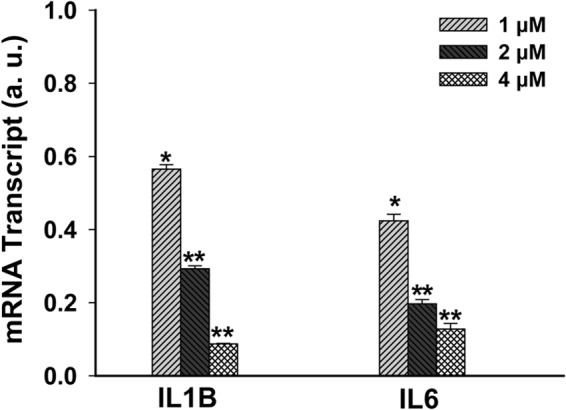
cPLA2 activity contributes to the inflammatory cytokine expression in NPC2-null fibroblasts. Cells were treated for 4 h with the indicated concentrations of cPLA2 inhibitor in the DMEM supplemented with 0.1% BSA. IL-1B and IL-6 mRNA levels were probed by real-time RT-PCR. Data were normalized to their respective untreated controls and shown as the mean ± S.E. * depicts p ≤ 0.05 as compared with control, and ** depicts p ≤ 0.05 as compared with the lowest inhibitor concentration (n = 3). a.u., arbitrary units.
NPC2 Overexpression in SF from Patient with RA Suppresses IL-1β-stimulated PGE2 Production
One of the main maladaptive features of SF in RA is their ability to robustly produce PGE2 upon stimulation with the inflammatory cytokines, such as IL-1β (45–47). Recently, our group has identified SF from RA patients (SF RA) as being NPC2-deficient, which was indicated by their significantly reduced (ranging from ∼2.5 to 5.0-fold) NPC2 mRNA expression as compared with normal human skin fibroblasts (control) (30). Therefore, we reasoned that the up-regulated PGE2 production by SF in RA could be linked to their NPC2 deficiency. To test this hypothesis as well as to independently verify NPC2 involvement in the negative regulation of PGE2 biosynthesis, we engineered SF RA cell line, RA-83/NPC2, where NPC2 mRNA expression was stably up-regulated by ∼1.6-fold as compared with control (Fig. 8A). Yet in comparison to the parental RA-83 cells, the IL-1β-stimulated PGE2 production in RA-83/NPC2 cells was reduced by ∼1.6 -fold (Fig. 8B). Altogether, the above data point strongly toward important NPC2 involvement in the negative regulation of PGE2 biosynthesis.
FIGURE 8.
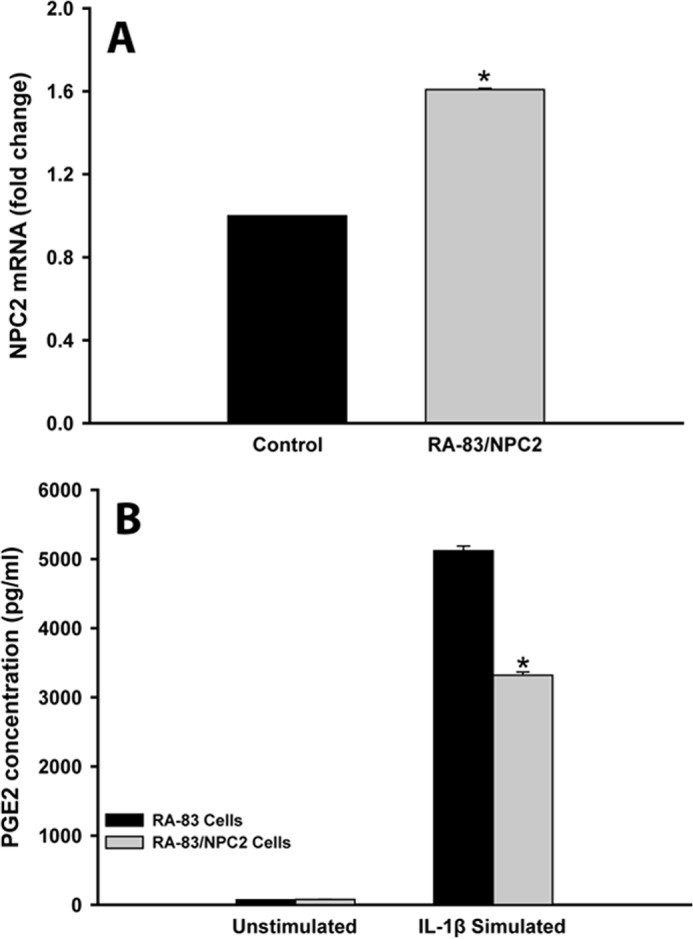
NPC2 overexpression in SF RA suppresses IL-1β-stimulated PGE2 production. A, shown is the NPC2 mRNA level in RA-83/NPC2 cells that stably overexpress NPC2 gene as compared with normal human skin fibroblasts (Control). B, shown is IL-1β-stimulated PGE2 production in SF RA cells as probed by PGE2 enzyme immunoassay. Data shown are the mean ± S.E. * depicts p ≤ 0.05 as compared with RA-83 parental cell line (n = 3).
