

Dephosphorylation of Atg18 increases its phosphoinositide-binding activity, thereby enhancing its association with the vacuolar membrane and inducing changes in vacuolar structure.
Abstract
The PROPPIN family member Atg18 is a phosphoinositide-binding protein that is composed of a seven β-propeller motif and is part of the conserved autophagy machinery. Here, we report that the Atg18 phosphorylation in the loops in the propellar structure of blade 6 and blade 7 decreases its binding affinity to phosphatidylinositol 3,5-bisphosphate in the yeast Pichia pastoris. Dephosphorylation of Atg18 was necessary for its association with the vacuolar membrane and caused septation of the vacuole. Upon or after dissociation from the vacuolar membrane, Atg18 was rephosphorylated, and the vacuoles fused and formed a single rounded structure. Vacuolar dynamics were regulated according to osmotic changes, oxidative stresses, and nutrient conditions inducing micropexophagy via modulation of Atg18 phosphorylation. This study reveals how the phosphoinositide-binding activity of the PROPPIN family protein Atg18 is regulated at the membrane association domain and highlights the importance of such phosphoregulation in coordinated intracellular reorganization.
Introduction
In eukaryotes, various biological processes are regulated by phosphoinositide (PI) signaling (Di Paolo and De Camilli, 2006). PIs contain inositol phosphate head groups that are specifically recognized by PI-binding domains on partner proteins, e.g., FYVE, PX, PH, and GRAM domains (Lemmon, 2008). However, the posttranslational regulation of lipid-binding activity in these domains has been scarcely investigated at this point.
PROPPIN family members, including Atg18, Atg21, and Hsv2, are proteins with a lipid-binding domain comprising a seven β-propeller motif that binds to phosphatidylinositol 3-monophosphate (PI(3)P) and/or phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2; Dove et al., 2009). Atg18 is one of the autophagy-related molecules responsible for autophagic processes, and is conserved from yeast to higher organisms, including the human proteins WIPI1–WIPI4 (Mizushima et al., 2011).
In autophagy, nutrient starvation leads to bulk degradation of intracellular components in a sequestering vesicle called the autophagosome. This remodeling is mediated by the TORC1 complex and PI(3)P signaling (Mizushima et al., 2011). Pexophagy, one type of selective autophagy involving peroxisomes, is classified into two distinct pathways: micropexophagy and macropexophagy (Manjithaya et al., 2010; Oku and Sakai, 2010). In the methylotrophic yeast, Pichia pastoris, the carbon source used for pexophagy induction determines which pathway is used: glucose and ethanol adaptation induce micropexophagy and macropexophagy, respectively. During micropexophagy, a vacuole engulfs the peroxisomal cluster by repeating septation, resulting in the formation of a vacuolar sequestering membrane (VSM). Simultaneously, a micropexophagy-specific membrane apparatus (MIPA) is necessary to complete the incorporation of peroxisomes into the vacuole. Similar to pexophagosome formation, Atg8 is localized to MIPA, and its assembly requires the conventional Atg proteins, Atg11 and Atg30 (Farré et al., 2008; Oku and Sakai, 2010). P. pastoris Atg18 (PpAtg18) is necessary for VSM formation (Guan et al., 2001); however, its involvement in MIPA formation has not yet been reported. In contrast, in the budding yeast, Saccharomyces cerevisiae, Atg18 is required for the regulation of vacuolar shape via its PI(3,5)P2-binding activity. Vacuolar fission is induced under certain environmental stresses, such as hyperosmotic pressure and high glucose concentrations. PI(3,5)P2, synthesized by the PI(3)P 5-kinase, Fab1, is required for this process (Dove et al., 1997, 2009). By regulating vacuolar shape, Atg18 is suggested to function as an effector of PI(3,5)P2 signaling (Dove et al., 2004).
Collectively, these studies suggested that Atg18 has dual physiological functions, i.e., autophagy regulation mediated by PI(3)P signaling, and control of vacuolar shape via PI(3,5)P2 signaling. However, the molecular mechanisms underlying Atg18’s role in these signaling pathways were not known. In this paper, two phosphorylation regions were found in the loops in the β-propeller structures. Based on the recently revealed molecular structure of the PROPPIN family member Hsv2, from the yeast Kluyveromyces lactis (KlHsv2), the two phosphorylation sites were thought to play critical roles in the protein’s affinity for phosphoinotides (Baskaran et al., 2012; Krick et al., 2012; Watanabe et al., 2012). However, phosphorylation of PpAtg18 was found to decrease the PI-binding activity of PpAtg18. Remarkably, the level of PpAtg18 phosphorylation in the cells correlated with the intracellular localization of PpAtg18. Further analyses revealed that the phosphoregulation of PpAtg18 governed vacuolar shape during adaption to various environmental stresses, including situations that induced micropexophagy.
Results
PpAtg18 is phosphorylated in P. pastoris
We overproduced GST-tagged PpAtg18 under the control of the constitutive GAPDH promoter in the original host P. pastoris. The purified fraction from a Glutathione Sepharose 4B (GS 4B) column exhibited two distinct bands corresponding to the molecular size of the GST-PpAtg18 protein as determined by Coomassie brilliant blue staining of SDS-PAGE (Fig. 1 A). Because phosphatase treatment abolished the upper band, leaving a single band on the gel, we concluded that the purified GST-PpAtg18 protein contained both phosphorylated and nonphosphorylated forms (Fig. 1, A and B).
Figure 1.

