

Background: In the central nervous system, the role of LCN2 as a chemokine inducer has been previously reported.
Results: Peripheral nerve injury increased LCN2 expression. Lcn2 deficiency attenuated pain hypersensitivity, microglial activation, and chemokine production. Chemokine expression and pain behavior were induced by LCN2.
Conclusion: The LCN2-chemokine axis plays a critical role in the pathogenesis of neuropathic pain.
Significance: LCN2 can be targeted for treatment of neuropathic pain.
Keywords: Chemokines, Glia, Nerve, Neuroinflammation, Pain, Neuropathic Pain, Spinal Cord
Abstract
Lipocalin 2 (LCN2), which is also known as 24p3 and neutrophil gelatinase-associated lipocalin (NGAL), binds small, hydrophobic ligands and interacts with cell surface receptor 24p3R to regulate diverse cellular processes. In the present study, we examined the role of LCN2 in the pathogenesis of neuropathic pain using a mouse model of spared nerve injury (SNI). Lcn2 mRNA levels were significantly increased in the dorsal horn of the spinal cord after SNI, and LCN2 protein was mainly localized in neurons of the dorsal and ventral horns. LCN2 receptor 24p3R was expressed in spinal neurons and microglia after SNI. Lcn2-deficient mice exhibited significantly less mechanical pain hypersensitivity during the early phase after SNI, and an intrathecal injection of recombinant LCN2 protein elicited mechanical pain hypersensitivity in naive animals. Lcn2 deficiency, however, did not affect acute nociceptive pain. Lcn2-deficient mice showed significantly less microglial activation and proalgesic chemokine (CCL2 and CXCL1) production in the spinal cord after SNI than wild-type mice, and recombinant LCN2 protein induced the expression of these chemokines in cultured neurons. Furthermore, the expression of LCN2 and its receptor was detected in neutrophils and macrophages in the sciatic nerve following SNI, suggesting the potential role of peripheral LCN2 in neuropathic pain. Taken together, our results indicate that LCN2 plays a critical role in the development of pain hypersensitivity following peripheral nerve injury and suggest that LCN2 mediates neuropathic pain by inducing chemokine expression and subsequent microglial activation.
Introduction
Neuropathic pain is a severe chronic pain caused by nerve damages such as nerve compression, nerve trauma, diabetes, infection with herpes zoster virus, autoimmune disease, or cancer (1). Growing evidence suggests that the activation of glial cells, such as astrocytes and microglia, in the central nervous system (CNS) plays critical roles in the pathogenesis of neuropathic pain (2, 3). Activated spinal microglia release a variety of proinflammatory mediators, such as, cytokines, chemokines, prostaglandins, and nitric oxide (NO) in the dorsal spinal cord (4–7). These mediators can enhance neuronal excitability and increase pain-associated neurotransmitter release from sensory afferents, leading to pain enhancement or central sensitization (8–11). In fact, it has been well demonstrated that chemokines play a critical role in the development of neuropathic pain (6, 12).
Lipocalin-2 (LCN2),2 also known as 24p3 or neutrophil gelatinase-associated lipocalin (NGAL), is a 25-kDa glycoprotein that was initially identified in neutrophil granules (13). LCN2 has various functions; for example, it is involved in the transport of fatty acids and iron and in the regulation of cell death/survival and innate immunity (14). LCN2 also protects against bacterial infections (15, 16) and kidney injury (17) and is an acute phase protein induced by injury, infection, or other inflammatory stimuli (18). Brain-type organic cation transporter (24p3R) has been shown to be a cell surface receptor for LCN2 (19). In one study, LCN2 was increased in the prefrontal cortex during inflammatory pain (20), and in another, LCN2 was found to regulate stress-induced changes in spine morphology, neuronal excitability, and anxiety (21). We previously reported that LCN2 protein is secreted by glial cells in culture and that it regulates cell death/survival, motility, and morphology of glia (22, 23) as well as neurons (24). More recently, we reported that LCN2 plays an important role as a chemokine inducer under inflammatory conditions of the CNS (25). Thus, based on these previous findings, we hypothesized that LCN2 might be involved in the development of pain hypersensitivity. This hypothesis was tested using a spared nerve injury (SNI) model. Our results based on Lcn2-deficient mice, an intrathecal injection of LCN2 protein, and cultured neurons suggest that the spinal LCN2-chemokine axis may contribute to the pathogenesis of neuropathic pain.
EXPERIMENTAL PROCEDURES
Animals
Wild-type C57BL/6 mice and Lcn2-deficient mice were obtained from Samtaco Inc. (Osan, South Korea) and Dr. Shizuo Akira (Osaka University, Japan), respectively. The absence of Lcn2 was confirmed by PCR analysis of genomic DNA. Animal experiments were approved by the Institutional Animal Care Committee of Kyungpook National University and were performed in accordance with the animal care guidelines of the National Institutes of Health. All efforts were made to minimize the number of animals used and to minimize animal suffering.
Animal Surgery
Surgery was performed on the left sides of mice under 2% isoflurane anesthesia; contralateral sides were left intact. For SNI, the three peripheral branches (the sural, common peroneal, and tibial nerves) of the sciatic nerve were exposed. The common peroneal and tibial nerves were then tightly ligated with 6-0 silk thread, and an ∼2-mm segment of the two nerves was removed (26, 27). Care was taken to avoid damage to the nearby sural nerve. After surgery, all wounds were irrigated with sterile saline and closed in layers. Non-operated animals were used as naive controls. The day of surgery was set as day 0.
Behavioral Testing
Animals were habituated to the experimental room for at least 1 h before behavioral testing, which was performed by two investigators. To quantify foot mechanical sensitivity, mice were placed under a transparent plastic box on a metal mesh floor, and a logarithmic series of calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL) was applied to left and right hind paws to determine the stimulus intensity threshold stiffness required to elicit hind paw withdrawal responses. The 50% paw withdrawal threshold was measured using Dixon's up-and-down method (28, 29). The time course of response was determined by stimulating the lateral region of left and right hind paws before and after SNI surgery or an intrathecal injection of recombinant LCN2 protein. Sensitivity of response to noxious mechanical stimuli was assessed using the withdrawal frequency method (30) by calculating the frequency of paw withdrawal for each monofilament (2.0 or 4.0 g). Thermal pain sensitivity was assessed using the tail immersion test. Mice were gently restrained in a 50-ml conical tube, and the distal third of the tail was immersed into a water bath at 50 or 55 °C. The tail flick latencies to response as indicated by vigorous tail flexion were calculated by averaging three measurements. A cutoff time of 15 s was used to prevent tissue damage. Open field test was conducted as described previously (31).
Purification of Recombinant LCN2 Protein
Bacterially expressed recombinant mouse LCN2 protein was prepared as described previously (23). In brief, recombinant mouse LCN2 protein was expressed as a glutathione S-transferase (GST) fusion protein in BL21 strain Escherichia coli, which does not synthesize siderophore. LCN2 protein was purified using glutathione-Sepharose 4B beads (GE Healthcare) by elution with either thrombin or glutathione. Denatured LCN2 protein, which was incubated for 10 min at 100 °C, was used as a control. The bacterially expressed LCN2 protein was used only for the cortical neuron culture experiment.
Primary Culture of Microglia and Cortical Neurons
Primary microglia cultures were prepared as described previously (32). Primary cultures of dissociated cerebral cortical neurons were prepared from embryonic day 20 ICR mice, as described previously (33, 34). Briefly, mouse embryos were decapitated, and brains were rapidly removed and placed in a culture dish containing cold phosphate-buffered saline (PBS). Cortices were then isolated and transferred to a culture dish containing 0.25% trypsin-EDTA (Life Technologies) in PBS for 30 min at 37 °C. After two washes in serum-free Neurobasal medium (Life Technologies), cortical tissues were mechanically dissociated by gentle pipetting. Dissociated cortical cells in Neurobasal medium containing 10% FBS, 0.5 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, N2 supplement (Life Technologies), and a B27 supplement (Life Technologies) were then seeded onto 6-well plates coated with poly-d-lysine (Falcon; BD Biosciences). The cells were cultured at 37 °C in a 5% CO2 humidified atmosphere; media were changed every 2–3 days.