DISCUSSION
The current report provides several important insights into the regulation of inflammation and innate immunity. First, using NPC2-null human skin fibroblasts, we identified NPC2 protein as a novel regulator of AA metabolism that specifically targets the cPLA2 → COX-2 → mPGES-1 → PGE2 biosynthetic pathway (Fig. 1). It appears that NPC2 exerts its function at multiple levels including regulation of cPLA2, COX1/2, and mPGES-1 gene expression as well as the enzyme post-translational modification through phosphorylation, as was seen for cPLA2. In addition, it is quite plausible that mPGES-1 may represent a selective target for NPC2 regulation because in the setting of NPC2 deficiency, which leads to a significant up-regulation of AKR1C3, mPGES-1, and PTGDS that metabolize PGH2 to yield, respectively, PGF2α, PGE2, and PGD2 (Fig. 1), only PGE2 production was significantly up-regulated, as demonstrated by our lipidomics analysis. Yet the same analysis has revealed the lipoxygenase- and cytochrome P450-dependent as well as non-enzymatic oxidative pathways within the AA metabolic cascade (Fig. 1) were efficiently suppressed in NPC2-null cells as evidenced by a significant reduction in the production of their respective products. Such a pattern of eicosanoid production in the setting of NPC2 deficiency is unique and to our best knowledge has not been previously reported in the literature.
Second, we further delineated a mechanism of NPC2-dependent regulation of the cPLA2-dependent PGE2 biosynthetic pathway in fibroblasts by demonstrating that for cPLA2 this mechanism does not include a subcellular component because cPLA2 activation was not accompanied by its translocation from cytosol to the perinuclear region and/or nucleus as shown earlier for the immune and endothelial cells (40–42). cPLA2 is now considered to be a central enzyme in mediating AA release for eicosanoid production, including PGE2 (48, 49). The most likely mechanism for constitutive cPLA2 activation and subsequent PGE2 production could include, in addition to its Ser-505 phosphorylation, the release of active cPLA2 monomers from inactive intracellular clusters (43, 44). Indeed, it has been previously reported that in fibroblasts, cPLA2 is present in small clusters that were identified as punctuate structures by immunofluorescence and immunogold electron microscopy (43). These cPLA2 clusters were randomly distributed throughout the cytosol and localized in the vicinity of major intracellular membranes, except for the Golgi apparatus. More importantly, upon cell stimulation with EGF or calcium ionophore A23187 that are known to activate cPLA2 and stimulate AA metabolism, the cPLA2 punctate localization pattern did not change (43), but the release of activated cPLA2 monomeric forms from inactive clusters and their binding to the nearing intracellular membranes were observed (44). The latter was accompanied by an induction of cPLA2 hydrolytic activity, indicating that the release of active cPLA2 monomers from the existent inactive cPLA2 clusters could be an important mechanism that controls cPLA2 activity in fibroblasts and quite possibly in other non-immune cells (44). Given that very similar punctate cPLA2 structures were observed in NPC2-null fibroblasts by immunofluorescence in the current study and that cPLA2 phosphorylation by ERK1/2 was proposed to be a prerequisite for cPLA2 release from the inactive clusters (44), one may suggest that RTK/GFR → ERK1/2 → cPLA2 phosphorylation axis could be a primary regulatory target for NPC2 within the AA metabolic pathway.