Phosphorylation of PpAtg18 modulates PI(3,5)P2-binding activity. (A) Evaluation of purified GST-PpAtg18 from P. pastoris by SDS-PAGE and Coomassie brilliant blue staining. Lane 1: Purified GST-PpAtg18 was eluted from a GS 4B column using the reduced form of glutathione. Lane 2: Purified GST-PpAtg18 was treated with λ-phosphatase on the GS 4B column. This sample was used for the lipid binding assay. (B) Phosphatase treatment of purified PpAtg18 from P. pastoris. Heat, λ-phosphatase was inactivated at 65°C for 1 h. (C) Phosphatase treatment of cell-free extracts from the strain expressing PpAtg18-5×Flag under the original PpATG18 promoter. The cell lysate was treated with λ-phosphatase at 37°C for 1 h. (D) PIP strip analysis. Membranes were incubated with 0.5 µg/ml protein and detected using the Light Capture II system (ATTO). GST-PpAtg18Wt and GST-Atg18Wt with λ-phosphatase were acquired simultaneously to ensure equal exposure times. (E) PIP array analysis. Membranes were incubated with 1.0 µg/ml protein and detected simultaneously to ensure equal exposure times. (F) Liposome pull-down assay. Purified GST-PpAtg18 (1.0 µg) was incubated with each liposome preparation and centrifuged at 16,000 g for 20 min. The pellets were washed twice, suspended in sample buffer, and analyzed by immunoblotting to detect GST-PpAtg18. No lipid, no liposomes; No PI, liposomes without PIs; PI(3)P, liposome containing PI(3)P; PI(3,5)P2, liposome containing PI(3,5)P2. (G) Liposome pull-down with dephosphorylated PpAtg18. Each protein (1.0 µg) was incubated with liposomes containing PI(3,5)P2 and analyzed by immunoblot to detect GST-PpAtg18. The bands were analyzed by densitometry, and band intensities were normalized to samples not treated with phosphatase. Error bars indicate mean ± SEM. **, P < 0.05.
To exclude the possibility that phosphorylation of PpAtg18 was caused by overexpression of the protein, we expressed functional PpAtg18 tagged with five repeats of the Flag peptide (5×Flag) under the control of the original PpATG18 promoter. Again, we observed two bands via immunoblot analysis of cell-free extracts from P. pastoris (grown in glucose). Phosphatase treatment of the cell-free extract yielded a single band that was cross-reactive with the Flag tag (Fig. 1 C). These experiments indicated that PpAtg18 was present in both phosphorylated and nonphosphorylated forms in P. pastoris under physiological conditions.
Phosphorylation of PpAtg18 inhibits PI(3,5)P2-binding activity
Atg18 proteins were reported to bind to PI(3)P and PI(3,5)P2 (Dove et al., 2004). Purified GST-PpAtg18 protein containing both phosphorylated and nonphosphorylated isoforms was subjected to a variety of lipid binding tests, including the phosphatidylinositol phosphate (PIP) strip assay, the PIP array assay, a liposome pull-down assay, and surface-plasmon resonance analysis to determine the specificity of lipid binding. The PIP strip, PIP array, and liposome pull-down assays identified significant PI(3,5)P2-binding and weaker PI(3)P-binding activities (Fig. 1, D–F). However, we could not detect significant PI(3)P binding activity using Biacore surface-plasmon resonance analysis (Fig. S2 B). These data were in accord with previous results obtained using Atg18 from S. cerevisiae (Dove et al., 2004).
Next, we treated the purified PpAtg18 fraction (including both phosphorylated and nonphosphorylated forms) with phosphatase (Fig. 1 A, lane 2), and PI-binding activities were compared between phosphatase-treated and nontreated samples. As determined using the PIP strip assay, phosphatase treatment dramatically increased PpAtg18 binding activity toward PI(3,5)P2 as compared with PI(3)P (Fig. 1 D). This increase in affinity to PI(3,5)P2 after dephosphorylation was confirmed using the PIP array and the liposome-binding assay (Fig. 1, E and G). These experiments suggested that the affinity of PpAtg18 toward PI(3,5)P2 is reduced by phosphorylation. However, the affinity of PpAtg18 toward PI(3)P was too weak to draw comparisons between phosphatase-treated and nontreated samples.
PpAtg18 has two distinct phosphorylation regions in the loops of blades 6 and 7 that affect PI binding activity
MS analysis of purified PpAtg18 protein identified two phosphorylated peptides, one with two putative phosphorylation sites. Further MS/MS analyses identified these as 387-SSTTS-391 and 492-SSTS-495 (Fig. S1, A and B). Next, we compared the sequence around the phosphorylated sites with those of other PROPPIN family members by BLAST analysis. The crystal structure of KlHsv2 revealed consensus sequences for each blade (Fig. 2 A; Baskaran et al., 2012; Krick et al., 2012). The phosphorylated region in blade 6 was found in a loop that is conserved in all PROPPIN family proteins, including WIPI1 and KlHsv2 (Fig. 2, A and B). This loop is thought to be associated with the membrane via a hydrophobic bond (Baskaran et al., 2012); therefore, phosphorylation in this loop is speculated to inhibit lipid binding. However, the region close to the C terminus (between β1 and β2 of blade 7) was conserved in the methylotrophic yeast Hansenula polymorpha, and partially conserved in S. cerevisiae, but was not retained in higher organisms.
Figure 2.

PpAtg18 is phosphorylated at two distinct sites, and the loop region in blade 6 is conserved among PROPPIN family members. (A) Sequence alignment of PROPPIN domains between KlHsv2p (K. lactis), PpAtg18 (P. pastoris), and WIPI1 (human). Each blade contains 4 β sheets marked by different colors. The arrowheads indicate phosphorylated regions of PpAtg18. (B) Sequences of phosphorylated regions in the blade 6 and 7 loop structures of PpAtg18. The predicted sites of phosphorylation determined from LC-MS/MS analyses are underlined, and amino acid residues critical for phosphorylation are shown in bold font.
To confirm the negative effect of phosphorylation in vivo, the putative phosphorylated residues of Ser and Thr were mutated to Ala and expressed in P. pastoris (Table 1). Cell-free extracts were subjected to normal and Phos-tag SDS-PAGE. Both PpAtg18 SA and AS mutations caused increased mobility of bands in both normal and Phos-tag SDS-PAGE; the PpAtg18 AA mutant produced the fastest migrating band among the tested mutants (Fig. 3, A and B). Therefore, we concluded that both the Ser and Thr residues at positions 387–391 and 491–495 were phosphorylated in P. pastoris. Similarly, we also constructed PpAtg18 mutants that mimicked phosphorylation by replacing Ser or Thr residues within these regions with Asp (Table 1).