Intrathecal Injection of LCN2 Protein
A single intrathecal injection was performed by direct lumbar puncture between the L5 and L6 vertebrae using a 25-μl Hamilton syringe with a 30-gauge needle, as described previously (35). Recombinant mouse LCN2 protein expressed in mammalian cells was purchased from R&D Systems (Minneapolis, MN) and diluted in PBS. Mice were injected intrathecally with 0.1 or 1 μg of LCN2 protein in a volume of 10 μl. Correct intrathecal localization was confirmed by a tail flick upon penetration. PBS was used as the vehicle control.
Immunohistochemistry
Mice were deeply anesthetized and perfused through the aorta with 0.1 m PBS followed by 4% paraformaldehyde fixative. The L4/5 spinal cord and the sciatic nerve were dissected out and postfixed in the same fixative overnight before specimens were cryoprotected in 30% sucrose in 0.1 m PBS overnight at 4 °C. A cryostat was used to prepare 10-μm sections of sciatic nerve, which were mounted on gelatin-coated slides, and 30-μm sections of spinal cord, which were placed in PBS and stained with immunoperoxidase, as described previously (36). Cryostat sections were blocked with 4% normal serum in 0.3% Triton X-100 for 1 h at room temperature and then hybridized with primary antibodies (rabbit anti-mouse Iba-1 antibody (1:1000; Wako, Osaka, Japan) and rabbit anti-phospho-p38 mitogen-activated protein kinase (MAPK) antibody (1:1000; Cell Signaling Technology, Beverly, MA)) overnight at 4 °C. Sections were then washed in PBS containing 0.1% Tween 20 (PBS-T) and incubated at room temperature with biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA) for 2 h at a dilution of 1:200. After several washes in PBS-T, sections were incubated for 1 h at room temperature in avidin-biotin-peroxidase complex (1:100 dilution; ABC Elite; Vector Laboratories). The horseradish peroxidase reaction was developed in 0.1 m Tris-buffered saline (pH 7.4) containing 0.05% 3,3′-diaminobenzidine. Sections were then dehydrated, mounted onto glass microscope slides, and cover-slipped. For immunofluorescence staining, sections were incubated with primary antibodies against LCN2 (goat, 1:500; R&D Systems), 24p3R (rabbit, 1:200; Sigma-Aldrich), NeuN (mouse, 1:200; Millipore, Billerica, MA), phospho-p38 MAPK (rabbit, 1:200; Cell Signaling Technology), GFAP (mouse, 1:200; BD Biosciences), Iba-1 (mouse, 1:200; Wako), or Ly6G (rat, 1:200; BD Biosciences) overnight at 4 °C. They were then incubated with FITC- or Cy3-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA). Slides were washed, cover-slipped with VECTASHIELD mounting medium (Vector Laboratories), and visualized under a fluorescence microscope.
Traditional Reverse Transcription-PCR and Real-time PCR
Mice were deeply anesthetized and perfused with PBS through the aorta to remove blood, and the lumbar spinal cord and dorsal root ganglion (DRG) were then rapidly dissected out. To isolate the dorsal horn of the spinal cord, the portion corresponding to segments L4–L6 was divided into four constituent quadrants; dorsal right, dorsal left, ventral right, and ventral left. These were then immediately frozen in liquid nitrogen and immediately homogenized in TRIzol reagent (Life Technologies) for total RNA isolation. Total RNA (2 μg) from each sample was reverse-transcribed into cDNA using a First Strand cDNA synthesis kit (MBI Fermentas, Hanover, Germany). PCR was performed by using a DNA Engine Tetrad Peltier thermal cycler (MJ Research, Waltham, MA). To analyze PCR products, 10 μl of each PCR was electrophoresed in 1% agarose gel and detected under UV light. GAPDH was used as an internal control. Real-time PCR was performed using the one-step SYBR® PrimeScriptTM RT-PCR kit (Perfect Real Time; Takara Bio Inc., Tokyo) and the ABI Prism® 7000 sequence detection system (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The 2−ΔΔCT method was used to calculate relative changes in gene expression determined by real-time PCR experiments (37). GAPDH was used as an internal control. The nucleotide sequences of the primers used are listed in Table 1.
TABLE 1.
Nucleotide sequences of the primers used in RT-PCR
F, forward primer; R, reverse primer.
| Mouse cDNA | RT-PCR methods | Primer sequences |
|---|---|---|
| BDNF | Real-time | F, 5′-CGG CGC CCA TGA AAG AAG TA-3′ |
| R, 5′-AGA CCT CTC GAA CCT GCC CT-3′ | ||
| Ctss (cathepsin S) | Real-time | F, 5′-CCG AAG CTT TCC AGT ACA TCA-3′ |
| R, 5′-TGA GTT ATA GTG ACA CTT TTC ATC CA-3′ | ||
| CCL2 | Real-time | F, 5′-TCA GCC AGA TGC AGT TAA CG-3′ |
| R, 5′-GAT CCT CTT GTA GCT CTC CAG C-3′ | ||
| CCL3 | Real-time | F, 5′-ACT GCC TGC TGC TTC TCC TAC A-3′ |
| R, 5′-AGG AAA ATG ACA CCT GGC TGG-3′ | ||
| CCL5 | Real-time | F, 5′-CTC ACC ATC ATC CTC ACT GCA-3′ |
| R, 5′-GCA CTT GCT GCT GGT GTA GAA A-3′ | ||
| CXCL1 | Real-time | F, 5′-CAC ACT CAA GAA TGG TCG CGA-3′ |
| R, 5′-TTG TCA GAA GCC AGC GTT CAC-3′ | ||
| CXCL10 | Real-time | F, 5′-AAG TGC TGC CGT CAT TTT CT-3′ |
| R, 5′-GTG GCA ATG ATC TCA ACA CG-3′ | ||
| GAPDH | Real-time | F, 5′-TGG GCT ACA CTG AGC ACC AG-3′ |
| R, 5′-GGG TGT CGC TGT TGA AGT CA-3′ | ||
| GAPDH | Traditional | F, 5′-ACC ACA GTC CAT GCC ATC AC-3′ |
| R, 5′-TCC ACC ACC CTG TTG CTG TA-3′ | ||
| IL-1β | Real-time | F, 5′-AAG TTG ACG GAC CCC AAA AGA T-3′ |
| R, 5′-TGT TGA TGT GCT GCT GCG A-3′ | ||
| IL-6 | Real-time | F, 5′-AGT TGC CTT CTT GGG ACT GA-3′ |
| R, 5′-TCC ACG ATT TCC CAG AGA AC-3′ | ||
| LCN2 | Traditional | F, 5′-ATG TCA CCT CCA TCC TGG TC-3′ |
| R, 5′-CAC ACT CAC CAC CCA TTC AG-3′ | ||
| TNF-α | Real-time | F, 5′-ATG GCC TCC CTC TCA GTT C-3′ |
| R, 5′-TTG GTG GTT TGC TAC GAC GTG-3′ |
LCN2 ELISA
The levels of LCN2 protein were measured by indirect ELISA using a goat anti-mouse LCN2 antibody as the capture antibody and HRP-conjugated anti-goat IgG antibody as a secondary antibody, respectively, followed by colorimetric detection.
Quantification and Statistical Analysis
The numbers of cells showing Iba-1 or phospho-p38 MAPK immunoreactivity in superficial laminae (100 × 100-μm2 rectangular field for laminae I–III) were quantified in three consecutive sections (taken at 200-μm intervals) per L4/L5 spinal segment. An observing field of 104 μm2 was placed on the areas of the lateral, central, and medial dorsal horn, and cells that stained positively for each marker were counted. All results are presented as means ± S.E. The effects of different treatments were compared using Student's t test. Differences with p values of < 0.05 were considered statistically significant.