Third, we demonstrated that constitutive cPLA2 activation is responsible for the increased basal production of proinflammatory cytokines in NPC2 null cells. Given that constitutive induction of cPLA2 gene expression and cPLA2 activation through phosphorylation is regulated by sustained RTK/GFR → ERK1/2 signaling (32–34) and that the latter was observed earlier by us in NPC2-null fibroblasts (30), the molecular mechanism of the NPC2-dependent negative regulation of fibroblast activation (30) could be further detailed to include cPLA2 activation step downstream from sustained ERK1/2 signaling.
Fourth, the fact that NPC2 deficiency results in co-stimulated induction of PGE2 (current study) and IL-6 (30) production and secretion could bear very important consequences for the regulation of the innate and adaptive immunity. Indeed, it has been recently reported (18) that the immunosuppressive tumor microenvironment of human cervical carcinoma could be linked to the ability of cancer cells to block dendritic cell differentiation and to skew monocyte-to-macrophage differentiation toward tolerogenic M2 phenotype. As the result, M2 macrophages had a significantly impaired ability to stimulate T cell proliferation and Th1 cell activation, thereby providing, along with the dendritic cell deficiency, a permissive environment for tumor growth. Characteristically, IL-6 and PGE2 had distinct functions in the above process; IL-6 blocks dendritic cell differentiation and PGE2 drives M2 macrophage polarization (18). Finally, using RA-83/NPC2 cells with the stable overexpression of NPC2 gene, we independently confirmed its important involvement in the negative regulation of PGE2 biosynthesis in fibroblasts as well as demonstrated, at least in vitro, a potential for the SF RA maladaptive phenotype correction by overcoming their NPC2 deficiency. The latter could provide a basis for the development of a novel therapeutic intervention in RA. In summary, all of these data point toward NPC2 as an important regulator of the phospholipid/AA metabolism, immunity, and inflammation.
Acknowledgments
We thank Daniel Ory (Washington University in St. Louis) for providing NPC2-null human fibroblasts. We also thank Mary Gail Engle and James Begley (Imaging Facility, University of Kentucky) for expert assistance with the confocal fluorescence microscopy.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1AR049010 (to L. J. C.). This work was also supported by American Heart Association Grant SDG 023017N (to A. F.). Partial support for analytical chemistry was provided by National Institutes of Health (NIH) Grant RO1 ES ES002710 (NIEHS) and NIEHS Superfund Research Program Grant P42 ES004699 and NIH Grant U24DK097154 (to B. D. H.).
- AF
- activated fibroblast
- NPC2
- Niemann-Pick type C2
- RTK
- receptor tyrosine kinase
- GFR
- growth factor receptor
- cPLA2
- cytosolic phospholipase A2
- COX-1/2
- cyclooxygenase-1/2 also known as PTGS1/2 (prostaglandin-endoperoxide synthase 1/2)
- PG
- prostaglandin
- PGD2
- prostaglandin D2
- PGF2α
- prostaglandin F2α
- 5-, 9-, 11, and 15-HETE
- 5-, 9-, 11-, and 15-hydroxyeicosatetraenoic acid
- 9,10-DiHOME
- (±)9,10-dihydroxy-12Z-octadecenoic acid
- 14,15-DiHETrE
- (±)14,15-dihydroxy-5Z,8Z,11Z-eicosatrienoic acid
- 9,10-EpOME
- (±)9,10-epoxy-12Z-octadecenoic acid
- 14,15-EpETrE
- (±)14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid
- SF
- synovial fibroblast
- AA
- arachidonic acid
- RA
- rheumatoid arthritis.