Table 1.
Designation of phosphorylation-mutants
| Name | Phosphorylated regions in: | |
| Blade 6 | Blade 7 | |
| Wt | 383PRRVSSTTSLGSYGSQES | 487DVDNSSSTSDSTF |
| AS | 383PRRVAAAAALGSYGSQES | 487DVDNSSSTSDSTF |
| SA | 383PRRVSSTTSLGSYGSQES | 487DVDNAAAAADSTF |
| AA | 383PRRVAAAAALGSYGSQES | 487DVDNAAAAADSTF |
| DS | 383PRRVDDDDDLGSYGSQES | 487DVDNSSSTSDSTF |
| SD | 383PRRVSSTTSLGSYGSQES | 487DVDNDDDDDDSTF |
| DD | 383PRRVDDDDDLGSYGSQES | 487DVDNDDDDDDSTF |
Wt, wild type.
Figure 3.

PpAtg18 is phosphorylated at two distinct sites in vivo. (A) The designated PpAtg18 phosphorylation mutants were grown in SD, collected at the exponential phase, and analyzed (5 µg) by immunoblotting. (B) Analysis of PpAtg18 variants using Phos-tag. The samples are the same as those used in Fig. 3 A. (C) PIP array analysis. Membranes were incubated with 1.0 µg/ml protein and detected simultaneously to ensure equal exposure times. (D) Liposome pull-down assay of PpAtg18 phosphorylation-defective mutants. The samples were subjected to immunoblotting and the bands were analyzed by densitometry. Error bars indicate mean ± SEM. ***, P < 0.1.
We purified the phosphorylation-defective PpAtg18 AA protein and the phosphorylation mimic PpAtg18 DD protein as GST fusion proteins, along with the wild-type PpAtg18 protein from P. pastoris cells. Molar equivalents of each sample were subjected to SDS-PAGE. The purified PpAtg18 DD protein migrated slower and the PpAtg18 AA protein faster, the former and the latter nearly corresponding to the mobility of the phosphorylated and dephosphorylated forms, respectively (Fig. S2 A). Next, these samples were subjected to lipid binding experiments (Fig. 1, D–G; and Fig. S2 B). Both the PIP array and the liposome pull-down assay revealed that the phosphorylation-defective mutant had a higher affinity to PI(3,5)P2, whereas the phosphorylation mimic PpAtg18 DD protein showed an affinity similar to the wild-type protein (Fig. 3, C and D). Similarly, we further assessed PI(3,5)P2 binding activity of PpAtg18 SA and PpAtg18 AS proteins in comparison with the PpAtg18 wild-type protein to see dephosphorylation in which loop structure was responsible to the regulation of the affinity (Fig. S2 C). Because both mutations increased the affinity, we concluded that dephosphorylation of either phosphorylated region of PpAtg18 increased the affinity toward PI(3,5)P2.
We mutated each of putative phosphorylated residues of serine/threonine residues to alanine, and examined the phosphorylation levels of the variant forms by the immunoblot analysis after SDS-PAGE with or without Phos-tag (Fig. S2 D). As a result, we identified four amino acid residues critical for Atg18 phosphorylation: two in the loop of blade 6, S388 and S391; and two in the loop of the C terminus, S492 and S495 (Fig. 2). Phosphorylation of PROPPIN, as observed for PpAtg18, may mediate the physiological signal specific to each PROPPIN by modulating its PI-binding activity.
Phosphorylation of PpAtg18 correlates with vacuolar shape under various environmental stresses
Atg18 was previously shown to regulate vacuolar shape during adaptation to various environmental stresses; e.g., nutrient and osmotic change. Furthermore, this regulation was mediated by the PI-binding activity of Atg18 (Dove et al., 2004; Krick et al., 2006; Efe et al., 2007; Obara et al., 2008). In S. cerevisiae, hypo-osmotic stress and carbon-source limitation lead to homotypic vacuolar fusion. In contrast, hyperosmotic stress and high glucose concentrations lead to vacuolar fission (Dove et al., 2009; Li and Kane, 2009).
The physiological significance of PpAtg18 phosphorylation was sought. Cells expressing PpAtg18-5×Flag were adapted to various environmental conditions, and the phosphorylation level of PpAtg18-5×Flag was assessed (Fig. 4 A). When cells were adapted to hyperosmotic stress (0.9 M NaCl) or high-glucose conditions (20% glucose), vacuolar fission occurred (Fig. 4 B), and the dephosphorylated form of PpAtg18 increased. When cells were subjected to hypo-osmotic stress (H2O) or glucose starvation (−glucose), vacuolar fusion occurred, and the phosphorylated form of PpAtg18 increased. The change from hyperosmotic stress to glucose starvation caused a drastic increase in phosphorylated PpAtg18, and adaptation in the opposite direction caused a decrease of phosphorylated PpAtg18 (Fig. 4 C). However, the phosphorylation levels did not change in a control experiment (transfer from synthetic dextrose [SD] medium to SD medium; Fig. 4 A).
Figure 4.

PpAtg18 phosphorylation correlates with the vacuolar dynamics observed under a variety of environmental conditions. (A) PpAtg18-5×Flag from cells grown under a variety of conditions was detected by immunoblotting with the anti-FLAG antibody. Cells were shifted from SD (0 min) to SD medium containing 0.9 M NaCl or 20% glucose, to water, to YNB medium without amino acids (−glucose), to methanol-containing medium, or (as a control) to SD medium for the indicated times (10, 20, 40, or 60 min). (B) Fluorescence microscopy analysis of PpAtg18 localization and vacuolar morphology. Growing conditions were the same as in Fig. 4 A. PpAtg18-tagged YFP was expressed under the original promoter and vacuoles were stained with FM 4-64. (C) Pulse-chase experiment. Cells were shifted from SD medium to either SD + 0.9 M NaCl or SD-glucose, and then shifted again into the opposite medium (see arrows). Analysis was performed by immunoblotting. (D) Oxidative stress induces PpAtg18 phosphorylation. t-BOOH, t-butylhydroperoxide. Analysis was performed by immunoblotting. (E) Morphological analysis and morphometric analysis of vacuoles in cells with or without oxidants. Cells grown on SD were shifted to SD (control) or to SD with 1 mM H2O2, 15 mM t-BOOH, or 4 mM diamide. Vacuoles were stained with FM 4-64. Error bars indicate mean ± SEM. *, P < 0.01 (compared with SD [control]). Bars, 2 µm.