RESULTS
LCN2 Expression in the Spinal Cord after SNI
To determine the role of LCN2 in neuropathic pain, we first examined the expression of LCN2 in the spinal cords of SNI-injured mice by RT-PCR. Lcn2 mRNA levels in the ipsilateral dorsal horn of the spinal cord were significantly increased at 1 or 3 days after SNI (Fig. 1A), and the levels of Lcn2 mRNA expression in the ipsilateral side at 3 days after SNI were higher than those in the contralateral side (Fig. 1B). Lcn2 mRNA expression was not significantly induced by SNI in dorsal root ganglia (DRGs) (Fig. 1C). We then examined the localization of LCN2 protein in spinal cords after SNI using an immunohistochemical method (Fig. 2). In the spinal cords of naive control mice, minimal LCN2 immunoreactivity (IR) was detected in the deep layer of the spinal dorsal horn and in motor neurons of the ventral horn. However, at 3 days after SNI, LCN2 IR was markedly increased in the motor neurons of the ipsilateral ventral horn, whereas LCN2 IR in the ipsilateral dorsal horn was slightly increased (Fig. 2). The SNI-induced increase in the LCN2 protein levels in the spinal cord was confirmed by ELISA: naive animals, 10.51 ± 4.46 ng/ml; 1 day after SNI, 23.77 ± 3.46 ng/ml; 3 days after SNI, 20.87 ± 0.90 ng/ml (n = 3, p < 0.05). To determine the cellular distribution of LCN2, we performed double immunostaining for LCN2 using different cell markers at 3 days after SNI. LCN2 IR in the dorsal and ventral horns after SNI was found to be co-localized with the neuronal marker NeuN (Fig. 2A), but not with the microglia marker Iba-1 or the astrocyte marker GFAP (Fig. 2B).
FIGURE 1.

Expression of Lcn2 mRNA in the spinal cord after SNI. A–C, the expression of Lcn2 mRNA in the spinal cord or DRG after SNI was assessed using traditional RT-PCR (A, left, and B and C) or real-time PCR (A, right). Lcn2 mRNA levels in ipsilateral region of the dorsal horn of the spinal cord were significantly increased at 1–3 days after SNI and then decreased to basal levels (A). The mRNA levels of Lcn2 in ipsilateral dorsal horn were significantly higher than those in contralateral sides in three animals (contra- or ipsi-1, -2, and -3) at 3 days after SNI (B). The mRNA levels of Lcn2 in the DRG were not significantly changed by SNI at any time point (C). Results are representative of either three independent experiments or means ± S.E. *, p < 0.05 versus the control group or contralateral versus ipsilateral side, n = 3. Results of densitometric analyses normalized to GAPDH are shown either below or to the right of the gels.
FIGURE 2.
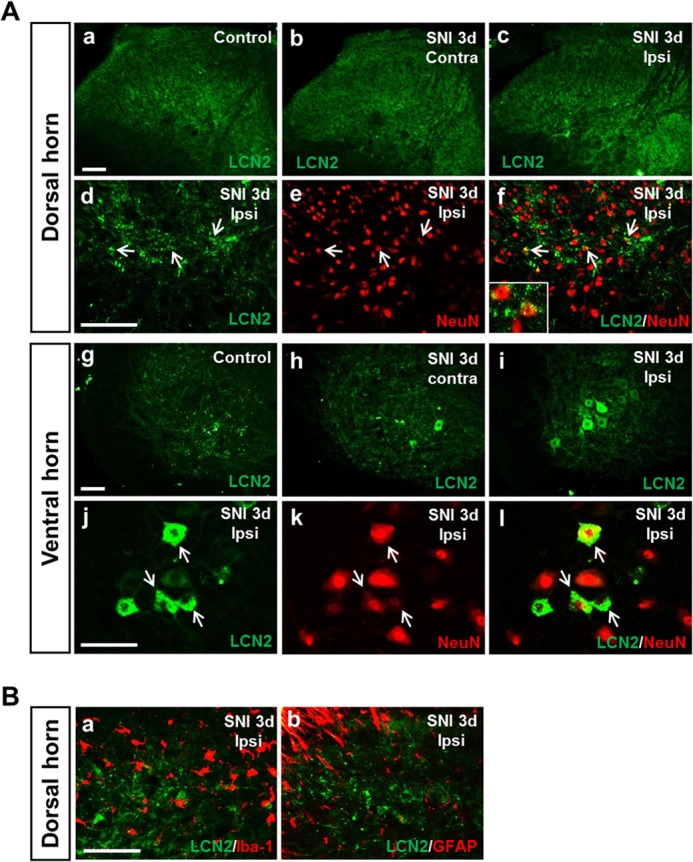
Immunolocalization of LCN2 in the spinal cord after SNI. A, a low level of LCN2 immunoreactivity was detected in the spinal cord of naive control mice (panels a and g). At 3 days after SNI, LCN2 immunoreactivity was detected in the contralateral (Contra) dorsal and ventral horn (panels b and h) and in the ipsilateral (Ipsi) dorsal and ventral horn (panels c and i). Scale bars, 100 μm. Double immunostaining showed that LCN2 (green) in the dorsal (panels d–f) and ventral horn (panels j–l) of the spinal cord was co-localized with NeuN (red), a neuronal marker. Arrows indicate examples of doubly labeled cells. Inset in panel f, high magnification image showing doubly labeled cells in the ipsilateral dorsal horn. Scale bars, 100 μm. B, double immunostaining showed that LCN2 (green) in the ipsilateral dorsal horn of the spinal cord at 3 days after SNI was not co-localized with Iba-1 (red) (panel a) or GFAP (red) (panel b). Scale bars, 100 μm. Results are a representative of more than three independent experiments.
Inhibition of Mechanical Nociceptive Behavior in Lcn2-deficient Mice
To investigate the role of LCN2 in the pathogenesis of pain hypersensitivity after peripheral nerve injury, we compared the pain behaviors of Lcn2-deficient mice and wild-type mice following SNI. Sensitivity to mechanical stimuli was measured in injured ipsilateral and uninjured contralateral hind paws before and for 10 days after surgery. Following SNI, wild-type mice developed pain hypersensitivity in the ipsilateral side as compared with either the contralateral side or the base-line values before surgery. However, SNI-induced mechanical allodynia was significantly attenuated at 1–3 days after SNI in Lcn2-deficient mice (Fig. 3A). The behavioral difference between wild-type and Lcn2-deficient mice was confirmed using the littermates obtained from mating of heterozygous animals (data not shown). To investigate the effect of exogenous LCN2 on pain sensitivity, we injected recombinant LCN2 protein (0.1 or 1 μg) intrathecally into wild-type mice. At the higher dose, a significant reduction in paw withdrawal threshold was detected within 3 h of injection, and this reduction was maintained throughout the 3-day testing period, whereas the paw withdrawal threshold of mice treated with the lower dose (0.1 μg) was reduced at 3 h to 1 day after injection and then returned to base-line values (Fig. 3B). No significant difference in pain sensitivity was detected in vehicle-injected control mice throughout the experiment. Intrathecal injection of LCN2 (1 μg) also induced pain behavior in Lcn2-deficient mice. As compared with wild-type animals, KO mice showed delayed LCN2-induced pain hypersensitivity (data not shown). To determine whether acute nociceptive pain behavior is altered in Lcn2-deficient mice, we compared responses of Lcn2-deficient mice and the wild type to acute mechanical and thermal stimuli. In the von Frey filament test, no significant difference in paw withdrawal frequency after mechanical stimulation (2.0 and 4.0 g) was found between the two groups. Similarly, the tail immersion test revealed no significant difference in latencies to thermal stimuli at 50 or 55 °C between wild-type and Lcn2 knock-out mice (data not shown). Given the expression of LCN2 in the ventral horn, we examined whether intrathecal LCN2 administration or genetic deletion of LCN2 affected locomotion. No significant difference was found in the locomotive activity in the open field test between WT and Lcn2 KO mice after SNI, excluding potentially confounding effects of LCN2 on locomotion that might affect reflexive responses (data not shown).