REFERENCES
- 1. Kalluri R., Zeisberg M. (2006) Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 [DOI] [PubMed] [Google Scholar]
- 2. Wynn T. A., Ramalingam T. R. (2012) Mechanisms of fibrosis. Therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhattacharyya S., Wei J., Varga J. (2012) Understanding fibrosis in systemic sclerosis. Shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol. 8, 42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coen M., Gabbiani G., Bochaton-Piallat M. L. (2011) Myofibroblast-mediated adventitial remodeling. An underestimated player in arterial pathology. Arterioscler. Thromb. Vasc. Biol. 31, 2391–2396 [DOI] [PubMed] [Google Scholar]
- 5. Noss E. H., Brenner M. B. (2008) The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol. Rev. 223, 252–270 [DOI] [PubMed] [Google Scholar]
- 6. Bartok B., Firestein G. S. (2010) Fibroblast-like synoviocytes. Key effector cells in rheumatoid arthritis. Immunol. Rev. 233, 233–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology. Paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otte J. M., Rosenberg I. M., Podolsky D. K. (2003) Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 124, 1866–1878 [DOI] [PubMed] [Google Scholar]
- 9. Saada J. I., Pinchuk I. V., Barrera C. A., Adegboyega P. A., Suarez G., Mifflin R. C., Di Mari J. F., Reyes V. E., Powell D. W. (2006) Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J. Immunol. 177, 5968–5979 [DOI] [PubMed] [Google Scholar]
- 10. Powell D. W., Pinchuk I. V., Saada J. I., Chen X., Mifflin R. C. (2011) Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol 73, 213–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seymour M. L., Zaidi N. F., Hollenberg M. D., MacNaughton W. K. (2003) PAR1-dependent and independent increases in COX-2 and PGE2 in human colonic myofibroblasts stimulated by thrombin. Am. J. Physiol. Cell Physiol 284, C1185–C1192 [DOI] [PubMed] [Google Scholar]
- 12. Hai L., Kawarabayashi Y., Imai Y., Honda A., Inoue R. (2011) Counteracting effect of TRPC1-associated Ca2+ influx on TNF-α-induced COX-2-dependent prostaglandin E2 production in human colonic myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G356–G367 [DOI] [PubMed] [Google Scholar]
- 13. Newberry R. D., McDonough J. S., Stenson W. F., Lorenz R. G. (2001) Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria. Directing the tone of the intestinal immune response. J. Immunol. 166, 4465–4472 [DOI] [PubMed] [Google Scholar]
- 14. Rudnick J. A., Arendt L. M., Klebba I., Hinds J. W., Iyer V., Gupta P. B., Naber S. P., Kuperwasser C. (2011) Functional heterogeneity of breast fibroblasts is defined by a prostaglandin secretory phenotype that promotes expansion of cancer-stem like cells. PLoS ONE 6, e24605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith R. J. (1977) Modulation of phagocytosis by and lysosomal enzyme secretion from guinea pig neutrophils. Effect of nonsteroid anti-inflammatory agents and prostaglindins. J. Pharmacol. Exp. Ther. 200, 647–657 [PubMed] [Google Scholar]
- 16. Qian X., Zhang J., Liu J. (2011) Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J. Biol. Chem. 286, 2111–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Németh K., Leelahavanichkul A., Yuen P. S., Mayer B., Parmelee A., Doi K., Robey P. G., Leelahavanichkul K., Koller B. H., Brown J. M., Hu X., Jelinek I., Star R. A., Mezey E. (2009) Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 15, 42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heusinkveld M., de Vos van Steenwijk PJ, Goedemans R., Ramwadhdoebe T. H., Gorter A., Welters M. J., van Hall T., van der Burg S. H. (2011) M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J. Immunol. 187, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 19. Okamura N., Kiuchi S., Tamba M., Kashima T., Hiramoto S., Baba T., Dacheux F., Dacheux J. L., Sugita Y., Jin Y. Z. (1999) A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim. Biophys. Acta 1438, 377–387 [DOI] [PubMed] [Google Scholar]