Next, we observed vacuolar morphology and the localization of PpAtg18 under these various environmental conditions. PpAtg18 tagged with the fluorescent protein YFP was expressed under its own promoter. Under vacuolar-fission conditions, the vacuole became fragmented, and PpAtg18-YFP was recruited to the vacuolar membrane, especially to the sites of vertex domains (Fig. 4 B). Alternatively, under vacuolar fusion conditions, PpAtg18-YFP was released from vacuolar membrane, and diffused throughout the cytosol. In the control experiment, PpAtg18 was weakly detected on the vacuolar membrane, and the level of vacuolar fission (counted as the vacuolar number) seemed to be intermediate between that observed under hyperosmotic and hypo-osmotic stress (Fig. 4 B). We also tested other stress conditions that induce changes in vacuolar morphology (e.g., heat shock and ethanol stress), and observed that PpAtg18 phosphorylation levels increased under vacuolar fusion conditions and decreased under fission conditions.
Interestingly, PpAtg18 was highly phosphorylated when cells were exposed to oxidative stresses that induced vacuolar fusion: the oxidants, H2O2, tBOOH, and diamide (Fig. 4, D and E). We further tested other environmental stresses (reducing agents, exogenous calcium levels) that did not change either PpAtg18 phosphorylation levels or vacuolar shape. These results were consistent with a correlation between PpAtg18 phosphorylation levels and vacuolar shape in P. pastoris.
Dephosphorylation of PpAtg18 regulates vacuolar fission by increasing PI(3,5)P2-binding activity
Fab1 synthesizes PI(3,5)P2 from PI(3)P. In Ppfab1Δ, PpAtg18-YFP, which is present on the vacuolar membrane in the wild-type strain when cells are cultured in glucose media or under hyperosmotic conditions, seemed to be released to the cytosol (Fig. 5 A). The finding that PpAtg18-YFP was recruited to the vacuolar membrane in a PI(3,5)P2-dependent manner led us to test whether PpAtg18 phosphorylation-defective mutants were localized differently as compared with wild-type protein. The phosphorylation-defective mutant PpAtg18AA-YFP showed stronger fluorescence at the vacuolar membrane in hypo-osmotic (SD + 0.9 M NaCl) or control (SD) conditions. In contrast, localization of the PpAtg18DD-YFP mutant, which retained PI(3,5)P2-binding activity, was similar to the wild-type PpAtg18 (Fig. 5 B). However, both PpAtg18 AA and DD mutant proteins were released into the cytosol under vacuolar fusion conditions (SD − glucose); the cells in these experiments displayed a single spherical vacuole. To investigate whether PpAtg18AA localized to the vacuolar membrane affected vacuolar shape, we next assessed the level of vacuolar fission as the number of vacuolar compartments per cell. As a result, PpAtg18AA displayed more vacuolar fission than the wild-type and PpAtg18DD when cultured both in SD media and under hyperosmotic conditions (Table 2). Together with the results from the in vitro PI(3,5)P2-binding experiment (Fig. 1 and Fig. 3), we concluded that dephosphorylation of PpAtg18 positively regulates vacuolar fission by increasing binding to PI(3,5)P2.
Figure 5.

Phosphorylation of PpAtg18 regulates vacuolar membrane dynamics via PI(3,5)P2 binding. (A) Localization of PpAtg18-YFP in Ppfab1Δ. YFP tagged at the C terminus with PpAtg18 was expressed under its own promoter, and vacuoles were stained with FM 4-64. (B) Localization of PpAtg18-YFP phosphorylation mutants. PpAtg18AA, a phosphorylation-defective mutant, and PpAtg18DD, a phosphorylation mimic mutant, appeared similar to the wild type as observed in Fig. 4 B. Bars, 2 µm. (C) Western blot analysis of PpAtg18Wt-5×Flag in Ppfab1Δ or PpAtg18FTTG-5×Flag in Ppatg18Δ.
Table 2.
Septation of vacuoles during hyperosmotic adaptation
| Medium | Straina | The number of vacuoles/cellb | ||||
| 1 | 2 | 3 | 4 | 5 | ||
| % | % | % | % | % | ||
| SD | Wt | 68.6 (±5.76) | 27.1 (±5.15) | 1.74 (±1.43) | 0 (±0) | 0 (±0) |
| AA | 45.6 (±10.6) | 42.4 (±7.25) | 6.44 (±2.43) | 1.06 (±0.495) | 0.292 (±0.413) | |
| DD | 57.0 (±5.62) | 36.5 (±5.50) | 3.65 (±0.943) | 0.960 (±0.698) | 0.273 (±0.387) | |
| SD + 0.9 M NaCl | Wt | 27.9 (±9.90) | 42.7 (±4.63) | 20.6 (±5.25) | 5.60 (±2.72) | 2.32 (±1.33) |
| AA | 17.4 (±2.93) | 42.1 (±7.15) | 25.4 (±2.58) | 9.56 (±3.26) | 4.73 (±4.95) | |
| DD | 35.2 (±9.76) | 39.5 (±3.80) | 17.6 (±3.82) | 4.68 (±1.60) | 1.60 (±1.71) | |
The difference in the average of total vacuoles per cell between PpAtg18Wt and PpATG18AA was marginally significant (P < 0.1). Wt, wild type.