FIGURE 3.
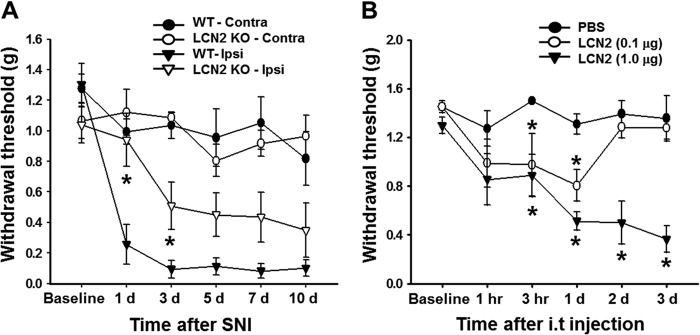
Effect of LCN2 on mechanical allodynia. A, SNI induced a significant decrease in paw withdrawal threshold in wild-type (WT) mice, whereas SNI-induced mechanical allodynia was significantly inhibited at 1–3 days in Lcn2-deficient (KO) mice. Data are means ± S.E. *, p < 0.05 as compared with the ipsilateral hind paw of wild-type mice, n = 5–6. Intrathecal injection of LCN2 protein (0.1 and 1 μg) into naive mice induced mechanical allodynia. Contra, contralateral side; Ipsi, ipsilateral side. B, mechanical allodynia increased rapidly 3 h after LCN2 (1 μg) administration and was maintained for 3 days. The results shown are means ± S.E. *, p < 0.05 versus the vehicle (PBS) group, n = 4–5.
Attenuation of Microglial Activation and p38 MAPK Phosphorylation in Lcn2-deficient Mice after SNI
We previously reported LCN2 protein is secreted by glial cells in culture and that it promotes their migration and causes morphological changes (22, 23). In the present study, we investigated the effect of LCN2 on the activation of spinal microglia in neuropathic pain states. Iba-1 IR was higher in the ipsilateral spinal cord at 3 days after SNI as compared with the contralateral side (Fig. 4A). However, this induction of Iba-1 IR in the ipsilateral spinal cord was significantly attenuated in Lcn2-deficient mice (Fig. 4A), whereas in the contralateral side, no significant difference in Iba-1-positive cell number was observed between Lcn2-deficient and wild-type mice (Fig. 4, A and B). p38 MAPK has been shown to be activated in spinal microglia after peripheral nerve injury (38, 39), and thus, we examined the effect of LCN2 on p38 MAPK activation in the spinal cord after SNI. As was observed for Iba-1-positive cell number, increased activation of p38 MAPK was detected in the ipsilateral spinal cord at 3 days after SNI (Fig. 4A), and this was significantly attenuated in Lcn2-deficient mice (Fig. 4A). Lcn2-deficient mice also showed less p38 MAPK activation in contralateral sides as compared with the wild type (Fig. 4, A and C). Furthermore, the co-localization of Iba-1 and p38 MAPK was confirmed by double immunostaining (data not shown), indicating that spinal microglia are indeed activated by peripheral nerve injury concurrent with the phosphorylation of p38 MAPK. Because p38 MAPK in microglia is known to drive the release of signaling molecules that play a key role in the development of neuropathic pain following peripheral nerve injury, we further explored the cellular consequence of LCN2-mediated activation of p38 MAPK for microglia-to-neuron signaling. Recombinant LCN2 protein induced the expression of TNF-α and IL-1β, but not IL-6, BDNF, or cathepsin S in primary microglia cultures (data not shown).
FIGURE 4.

Effect of LCN2 on the activations of microglia and p38 MAPK after SNI. A, Iba-1 and phospho-p38 MAPK immunoreactivities were detected in the spinal dorsal (panels a–d) and ventral horn (panels i–l) of wild-type (WT) mice and in the spinal dorsal (panels e–h) and ventral horn (panels m–p) of Lcn2-deficient (KO) mice at 3 days after SNI. Scale bar, 100 μm. Results are representative of more than three independent experiments. B and C, quantified immunohistochemical data for Iba-1 and phospho-p38 MAPK IR are shown in the bar graphs, which display the number of Iba-1-positive cells (B) or phospho-p38 MAPK-positive cells (C) in the dorsal and ventral horn of the spinal cord at 3 days after SNI. Results are presented as means ± S.E. *, p < 0.05 versus the contralateral side; #, p < 0.05, the ipsilateral side of WT versus KO mice, n = 3. Contra, contralateral side; Ipsi, ipsilateral side.
Our previous studies suggested that LCN2 is a chemokine inducer in the brain (25), and therefore, we investigated how LCN2 participates in the regulation of the expression of the pain-associated chemokines, CCL2, CXCL1, CCL3, and CCL5, in the spinal cord following SNI (Fig. 5). Two days after SNI, the expression of CCL2 and CXCL1 was significantly increased in the ipsilateral spinal cords of wild-type mice (Fig. 5), but no such difference was observed in the expression of CCL3 or CCL5 (data not shown). Furthermore, the induction of CCL2 and CXCL1 following SNI was significantly reduced in Lcn2-deficient mice as compared with wild-type mice after SNI (Fig. 5). However, no significant difference was observed between the contralateral expression of CCL2 and CXCL1 in Lcn2-deficient and wild-type mice. To further examine the role of proalgesic chemokine CCL2 in the LCN2-induced pain hypersensitivity, we employed anti-CCL2-neutralizing antibody. Intrathecal injection of anti-CCL2 antibody (1 μg) significantly reduced the pain behavior following recombinant LCN2 protein administration, as determined by paw withdrawal threshold (n = 3, p < 0.05).
FIGURE 5.
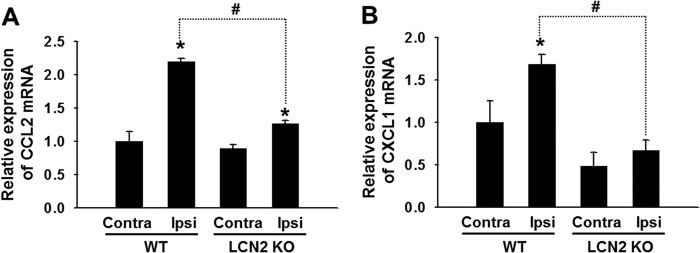
Effect of LCN2 on chemokine expression after SNI. A and B, relative mRNA expression of Ccl2 (A) and Cxcl1 (B) in the contralateral or ipsilateral side of the spinal cord at 2 days after SNI was evaluated by real-time RT-PCR. The expression of both chemokines was significantly attenuated in the spinal cord of Lcn2-deficient (KO) mice after SNI. Results are presented as means ± S.E. *, p < 0.05 versus the contralateral side (Contra); #, p < 0.05, the ipsilateral side (Ipsi) of WT versus KO mice, n = 3.
LCN2 Receptor (24p3R) Expression in the Spinal Cord after SNI
We next examined the expression of 24p3R in spinal cords after SNI (Fig. 6). 24p3R was detected at high levels in the spinal cords of naive control mice, mainly on neuron cell surfaces (Fig. 6, A and G). Following SNI, 24p3R expression was slightly different in control and SNI-injured mice. To determine the cellular distribution of 24p3R at 3 days after SNI, we performed double immunostaining for 24p3R using different cell markers. 24p3R IR in the dorsal horn of the spinal cord after SNI was co-localized with NeuN or Iba-1, but not with GFAP (Fig. 6, D–F). Similarly, in the ventral horn, 24p3R was expressed in NeuN- or Iba-1-positive cells (Fig. 6, J–L). To determine whether neurons co-express 24p3R and LCN2, double immunostaining was also performed for these two proteins. 24p3R and LCN2 were found to be co-localized in dorsal and ventral horns (data not shown). The results indicate that 24p3R is mainly expressed in LCN2-expressing neurons or microglia in the spinal cord. Alternatively, LCN2 may be made elsewhere and then taken up by endocytosis once it has bound to 24p3R target cells. This possibility has yet to be tested.