- 20. Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Jadot M., Lobel P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290, 2298–2301 [DOI] [PubMed] [Google Scholar]
- 21. Friedland N., Liou H. L., Lobel P., Stock A. M. (2003) Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2512–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gruber A., Mancek M., Wagner H., Kirschning C. J., Jerala R. (2004) Structural model of MD-2 and functional role of its basic amino acid clusters involved in cellular lipopolysaccharide recognition. J. Biol. Chem. 279, 28475–28482 [DOI] [PubMed] [Google Scholar]
- 23. Klein A., Amigo L., Retamal M. J., Morales M. G., Miquel J. F., Rigotti A., Zanlungo S. (2006) NPC2 is expressed in human and murine liver and secreted into bile. Potential implications for body cholesterol homeostasis. Hepatology 43, 126–133 [DOI] [PubMed] [Google Scholar]
- 24. Ko D. C., Binkley J., Sidow A., Scott M. P. (2003) The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. U.S.A. 100, 2518–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Storch J., Xu Z. (2009) Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta 1791, 671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 27. Du X., Kazim A. S., Brown A. J., Yang H. (2012) An essential role of Hrs/Vps27 in endosomal cholesterol trafficking. Cell Rep. 1, 29–35 [DOI] [PubMed] [Google Scholar]
- 28. Heo K., Jariwala U., Woo J., Zhan Y., Burke K. A., Zhu L., Anderson W. F., Zhao Y. (2006) Involvement of Niemann-Pick type C2 protein in hematopoiesis regulation. Stem Cells 24, 1549–1555 [DOI] [PubMed] [Google Scholar]
- 29. Schrantz N., Sagiv Y., Liu Y., Savage P. B., Bendelac A., Teyton L. (2007) The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J. Exp. Med. 204, 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csepeggi C., Jiang M., Kojima F., Crofford L. J., Frolov A. (2011) Somatic cell plasticity and Niemann-Pick type C2 protein. Fibroblast activation. J. Biol. Chem. 286, 2078–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Csepeggi C., Jiang M., Frolov A. (2010) Somatic cell plasticity and Niemann-pick type C2 protein. Adipocyte differentiation and function. J. Biol. Chem. 285, 30347–30354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sa G., Murugesan G., Jaye M., Ivashchenko Y., Fox P. L. (1995) Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 270, 2360–2366 [DOI] [PubMed] [Google Scholar]
- 33. Hwang M. K., Bode A. M., Byun S., Song N. R., Lee H. J., Lee K. W., Dong Z. (2010) Cocarcinogenic effect of capsaicin involves activation of EGFR signaling but not TRPV1. Cancer Res. 70, 6859–6869 [DOI] [PubMed] [Google Scholar]
- 34. Nah S. S., Won H. J., Ha E., Kang I., Cho H. Y., Hur S. J., Lee S. H., Baik H. H. (2010) Epidermal growth factor increases prostaglandin E2 production via ERK1/2 MAPK and NF-κB pathway in fibroblast like synoviocytes from patients with rheumatoid arthritis. Rheumatol. Int. 30, 443–449 [DOI] [PubMed] [Google Scholar]
- 35. Petrovic N., Knight D. A., Bomalaski J. S., Thompson P. J., Misso N. L. (2006) Concomitant activation of extracellular signal-regulated kinase and induction of COX-2 stimulates maximum prostaglandin E2 synthesis in human airway epithelial cells. Prostaglandins Other Lipid Mediat. 81, 126–135 [DOI] [PubMed] [Google Scholar]