PpAtg18Wt, AA, or DD tagged 5×Flag were expressed under the PpATG18 promoter in Ppatg18Δ. Cells were grown in SD medium, and shifted to SD or SD + 0.9 M NaCl for 1 h.
Vacuolar fission indicated as the population of the cells (%) having the indicated number of vacuoles in a cell.
In the wild-type strain, the level of PpAtg18 phosphorylation increased under vacuolar fusion conditions, and decreased under vacuolar fission conditions. Interestingly, the level of PpAtg18 phosphorylation was lower in the Ppfab1Δ strain and the PpAtg18 FTTG mutant (Dove et al., 2004; Krick et al., 2006; Obara et al., 2008) as compared with the wild-type protein (Fig. 5 C). In these mutants, however, the level of dephosphorylation was comparable to that observed in wild type (Fig. 5 C). Because the PI(3,5)P2-binding activity of PpAtg18 increased its vacuolar association, this result may represent the fact that phosphorylated PpAtg18 is mainly localized to the vacuolar membrane.
Phosphoregulation of PpAtg18 during adaptation to methanol medium, and during micro- and macropexophagy
Regulation of vacuolar shape.
Previously, we reported that vacuoles fused to form a single spherical vacuole in a PpAtg8-dependent manner during early periods of adaptation from glucose to methanol medium concomitant with peroxisome proliferation (Tamura et al., 2010). During subsequent adaptation to ethanol medium that induced macropexophagy, PpAtg18 was expected to be responsible for pexophagosome formation, but vacuolar shape did not change significantly. However, during adaptation to glucose medium that induced micropexophagy, PpAtg18 may have another function in addition to the formation of MIPA (Fig. 6 A). Specifically, because PpAtg18 is reported to be required for the early stage of micropexophagy, we speculate that PpAtg18 is involved in the formation of VSM concomitant with vacuolar fission (Guan et al., 2001). Also, the two distinct processes, i.e., VSM formation and MIPA formation, should be coordinately regulated. Therefore, we wanted to understand the mechanisms underlying PpAtg18 regulation of vacuolar dynamics, including the formation of autophagic membranes during methanol adaptation and pexophagic conditions.
Figure 6.

Phosphorylation of PpAtg18 regulates VSM formation in micropexophagy. (A) Intracellular structures observed during micropexophagy and macropexophagy. MIPA, micropexophagy-specific membrane apparatus; VSM, vacuolar sequestering membrane. (B) Fluorescence microscopy analysis during micropexophagy. Cells were shifted from methanol medium to glucose medium for 30–60 min to induce micropexophagy. Vacuoles were stained with FM 4-64, and MIPA was visualized by YFP-tagged PpAtg8 expressed under the PpATG8 promoter. (C) Intracellular localization of PpAtg18. YFP C-terminally tagged with PpAtg18 was expressed in Ppatg18Δ under the PpATG18 promoter. Cells were shifted from methanol medium to ethanol or glucose medium for 30–60 min to induce macropexophagy or micropexophagy, respectively. (D) Immunoblot detection of PpAtg18-5×Flag. PpAtg18-5×Flag was expressed in Ppatg18Δ under the PpATG18 promoter. Cells were shifted from methanol medium to ethanol or glucose medium to induce macropexophagy or micropexophagy, respectively. This blot was incubated with the anti-FLAG antibody. (E) Intracellular localization of PpAtg18 AA and DD during micropexophagy. YFP-tagged PpAtg18 mutants were expressed under the PpATG18 promoter. Cells were shifted from methanol medium to glucose medium for 60 min to induce micropexophagy. Bars, 2 µm. (F) PpPex12 degradation assay to assess micropexophagy and macropexophagy activity in PpAtg18 mutants. Strains were grown in methanol medium and then shifted to glucose or ethanol medium to induce micropexophagy or macropexophagy, respectively. Cell-free extracts were prepared and analyzed by immunoblotting with anti-PpPex12 or anti-Pgk1 antibodies. Optical density measurements showed no significant difference in growth between these mutants.
First, we examined MIPA and VSM formation during micropexophagy in the Ppatg18Δ strain (Fig. 6 B). In the wild-type strain, VSMs labeled with FM 4-64 budded from maternal vacuoles and YFP-PpAtg8 formed a cuplike structure representing MIPA formation. In contrast, neither VSM nor MIPA were observed in the Ppatg18Δ strain. In the Ppfab1Δ cells, MIPA-like structures were formed, but VSM formation was not observed. We performed morphometric analysis of VSM formation by counting the number of vacuoles during micropexophagy (unpublished data). Vac14 and Fab1 are components of the PI(3)P 5-kinase complex. Both kinase component mutants, PpFab1Δ and PpVac14Δ, along with the PpAtg18 mutant that was impaired in PI(3,5)P2-binding activity, showed a drastic decrease in VSM formation (unpublished data).
Next, we investigated the intracellular localization of PpAtg18-YFP. In cells grown in methanol medium (at both at late log phase and early lag phase) or under macropexophagic conditions, PpAtg18-YFP localized to the cytosol (Fig. 6 C). In contrast, during micropexophagy, PpAtg18-YFP was recruited to the vacuolar membrane. This vacuolar recruitment was lost in the PI(3)P 5-kinase–defective Ppfab1Δ strain (Fig. 6 C), which indicates that PpAtg18 was recruited to the vacuolar membrane in a PI(3,5)P2-dependent manner during micropexophagy. Collectively, these data suggest that VSM formation during micropexophagy requires the PI(3,5)P2-dependent recruitment of PpAtg18 to the vacuolar membrane.
Then, we examined the phosphorylation levels of PpAtg18 under various conditions. At early phases of methanol adaptation (1–2 h), the phosphorylated form of PpAtg18 became dominant concomitant with vacuolar fusion, and PpAtg18-YFP diffused into the cytosol (Fig. 4, A and B). After 8–12 h of methanol adaptation, we could detect both phosphorylated and dephosphorylated forms of PpAtg18 and observed huge peroxisomes (Fig. 6 D). Therefore, although PpAtg18 was phosphorylated upon vacuolar fusion at early log phase, part of the phosphorylated PpAtg18 became dephosphorylated during cellular proliferation. This may reflect the fact that vacuolar inheritance from mother to daughter cells includes both vacuolar fission and fusion, which may be controlled by phosphoregulation of PpAtg18 (Weisman, 2006; Tamura et al., 2010).