FIGURE 6.
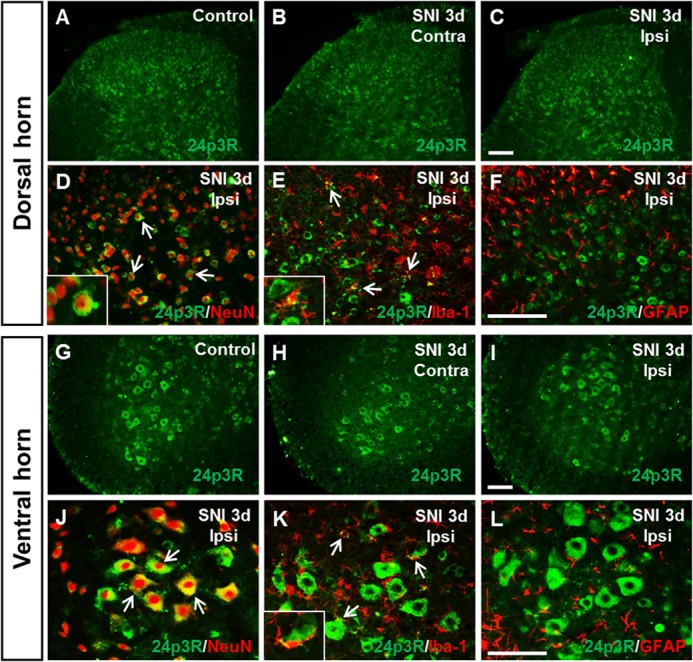
Immunolocalization of 24p3R (the LCN2 receptor) in spinal cord after SNI. A–C and G–I, 24p3R immunoreactivity in naive control animals (A and G), in the contralateral (Contra) dorsal and ventral horn of the spinal cord (B and H) and in the ipsilateral (Ipsi) dorsal and ventral horn (C and I) of the spinal cord at 3 days after SNI. Scale bars, 100 μm. D–F and J–L, double immunostaining showed that 24p3R (green) in the dorsal (D--F) and ventral horn (J–L) of the spinal cord was co-localized with NeuN (red) or Iba-1 (red), but not with GFAP (red). High magnification images (insets in D, E, and K) indicate doubly labeled cells in the ipsilateral dorsal (D and E) or ventral horn (K), respectively. Arrows indicate examples of doubly labeled cells. Scale bars, 100 μm. Results are representative of more than three independent experiments.
LCN2 Induction of Chemokine Expression in Primary Cortical Neuron Cultures
We determined whether LCN2 affected the expression of pain-associated chemokines, such as CCL2, CXCL1, CCL3, CCL5, and CXCL10, in primary cultures of cortical neurons (Fig. 7). Cultured neuronal cells were incubated with recombinant LCN2 protein (1 and 10 μg/ml) for 3, 6, or 12 h, and the mRNA levels of these chemokines were assessed by real-time RT-PCR. The mRNA levels of Ccl2 and Cxcl1 were dose- and time-dependently increased by LCN2 protein, and the expression of Ccl3, Ccl5, and Cxcl10 was up-regulated in cultured neuronal cells at 12 h after treatment. Denatured LCN2 protein (the control) had no effect.
FIGURE 7.

LCN2 induction of chemokine gene expression in primary cortical neurons. Primary cortical neurons were incubated with recombinant LCN2 protein (1 or 10 μg/ml) for 3, 6, or 12 h, and total RNA was isolated for real-time RT-PCR. A–E, the mRNA levels of chemokines, such as Ccl2 (A), Ccl3 (B), Ccl5 (C), Cxcl1 (D), and Cxcl10 (E) were determined. Results are means ± S.E. *, p < 0.05 versus the denatured LCN2 protein-treated group (Control) at each time point (n = 3).
LCN2 Expression in the Sciatic Nerve after SNI
We next examined the localization of LCN2 protein in the sciatic nerve after SNI (Fig. 8). Three sites of the sciatic nerve were examined: the site of ligature; a site 2 mm proximal to the ligature; and a site about 20 mm distal to the ligature at the peripheral branch (common peroneal and tibial nerves) of the sciatic nerve. No LCN2 IR was detected in the sciatic nerve of control animals (data not shown). LCN2 IR was markedly increased at the site of ligature of the ipsilateral sciatic nerve 3 days after SNI (Fig. 8, A and E), whereas LCN2 IR in the contralateral side of the sciatic nerve was not significantly different from that of control animals (data not shown). To determine the cellular distribution of LCN2, we performed double immunostaining for LCN2 using different cell type-specific markers at 3 days after SNI. LCN2 IR in the sciatic nerve after SNI was co-localized with the neutrophil marker Ly6G (Fig. 8, C, G, and K), but not with the macrophage marker Iba-1 (Fig. 8, B, F, and J). These results suggest that neutrophils are the major cellular source of LCN2 in the injured sciatic nerve. We also examined the expression of 24p3R in the sciatic nerve after SNI (Fig. 8, D, H, and L). 24p3R was detected at low levels in the sciatic nerve of naive control mice, whereas 24p3R expression was markedly increased at the site of ligature in the sciatic nerve 3 days following SNI (Fig. 8). 24p3R IR in the injured sciatic nerve was co-localized with Iba-1 or partly with Ly6G (data not shown), indicating that both macrophages and neutrophils express LCN2 receptor in the periphery.
FIGURE 8.
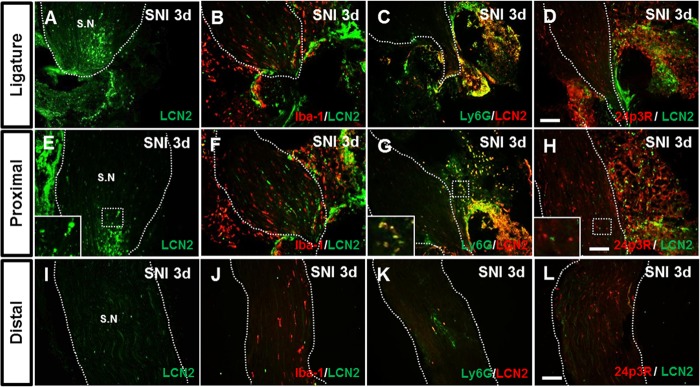
Immunolocalization of LCN2 in the sciatic nerve after SNI. A–L, LCN2 immunoreactivity was examined at the site of ligature (A–D) or proximal (E–H) and distal (I–L) sites of the sciatic nerve injury at 3 days after SNI. High magnification image (inset in E) indicates LCN2-positive cells in the sciatic nerve. Double immunostaining showed that LCN2 (green or red) in the sciatic nerve was co-localized with Ly6G (green; C, G, and K), but not with Iba-1 (red; B, F, and J) or 24p3R (red; D, H, and L). High magnification image (inset in G) indicates doubly labeled cells in the sciatic nerve. S.N, sciatic nerve. Scale bars, 100 μm. Results are representative of more than three independent experiments.
DISCUSSION
In the present study, we investigated whether LCN2 is involved in the pathogenesis of neuropathic pain following peripheral nerve injury. After SNI, Lcn2 mRNA was up-regulated in the spinal cord, and LCN2 protein was localized in spinal neurons. SNI-induced mechanical allodynia was remarkably attenuated during the early stage in the Lcn2-deficient mice as compared with wild-type mice. In addition, an intrathecal injection of recombinant LCN2 protein elicited mechanical hypersensitivity. Furthermore, the activation of microglia and p38 MAPK and the expression of proalgesic chemokines (CCL2 and CXCL1) were lower in Lcn2-deficient mice. These results strongly suggest that LCN2 in the spinal cord plays a pivotal role in the development of neuropathic pain following peripheral nerve injury (Fig. 9). Moreover, our subsequent studies revealed that the LCN2 receptor 24p3R is mainly expressed in spinal neurons or microglia. In a previous study and in the present study, recombinant LCN2 protein was found to induce chemokine expression in cultured neurons and microglia (25). Our results suggest that LCN2 expressed in spinal neurons contributes to the development of pain hypersensitivity following peripheral nerve injury by inducing the expression of proalgesic chemokines.