- 36. Frolov A., Srivastava K., Daphna-Iken D., Traub L. M., Schaffer J. E., Ory D. S. (2001) Cholesterol overload promotes morphogenesis of a Niemann-Pick C (NPC)-like compartment independent of inhibition of NPC1 or HE1/NPC2 function. J. Biol. Chem. 276, 46414–46421 [DOI] [PubMed] [Google Scholar]
- 37. Frolov A., Zielinski S. E., Crowley J. R., Dudley-Rucker N., Schaffer J. E., Ory D. S. (2003) NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 278, 25517–25525 [DOI] [PubMed] [Google Scholar]
- 38. Charles R. L., Burgoyne J. R., Mayr M., Weldon S. M., Hubner N., Dong H., Morisseau C., Hammock B. D., Landar A., Eaton P. (2011) Redox regulation of soluble epoxide hydrolase by 15-deoxy-δ-prostaglandin J2 controls coronary hypoxic vasodilation. Circ. Res. 108, 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin L. L., Wartmann M., Lin A. Y., Knopf J. L., Seth A., Davis R. J. (1993) cPLA2 is phosphorylated and activated by MAP kinase. Cell 72, 269–278 [DOI] [PubMed] [Google Scholar]
- 40. Glover S., de Carvalho M. S., Bayburt T., Jonas M., Chi E., Leslie C. C., Gelb M. H. (1995) Translocation of the 85-kDa phospholipase A2 from cytosol to the nuclear envelope in rat basophilic leukemia cells stimulated with calcium ionophore or IgE/antigen. J. Biol. Chem. 270, 15359–15367 [DOI] [PubMed] [Google Scholar]
- 41. Grewal S., Herbert S. P., Ponnambalam S., Walker J. H. (2005) Cytosolic phospholipase A2-α and cyclooxygenase-2 localize to intracellular membranes of EA.hy. 926 endothelial cells that are distinct from the endoplasmic reticulum and the Golgi apparatus. FEBS J. 272, 1278–1290 [DOI] [PubMed] [Google Scholar]
- 42. Freeman E. J., Ruehr M. L., Dorman R. V. (1998) ANG II-induced translocation of cytosolic PLA2 to the nucleus in vascular smooth muscle cells. Am. J. Physiol. 274, C282–C288 [DOI] [PubMed] [Google Scholar]
- 43. Bunt G., de Wit J., van den Bosch H., Verkleij A. J., Boonstra J. (1997) Ultrastructural localization of cPLA2 in unstimulated and EGF/A23187-stimulated fibroblasts. J. Cell Sci. 110, 2449–2459 [DOI] [PubMed] [Google Scholar]
- 44. Bunt G., van Rossum G. S., Boonstra J., van Den Bosch H., Verkleij A. J. (2000) Regulation of cytosolic phospholipase A2 in a new perspective. Recruitment of active monomers from an inactive clustered pool. Biochemistry 39, 7847–7850 [DOI] [PubMed] [Google Scholar]
- 45. Leizer T., Clarris B. J., Ash P. E., van Damme J., Saklatvala J., Hamilton J. A. (1987) Interleukin-1β and interleukin-1α stimulate the plasminogen activator activity and prostaglandin E2 levels of human synovial cells. Arthritis Rheum. 30, 562–566 [DOI] [PubMed] [Google Scholar]
- 46. Leclerc P., Wähämaa H., Idborg H., Jakobsson P. J., Harris H. E., Korotkova M. (2013) IL-1β/HMGB1 complexes promote the PGE2 biosynthesis pathway in synovial fibroblasts. Scand. J. Immunol. 77, 350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chi P. L., Luo S. F., Hsieh H. L., Lee I. T., Hsiao L. D., Chen Y. L., Yang C. M. (2011) Cytosolic phospholipase A2 induction and prostaglandin E2 release by interleukin-1β via the myeloid differentiation factor 88-dependent pathway and cooperation of p300, Akt, and NF-κB activity in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 63, 2905–2917 [DOI] [PubMed] [Google Scholar]
- 48. Ghosh M., Tucker D. E., Burchett S. A., Leslie C. C. (2006) Properties of the group IV phospholipase A2 family. Prog. Lipid Res. 45, 487–510 [DOI] [PubMed] [Google Scholar]
- 49. Burke J. E., Dennis E. A. (2009) Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 50, S237–S242 [DOI] [PMC free article] [PubMed] [Google Scholar]