Cells cultured in methanol at late-log phase were induced to undergo micro- and macropexophagy by transfer to glucose and ethanol medium, respectively. Then, PpAtg18 phosphorylation and localization (using PpAtg18-YFP) were observed. When cells were shifted to ethanol medium to induce macropexophagy, the phosphorylated form became dominant and the dephosphorylated form decreased significantly, suggesting that the phosphorylated form of PpAtg18 functioned in pexophagosome formation (Fig. 6 D). In contrast, both the phosphorylated and dephosphorylated forms of PpAtg18 were observed during micropexophagy, which included both MIPA formation and vacuolar fission for VSM formation (Fig. 6 D).
Regulation of autophagy.
As shown in Fig. 6 B, PpAtg18 was necessary for MIPA formation in micropexophagy. In nutrient starvation–induced autophagy, PpAtg18 was reported to be recruited to the preautophagosomal structure (PAS), represented by PpAtg8 dots, and this localization required Atg2 and PI(3)P (Suzuki et al., 2007). To visualize such colocalization in P. pastoris, we expressed CFP-tagged PpAtg18 under the PpACT1 promoter, and coexpressed YFP-PpAtg8 under its original promoter. Fluorescence microscopy revealed that CFP-PpAtg18 was colocalized with the PAS (Fig. S3 A) during micropexophagy. However, we also observed that PpAtg26, which is necessary for pexophagy, was dispensable for the recruitment of PpAtg18 to the PAS.
We compared pexophagy in cells bearing either wild-type or mutant PpAtg18 by following the degradation of two peroxisomal proteins: the abundant matrix protein, alcohol oxidase (AOX); and the less abundant peroxisomal membrane protein, PpPex12. We reasoned that observing changes in the less abundant PpPex12 would provide a more sensitive assay than could be obtained by following AOX. Although degradation of both AOX and PpPex12 was impaired in the Ppatg18Δ strain, we could not detect significant defects in AOX degradation by micro- and macropexophagy in Ppfab1Δ strain, PpAtg18 AA, and PpAtg18 DD mutants. However, PpPex12 degradation was retarded in the PpAtg18 AA mutant under micropexophagic conditions as compared with the wild-type and the PpAtg18 DD mutant, even though VSM formation was enhanced (Fig. 6, E and F). However, PpPex12 degradation was enhanced in the PpAtg18 DD mutant during macropexophagy when compared with the wild type and the PpAtg18 AA mutant (Fig. 6 F). These results indicated that phosphoregulation of PpAtg18 was involved in pexophagic activity.
Next, we monitored the intracellular localization of the phosphorylation-defective PpAtg18AA-YFP and the phosphorylation-mimic PpAtg18DD-YFP mutants during the pexophagic process. Although PpAtg18DD-YFP showed a similar localization to the wild-type protein, PpAtg18AA-YFP was recruited to the vacuolar membrane more efficiently than was the wild-type protein, which resulted in enhanced VSM formation (Table 3 and Fig. 6 E). These data indicated that phosphoregulation of PpAtg18 was required for optimum activity of both micro- and macropexophagy by regulating the intracellular distribution of PpAtg18.
Table 3.
VSM formation during micropexophagy in PpAtg18 AA and DD mutant strains
| Straina | VSM formationb | ||||
| 1 | 2 | 3 | 4 | 5 | |
| % | % | % | % | % | |
| Wt | 55.5 (±5.14) | 30.1 (±3.95) | 10.7 (±1.54) | 2.94 (±1.03) | 0.178 (±0.182) |
| AA | 40.3 (±4.81) | 36.7 (±2.84) | 16.9 (±2.25) | 4.11 (±1.02) | 0.986 (±0.632) |
| DD | 45.1 (±6.45) | 36.6 (±1.26) | 13.6 (±4.27) | 3.09 (±1.76) | 0.260 (±0.261) |
The difference in the average of VSMs per cell between PpAtg18Wt and PpATG18AA was significant (P < 0.05). Wt, wild type.
PpAtg18Wt, AA, or DD tagged 5×Flag were expressed under the PpATG18 promoter in Ppatg18Δ. Micropexophagy was induced by adapting cells from methanol to glucose medium for 30 min
VSM formation is indicated as the population of the cells (%) having the indicated number of vacuoles in a cell.
We evaluated how PpAtg18 phosphorylation levels affected nitrogen starvation–induced autophagy using the YFP-PpAtg8 procession assay (Yamashita et al., 2009). During nitrogen starvation, the phosphorylation levels of PpAtg18 (Fig. S3, B and C) and the vacuolar shape did not change significantly. Although the Ppatg18Δ strain was autophagy defective, neither the PpAtg18 AA nor the DD mutants were defective. These data suggested that mutations in the phosphorylated regions did not influence nitrogen starvation–induced autophagic activity.
Atg2, which binds Atg18, is an essential protein for all autophagic processes. Next, we investigated the role of PpAtg2 during micropexophagy and in the phosphorylation of PpAtg18. In contrast to the Ppfab1Δ strain, MIPA was not formed but VSM formation was normal in the Ppatg2Δ strain (Fig. 6 B). Deletion of PpAtg2 had no effect on VSM formation (Fig. S3 A) or on Atg18 phosphorylation (Fig. S3 D). Also, the association of PpAtg18 to the vacuolar membrane was normal. Because both PpAtg18 AA and DD mutants exhibited autophagic activities in starvation-induced autophagy and pexophagy, phosphoregulation of PpAtg18 is unlikely to be involved in the formation of the Atg18–Atg2 complex.