FIGURE 9.
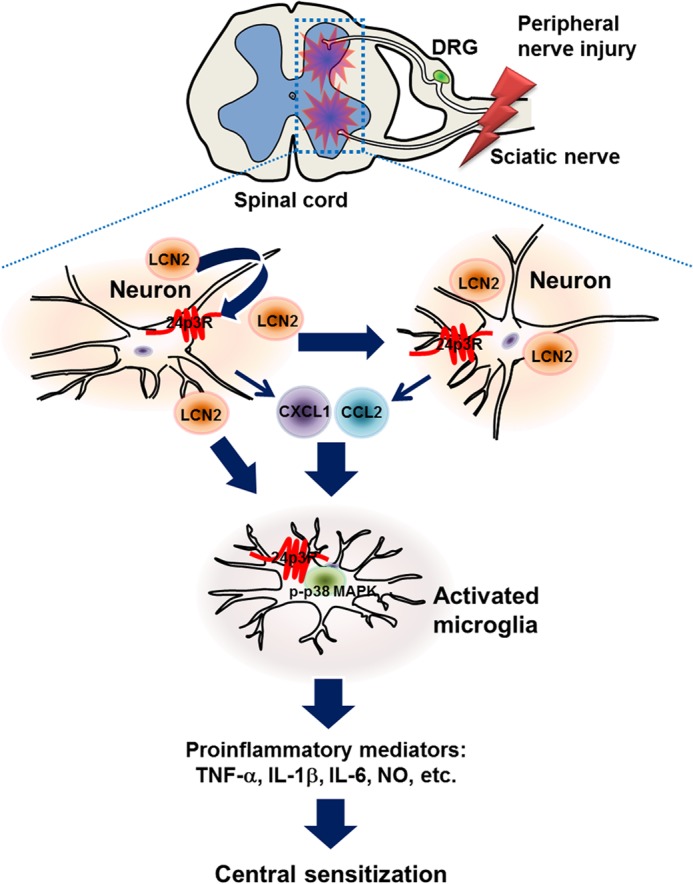
The involvement of LCN2 in the development of pain hypersensitivity following peripheral nerve injury. LCN2 synthesized in spinal neurons after peripheral nerve injury may be released into the extracellular space and bind to 24p3R, which is expressed on the surfaces of neurons and microglia (and astrocytes) in the spinal cord. We surmise that after binding to its receptor, LCN2 could induce the expression and release of chemokines from spinal neurons or other cell types. These chemokines may in turn activate spinal microglia, and thus, facilitate neuroinflammation and the trafficking of other glial and inflammatory cells, ultimately leading to pain sensitization in the spinal cord. Taken together, our findings suggest that the LCN2-chemokine axis plays a central role in the development of pain hypersensitivity under conditions such as neuropathic pain.
Following SNI, mice exhibited robust and prolonged mechanical allodynia as represented by decreases in paw withdrawal thresholds (Fig. 3), and this effect persisted for about 2 weeks after SNI. Lcn2 mRNA expression in the ipsilateral dorsal horn of the spinal cord was significantly increased for up to 3 days after SNI and then declined, despite the marked persistence of mechanical allodynia-like behavior. Furthermore, Lcn2-deficient mice exhibited significantly less mechanical allodynia in ipsilateral hind paws during the early phase after SNI. These results suggest that endogenous LCN2 plays an important role in the development of neuropathic pain, rather than in the maintenance of neuropathic pain. In addition, we also observed elevated LCN2 expression in the contralateral dorsal horn of spinal cord after SNI. Explanation for these observations remains to be done, but previous studies indicate that peripheral nerve injury can affect contralateral non-injured neurons, and thus, elicit mechanical allodynia in contralateral regions (29, 40). In the present study, immunofluorescence staining indicated elevated LCN2 levels in the neurons of the ipsilateral ventral horn. Because LCN2 is a secreted protein that is elevated in the cerebrospinal fluid of patients with neuroinflammatory disorders (41), LCN2 produced by ventral horn neurons may diffuse to the dorsal horn, and thus, influence pain hypersensitivity. Alternatively, LCN2-induced chemokines in the ventral horn may travel across the spinal cord to induce central sensitization in the dorsal horn. However, there is no direct evidence to support the “LCN2/chemokine diffusion” model yet. Further mechanistic studies are required to precisely understand the role and mechanism of LCN2 actions in pain.
We previously reported that LCN2 protein is secreted by glial cells in vitro and that it regulates glial cell death/survival, motility, and morphological phenotypes in an autocrine or paracrine manner (22, 23). In the present study, Lcn2-deficient mice showed markedly less microglial activation in the spinal cord after SNI. In the presence of neuropathic pain, microglia are activated by neurotransmitters and proinflammatory mediators, such as glutamate, ATP, and substance P, which are released from spinal neurons or peripheral afferent terminals (2, 4), and these activated microglia then help initiate and maintain enhanced pain signaling by releasing pro-nociceptive mediators (42, 43). However, the factors that activate spinal microglia in the neuropathic pain have not been clearly identified. p38 MAPK is activated in microglia after peripheral nerve injury and enhances proinflammatory cytokine production, and thus, promotes neuropathic pain (38, 44). In the present study, we found that after SNI spinal microglia and p38 MAPK in microglia were activated, the activation was significantly lower in Lcn2-deficient mice. These results strongly suggest that LCN2 contributes to the development of neuropathic pain by regulating microglial activation.
In addition, our observations that the expression of CCL2 and CXCL1 in the injured spinal cord was down-regulated in Lcn2-deficient animals and that recombinant LCN2 protein up-regulated the expression of these chemokines in cultured neurons indicate that LCN2 regulated the expression of CCL2 and CXCL1 in the spinal cord after SNI. CCL2 has been previously documented to trigger the activation of spinal astrocytes and microglia after peripheral nerve injury (45, 46), and treatment with anti-CCL2-neutralizing antibody or CCL2 receptor (CCR2) deficiency has been reported to reduce microglial activation after peripheral nerve injury (46–48). Consistently, anti-CCL2-neutralizing antibody administration reduced the LCN2-induced pain sensitivity in our study. CXCL1 exhibited potent chemotactic activity for the recruitment of neutrophils into both the CNS and the peripheral tissues (49, 50). Furthermore, several lines of evidence indicate that neutrophils infiltrate sites of peripheral nerve injury and contribute to the development of neuropathic pain (2, 51). Thus, chemokines, such as CCL2 and CXCL1, may be produced in response to LCN2 in the spinal cord and mediate microglial activation and ensuing neuropathic pain after peripheral nerve injury.
Consistent with this notion, our in vitro studies showed that treatment with LCN2 protein enhanced the expression of an array of chemokines, including CCL2, CCL3, CCL5, CXCL1, and CXCL10, in cultured neuronal cells. LCN2 has been reported to act as a chemokine inducer in glia and neurons in culture (25), and chemokines are known to have a role in the development and trafficking of leukocytes during immune and inflammatory responses (52, 53). Recent evidence indicates that chemokines can act as pro-nociceptive mediators following tissue injury and disease in the nervous system (54). After nerve injury, chemokines are released from injured nerve fibers and nearby immune cells (2) and from the DRGs of injured nerves (55). Within the spinal cord, the central terminals of primary afferents and spinal neurons are known to release chemokines, such as CCL2 and CX3CL1, which play important roles in the regulation of nociceptive response and in the establishment of neuropathic pain (46, 56, 57). CCL3 is also up-regulated in the macrophages and Schwann cells of injured sciatic nerves (58, 59), and the intraplantar injection of CCL3 or CCL5 induced tactile allodynia (60). These findings suggest that many different types of chemokines are involved in pain behavior and further support that the LCN2-chemokine axis contributes to the pathogenesis of neuropathic pain.