Among various autophagic processes, PpAtg18 and its phosphoregulation were found to play a critical role during micropexophagy, i.e., the autophagic process concomitant with dynamic changes in vacuolar shape and MIPA formation. The dephosphorylated PpAtg18 with higher affinity for PI(3,5)P2 was necessary for association of PpAtg18 with the vacuolar membrane and VSM formation that mediates incorporation of peroxisomes to the vacuole. Conversely, cytosolic phosphorylated PpAtg18 with decreased affinity for PI(3,5)P2 was suggested to be responsible for MIPA formation after complexing with PpAtg2 at the PAS.
Discussion
In this study, we observed phosphorylation of PpAtg18 in the yeast P. pastoris, and biochemically characterized the molecular function of these phosphate groups with respect to PI binding and the intracellular localization of PpAtg18. We further showed the physiological roles of this phosphoregulation of PpAtg18 by using various mutant strains that mimicked PpAtg18-phosphorylated or -dephosphorylated forms and were grown under various environmental stresses and conditions.
Phosphorylation of PpAtg18 interferes with the interactions of PpAtg18 and PI(3,5)P2 in vitro
One of the important findings in this study is that the dephosphorylation of PpAtg18 increases its affinity for PI. Atg18 is a member of the PROPPIN family of proteins that bind to PI(3)P and PI(3,5)P2 via a seven β-propeller motif (Dove et al., 2004). Recently, the crystal structure of KlHsv2 was solved, representing the first PROPPIN family member to be molecularly characterized at that level. Hsv2 binds PIs specifically at two distinct sites: site 1 on blade 5 and site 2 on blade 6 (Baskaran et al., 2012; Krick et al., 2012; Watanabe et al., 2012). Specificity to PI was mediated by ionic interactions between the negative charge of the PI head group and the basic amino acid residues of sites 1 or 2. Furthermore, the hydrophobic loop of blade 6, conserved in all PROPPINs, was able to embed in the membrane, thus facilitating the interactions between PROPPIN and the membrane (Baskaran et al., 2012).
Our results indicated that dephosphorylation of either region, in blade 6 or blade 7, was sufficient to increase the PI-binding activity (Fig. S2 C). The higher affinity of PpAtg18 AA protein toward PI(3,5)P2 compared with PpAtg18 DD protein (Fig. 3) suggested that the charged moieties of the phosphorylated regions inhibited the lipid-binding activity. One plausible explanation is that the negative charge of the phosphorylated residues simply impeded the hydrophobic interactions between the hydrophobic loop of blade 6 and the membrane (Fig. 7). Although the lipid-binding PH domain of protein kinase B was known to be phosphoregulated (Powell et al., 2003), to our knowledge, this is the first study that addresses, at molecular and biochemical levels, how phosphorylation of a protein modulates its binding to PIs.
Figure 7.
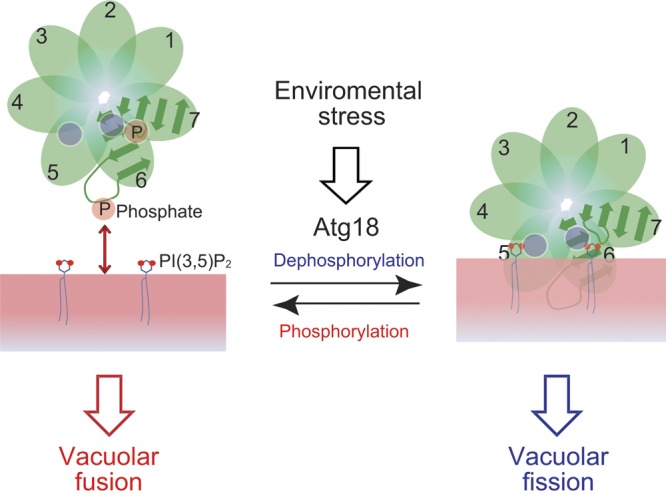
Phosphoregulation of PpAtg18 for maintenance of vacuolar shape.
Role of PpAtg18 phosphoregulation in the maintenance of vacuolar shape
When the dephosphorylated form of PpAtg18 increased and PpAtg18 was recruited to the vacuolar membrane by its PI(3,5)P2-binding activity, vacuolar fission occurred. Phosphorylation-defective PpAtg18 AA mutants exhibited more septated vacuoles than did the cells expressing wild-type PpAtg18. In contrast, when the phosphorylated form of PpAtg18 increased and PpAtg18 dissociated from the vacuolar membrane, the vacuoles fused and formed a single rounded compartment. From these results, we conclude that the phosphorylation of PpAtg18 is correlated to its intracellular localization and vacuolar shape.
Atg18 is thought to be a negative regulator of PI(3,5)P2 synthesis, and the localization cycle of Atg18 between the vacuolar membrane and the cytosol regulates PI(3,5)P2 levels at the vacuolar membrane (Dove et al., 2004, 2009; Efe et al., 2007). Under hyperosmotic conditions, the cytosolic phosphorylated form of PpAtg18 was dephosphorylated, and recruited to the vacuolar membrane by binding vacuolar PI(3,5)P2. Once PpAtg18 is recruited to the vacuolar membrane, the PI(3,5)P2 levels on the vacuolar membrane may be decreased. When the levels of PI(3,5)P2 on the vacuolar membrane are decreased and/or PpAtg18 is phosphorylated, PpAtg18 will be released from the vacuolar membrane. We think that dephosphorylation of PpAtg18 mediates a signal inducing vacuolar fission, as PpAtg18 AA mutation was sufficient to induce and enhance the vacuolar fission. This result also indicated that the vacuolar PI(3,5)P2 level is sufficient to recruit PpAtg18 (Dove et al., 2004; Krick et al., 2006; Efe et al., 2007). However, for dissociation of PpAtg18 from the vacuolar membrane to occur, their interactions should weaken. It was difficult to determine whether the phosphorylation of PpAtg18 or the decrease in vacuolar PI(3,5)P2 was the key event allowing the dissociation of PpAtg18 and the signaling of vacuolar fusion, because low vacuolar PI(3,5)P2 levels were difficult to follow experimentally, and the phosphorylation mimic PpAtg18 DD mutant behaved like the wild type, and did not induce vacuolar fusion. Although the detailed mechanism for vacuolar fission itself should be investigated further, this study first clarified that vacuolar fission is regulated by phosphoregulation of PpAtg18 through its association to the vacuolar membrane.