Recently, LCN2 and its receptor 24p3R were shown to be expressed in the neutrophils, astrocytes, microglia/macrophages, and neurons of the spinal cord following experimental autoimmune encephalomyelitis or spinal cord injury (61, 62). 24p3R, which was initially described as a brain type organic cation transporter (BOCT), is a cell surface receptor for LCN2 and is expressed in various organs (19, 25). Furthermore, 24p3R mediates the cellular uptake of LCN2 and diverse physiological processes (19). Here, we found that 24p3R was present at high levels in the normal spinal cord and localized to the neuronal cell surfaces. However, after SNI, 24p3R was expressed in spinal cord microglia as well as neurons. We also found that 24p3R was largely localized in spinal neurons that expressed LCN2 after SNI. In a recent study, it was suggested that 24p3R-expressing spinal neurons might be sensitive to LCN2 (62), and in a previous in vitro study, we suggested that LCN2 is capable of regulating diverse phenotypes of neurons in culture by acting on 24p3R (24). Together, these findings show that 24p3R, predominantly expressed in neurons and to some extent in microglia, may mediate the actions of LCN2 under the neuropathic pain condition. Furthermore, in our preliminary study, we found that 24p3R was also expressed in astrocytes in the white matter of the spinal cord,3 which suggests that 24p3R expression is rather widespread and occurs in the glia of gray and white matter of the spinal cord as well as in neurons.
Neutrophils infiltrating into the damaged peripheral nerves participate in the development of neuropathic pain and can release various inflammatory mediators such as lipoxygenase products, nitric oxide, cytokines, and chemokines (51, 63–65). Recently, locally accumulated neutrophils have been shown to exacerbate neuronal injury and to increase neuronal activity and excitability (66). In the present study, LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL) (13), was markedly increased in neutrophils in the damaged sciatic nerves, suggesting that LCN2 may also be involved in the early inflammatory response in the peripheral tissues after nerve injury. In addition, 24p3R was expressed in the infiltrated neutrophils and macrophages in the damaged sciatic nerve. Taken together, LCN2 expression in the damaged peripheral nerve may also contribute to the initiation of neuropathic pain and may play an important role in the recruitment of neutrophils and macrophages.
In conclusion, our results suggest that LCN2 synthesized by and secreted from spinal neurons and peripheral neutrophils following nerve injury is critically involved in the development of pain hypersensitivity by acting on its receptor, 24p3R, which is widely expressed in the spinal cord and peripheral tissues. Furthermore, our mechanistic studies indicate that LCN2 appears to cause spinal microglial activation by inducing the expression of proalgesic chemokines. These findings indicate that the LCN2-chemokine axis should be regarded a target for the treatment of neuropathic pain.
This work was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A111345), by National Research Foundation (NRF) grants funded by the Ministry of Education, Science and Technology (MEST) of the Korean government (Grant 2012-0009328), and by the Kyungpook National University Research Fund, 2012.
S. Jeon, M. K. Jha, J. Ock, J. Seo, M. Jin, H. Cho, W.-H. Lee, and K. Suk, unpublished data.
- LCN2
- lipocalin-2
- CCL
- chemokine (CC motif) ligand
- CXCL
- chemokine (CXC motif) ligand
- Iba-1
- ionized calcium binding adaptor molecule 1
- GFAP
- glial fibrillary acidic protein
- NeuN
- neuronal nuclei
- DRG
- dorsal root ganglion
- SNI
- spared nerve injury
- IR
- immunoreactivity.
REFERENCES
- 1. Campbell J. N., Meyer R. A. (2006) Mechanisms of neuropathic pain. Neuron 52, 77–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scholz J., Woolf C. J. (2007) The neuropathic pain triad: neurons, immune cells, and glia. Nat. Neurosci. 10, 1361–1368 [DOI] [PubMed] [Google Scholar]
- 3. Watkins L. R., Milligan E. D., Maier S. F. (2001) Glial activation: a driving force for pathological pain. Trends Neurosci. 24, 450–455 [DOI] [PubMed] [Google Scholar]
- 4. Milligan E. D., Watkins L. R. (2009) Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inoue K., Tsuda M. (2009) Microglia and neuropathic pain. Glia 57, 1469–1479 [DOI] [PubMed] [Google Scholar]
- 6. Gao Y. J., Ji R. R. (2010) Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 126, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raghavendra V., Tanga F., Rutkowski M. D., DeLeo J. A. (2003) Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain 104, 655–664 [DOI] [PubMed] [Google Scholar]
- 8. Clark A. K., Gentry C., Bradbury E. J., McMahon S. B., Malcangio M. (2007) Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur. J. Pain. 11, 223–230 [DOI] [PubMed] [Google Scholar]
- 9. Tawfik V. L., Nutile-McMenemy N., Lacroix-Fralish M. L., Deleo J. A. (2007) Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav. Immun. 21, 238–246 [DOI] [PubMed] [Google Scholar]
- 10. Biber K., Tsuda M., Tozaki-Saitoh H., Tsukamoto K., Toyomitsu E., Masuda T., Boddeke H., Inoue K. (2011) Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. 30, 1864–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ledeboer A., Gamanos M., Lai W., Martin D., Maier S. F., Watkins L. R., Quan N. (2005) Involvement of spinal cord nuclear factor κB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur. J. Neurosci. 22, 1977–1986 [DOI] [PubMed] [Google Scholar]
- 12. White F. A., Jung H., Miller R. J. (2007) Chemokines and the pathophysiology of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 104, 20151–20158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kjeldsen L., Bainton D. F., Sengeløv H., Borregaard N. (1994) Identification of neutrophil gelatinase-associated lipocalin as a novel matrix protein of specific granules in human neutrophils. Blood 83, 799–807 [PubMed] [Google Scholar]
- 14. Flower D. R. (1996) The lipocalin protein family: structure and function. Biochem. J. 318, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goetz D. H., Holmes M. A., Borregaard N., Bluhm M. E., Raymond K. N., Strong R. K. (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10, 1033–1043 [DOI] [PubMed] [Google Scholar]
- 16. MacManus J. P., Graber T., Luebbert C., Preston E., Rasquinha I., Smith B., Webster J. (2004) Translation-state analysis of gene expression in mouse brain after focal ischemia. J. Cereb. Blood Flow Metab. 24, 657–667 [DOI] [PubMed] [Google Scholar]
- 17. Mori K., Nakao K. (2007) Neutrophil gelatinase-associated lipocalin as the real-time indicator of active kidney damage. Kidney Int. 71, 967–970 [DOI] [PubMed] [Google Scholar]
- 18. Nilsen-Hamilton M., Liu Q., Ryon J., Bendickson L., Lepont P., Chang Q. (2003) Tissue involution and the acute phase response. Ann. N.Y. Acad. Sci. 995, 94–108 [DOI] [PubMed] [Google Scholar]
- 19. Devireddy L. R., Gazin C., Zhu X., Green M. R. (2005) A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123, 1293–1305 [DOI] [PubMed] [Google Scholar]
- 20. Poh K. W., Yeo J. F., Stohler C. S., Ong W. Y. (2012) Comprehensive gene expression profiling in the prefrontal cortex links immune activation and neutrophil infiltration to antinociception. J. Neurosci. 32, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mucha M., Skrzypiec A. E., Schiavon E., Attwood B. K., Kucerova E., Pawlak R. (2011) Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc. Natl. Acad. Sci. U.S.A. 108, 18436–18441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee S., Lee J., Kim S., Park J. Y., Lee W. H., Mori K., Kim S. H., Kim I. K., Suk K. (2007) A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J. Immunol. 179, 3231–3241 [DOI] [PubMed] [Google Scholar]
- 23. Lee S., Park J. Y., Lee W. H., Kim H., Park H. C., Mori K., Suk K. (2009) Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J. Neurosci. 29, 234–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee S., Lee W. H., Lee M. S., Mori K., Suk K. (2012) Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J. Neurosci. Res. 90, 540–550 [DOI] [PubMed] [Google Scholar]
- 25. Lee S., Kim J. H., Kim J. H., Seo J. W., Han H. S., Lee W. H., Mori K., Nakao K., Barasch J., Suk K. (2011) Lipocalin-2 is a chemokine inducer in the central nervous system: role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J. Biol. Chem. 286, 43855–43870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourquin A. F., Süveges M., Pertin M., Gilliard N., Sardy S., Davison A. C., Spahn D. R., Decosterd I. (2006) Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 122, 14 e11–14 [DOI] [PubMed] [Google Scholar]
- 27. Decosterd I., Woolf C. J. (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 [DOI] [PubMed] [Google Scholar]
- 28. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 [DOI] [PubMed] [Google Scholar]
- 29. Milligan E. D., Twining C., Chacur M., Biedenkapp J., O'Connor K., Poole S., Tracey K., Martin D., Maier S. F., Watkins L. R. (2003) Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J. Neurosci. 23, 1026–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mansikka H., Zhao C., Sheth R. N., Sora I., Uhl G., Raja S. N. (2004) Nerve injury induces a tonic bilateral μ-opioid receptor-mediated inhibitory effect on mechanical allodynia in mice. Anesthesiology 100, 912–921 [DOI] [PubMed] [Google Scholar]
- 31. Jang E., Lee S., Kim J. H., Kim J. H., Seo J. W., Lee W. H., Mori K., Nakao K., Suk K. (2013) Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 27, 1176–1190 [DOI] [PubMed] [Google Scholar]
- 32. Jeon H., Kim J. H., Kim J. H., Lee W. H., Lee M. S., Suk K. (2012) Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J. Neuroinflammation 9, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Enokido Y., Akaneya Y., Niinobe M., Mikoshiba K., Hatanaka H. (1992) Basic fibroblast growth factor rescues CNS neurons from cell death caused by high oxygen atmosphere in culture. Brain Res. 599, 261–271 [DOI] [PubMed] [Google Scholar]
- 34. Araki W., Yuasa K., Takeda S., Shirotani K., Takahashi K., Tabira T. (2000) Overexpression of presenilin-2 enhances apoptotic death of cultured cortical neurons. Ann. N.Y. Acad. Sci. 920, 241–244 [DOI] [PubMed] [Google Scholar]
- 35. Hylden J. L., Wilcox G. L. (1980) Intrathecal morphine in mice: a new technique. Eur. J. Pharmacol. 67, 313–316 [DOI] [PubMed] [Google Scholar]
- 36. Jeon S. M., Lee K. M., Cho H. J. (2009) Expression of monocyte chemoattractant protein-1 in rat dorsal root ganglia and spinal cord in experimental models of neuropathic pain. Brain Res. 1251, 103–111 [DOI] [PubMed] [Google Scholar]
- 37. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 38. Jin S. X., Zhuang Z. Y., Woolf C. J., Ji R. R. (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 23, 4017–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsuda M., Mizokoshi A., Shigemoto-Mogami Y., Koizumi S., Inoue K. (2004) Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45, 89–95 [DOI] [PubMed] [Google Scholar]
- 40. Koltzenburg M., Wall P. D., McMahon S. B. (1999) Does the right side know what the left is doing? Trends Neurosci. 22, 122–127 [DOI] [PubMed] [Google Scholar]
- 41. Choi J., Lee H. W., Suk K. (2011) Increased plasma levels of lipocalin 2 in mild cognitive impairment. J. Neurol. Sci. 305, 28–33 [DOI] [PubMed] [Google Scholar]
- 42. DeLeo J. A., Yezierski R. P. (2001) The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 90, 1–6 [DOI] [PubMed] [Google Scholar]
- 43. Watkins L. R., Hutchinson M. R., Ledeboer A., Wieseler-Frank J., Milligan E. D., Maier S. F. (2007) Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav. Immun. 21, 131–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ji R. R., Suter M. R. (2007) p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang J., De Koninck Y. (2006) Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 97, 772–783 [DOI] [PubMed] [Google Scholar]
- 46. Thacker M. A., Clark A. K., Bishop T., Grist J., Yip P. K., Moon L. D., Thompson S. W., Marchand F., McMahon S. B. (2009) CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain 13, 263–272 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J., Shi X. Q., Echeverry S., Mogil J. S., De Koninck Y., Rivest S. (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 27, 12396–12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abbadie C., Lindia J. A., Cumiskey A. M., Peterson L. B., Mudgett J. S., Bayne E. K., DeMartino J. A., MacIntyre D. E., Forrest M. J. (2003) Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. U.S.A. 100, 7947–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson E. A., Dao T. L., Guignet M. A., Geddes C. E., Koemeter-Cox A. I., Kan R. K. (2011) Increased expression of the chemokines CXCL1 and MIP-1α by resident brain cells precedes neutrophil infiltration in the brain following prolonged soman-induced status epilepticus in rats. J. Neuroinflammation 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaftel S. S., Carlson T. J., Olschowka J. A., Kyrkanides S., Matousek S. B., O'Banion M. K. (2007) Chronic interleukin-1β expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J. Neurosci. 27, 9301–9309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perkins N. M., Tracey D. J. (2000) Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 101, 745–757 [DOI] [PubMed] [Google Scholar]
- 52. Asensio V. C., Campbell I. L. (1999) Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 22, 504–512 [DOI] [PubMed] [Google Scholar]
- 53. Rossi D., Zlotnik A. (2000) The biology of chemokines and their receptors. Annu. Rev. Immunol. 18, 217–242 [DOI] [PubMed] [Google Scholar]
- 54. Miller R. J., Jung H., Bhangoo S. K., White F. A. (2009) Cytokine and chemokine regulation of sensory neuron function. Handb. Exp. Pharmacol. 194, 417–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanaka T., Minami M., Nakagawa T., Satoh M. (2004) Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci. Res. 48, 463–469 [DOI] [PubMed] [Google Scholar]
- 56. Clark A. K., Yip P. K., Malcangio M. (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 29, 6945–6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gao Y. J., Zhang L., Samad O. A., Suter M. R., Yasuhiko K., Xu Z. Z., Park J. Y., Lind A. L., Ma Q., Ji R. R. (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 29, 4096–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kiguchi N., Kobayashi Y., Maeda T., Saika F., Kishioka S. (2010) CC-chemokine MIP-1α in the spinal cord contributes to nerve injury-induced neuropathic pain. Neurosci. Lett. 484, 17–21 [DOI] [PubMed] [Google Scholar]
- 59. Kiguchi N., Maeda T., Kobayashi Y., Fukazawa Y., Kishioka S. (2010) Macrophage inflammatory protein-1α mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1β up-regulation. Pain 149, 305–315 [DOI] [PubMed] [Google Scholar]
- 60. Oh S. B., Tran P. B., Gillard S. E., Hurley R. W., Hammond D. L., Miller R. J. (2001) Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 21, 5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Berard J. L., Zarruk J. G., Arbour N., Prat A., Yong V. W., Jacques F. H., Akira S., David S. (2012) Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia 60, 1145–1159 [DOI] [PubMed] [Google Scholar]
- 62. Rathore K. I., Berard J. L., Redensek A., Chierzi S., Lopez-Vales R., Santos M., Akira S., David S. (2011) Lipocalin 2 plays an immunomodulatory role and has detrimental effects after spinal cord injury. J. Neurosci. 31, 13412–13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brack A., Stein C. (2004) Potential links between leukocytes and antinociception. Pain 111, 1–2 [DOI] [PubMed] [Google Scholar]
- 64. Marchand F., Perretti M., McMahon S. B. (2005) Role of the immune system in chronic pain. Nat. Rev. Neurosci. 6, 521–532 [DOI] [PubMed] [Google Scholar]
- 65. Haraguchi K., Kawamoto A., Isami K., Maeda S., Kusano A., Asakura K., Shirakawa H., Mori Y., Nakagawa T., Kaneko S. (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 32, 3931–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shaw S. K., Owolabi S. A., Bagley J., Morin N., Cheng E., LeBlanc B. W., Kim M., Harty P., Waxman S. G., Saab C. Y. (2008) Activated polymorphonuclear cells promote injury and excitability of dorsal root ganglia neurons. Exp. Neurol. 210, 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]