We tried to identify the upstream kinases that were responsible for PpAtg18 phosphorylation. Our results suggested that phosphorylation occurred at the vacuolar membrane. Specifically, both the Ppfab1Δ mutant and the PpAtg18 FTTG mutant, in which PpAtg18 was not recruited to the vacuolar membrane, had dephosphorylated PpAtg18 under low glucose conditions (Fig. 5 C), whereas wild-type cells had only phosphorylated PpAtg18 under the same conditions (Fig. 4 A). P. pastoris mutants with a single disruption of the kinases PpAtg1, PpTor1, PpHog1, or PpVps15 retained phosphorylation of PpAtg18 (Fig. S3 E). Depletion of yeast casein kinase PpYck3 caused a partial defect in PpAtg18 phosphorylation. It is noteworthy that, in contrast to PpAtg18, in S. cerevisiae, Vps41, a component of a tethering complex for vacuolar homotypic fusion (HOPS family), is phosphorylated by Yck3 under vacuolar fission conditions and released into the cytosol (LaGrassa and Ungermann, 2005; Cabrera et al., 2009). Vps41 contains an amphipathic lipid-packing sensor (ALPS) motif, which interacts with highly curved membranes, and the phosphorylation of this motif prevents membrane recruitment of Vps41 (Cabrera et al., 2010). Vps41 seems to serve as a counterpart to Atg18 in regulating vacuolar shape.
Role of PpAtg18 phosphoregulation in autophagic processes
Atg2, a binding partner of Atg18, was not involved in the phosphorylation of PpAtg18. Structural analysis revealed that Atg2 bound to blades 1 and 2 of Atg18, which are at opposite faces relative to the lipid binding sites and the blade 6 loop involved in membrane association (Watanabe et al., 2012; Rieter et al., 2013). During autophagic processes, Atg18 is thought to recruit and complex with Atg2 at the PAS. This interaction does not affect the lipid-binding activity of Atg18 (Kobayashi et al., 2012; Rieter et al., 2013). Because macroautophagy proceeds despite the phosphorylation defect in the PpAtg18 AA mutant, phosphoregulation in the loop of PpAtg18, which affects the PI(3,5)P2-binding activity necessary for its recruitment to the vacuolar membrane, does not seem to directly affect the PI(3)P-binding activity of PpAtg18 necessary for autophagy. It may be noteworthy to point out that the phosphorylated form PpAtg18 still binds to PIs, which may be sufficient to execute macroautophagy.
Phosphoregulation of PpAtg18 was necessary for optimal execution of micropexophagy, which involved formation of both the VSM and MIPA (Fig. 6 A), but was not necessary for other autophagic processes. During micropexophagy, although VSM formation was enhanced in the phosphorylation-defective PpAtg18 AA mutant, PpPex12 degradation was retarded. This result likely reflected the fact that efficient recruitment of the PpAtg18 AA mutant to the vacuolar membrane reduced the amount of PpAtg18 in the PAS, which is necessary for MIPA formation. The efficiency of MIPA formation could not be assessed using the PpAtg18 AA mutant, as macropexophagy was induced after glucose adaptation and nascent pexophagosomes could not be distinguished from MIPA. We suggest that, during micropexophagy, dephosphorylated and phosphorylated forms of PpAtg18 function in VSM and MIPA formation, respectively, and that PpAtg18 phosphorylation is regulated accordingly. Although a defect caused by impaired phosphoregulation was not observed in macroautophagy, PpAtg18 was observed to be mainly in a phosphorylated form, which suggests that the dissociation of PpAtg18 from the vacuolar membrane enhances the recruitment of PpAtg18 to PAS for autophagosome formation.
Phosphoregulation of PpAtg18 governs the intracellular rearrangement and coordinated dynamics of multiple organelles, i.e., vacuoles, autophagosomes, MIPA, and peroxisomes, in response to various environmental conditions and stresses. An example of such a highly coordinated event may be seen in fungi living in nature. Previously, we showed that the proper execution of pexophagy was necessary for fungal cells to proliferate on the leaves of living plants by showing that the pexophagy-defective PpAtg26 and PpAtg30 mutants lose normal growth and differentiation properties (Asakura et al., 2009; Kawaguchi et al., 2011). In nature, cells have to adapt not only to nutrient stresses leading to autophagy, but also to oxidative and osmotic stresses leading to changes in vacuolar shape. It is likely that, in P. pastoris, micropexophagy is the biological process through which cells adapt simultaneously to both nutritional and environmental stresses, whereby phosphoregulation of Atg18 plays a critical role under natural conditions.
Materials and methods
Strains, media, and antibodies
The strains used in this study are listed in Table S1. Yeast extract/peptone/dextrose (YPD) medium consisted of 1% yeast extract, 2% Bacto Peptone, and 2% glucose. SD medium contained 0.67% yeast nitrogen base without amino acids and 2% glucose. Synthetic methanol (SM) medium contained 0.67% yeast nitrogen base without amino acids and 0.8% methanol. These synthetic media were supplemented with the appropriate amino acids (100 µg/ml arginine and 100 µg/ml histidine) for auxotrophic strains. Anti-FLAG (Wako), anti-GFP (Molecular Probes), anti-GST (GE Healthcare), anti-Pgk1 (Molecular Probes), anti–mouse-HRP (EMD Millipore), anti–goat-HRP (Abcam), and anti–rabbit-HRP antibodies (Rockland Immunochemicals, Inc.) were used in this study. The anti-PpPex12 antibody was a generous gift from S. Subramani (University of California, San Diego, La Jolla, CA).
Plasmid construction
The primers are listed in Table S2. The 5×Flag tag was amplified by using FLAGORF(+ATG)FwXhoI and FLAGORF(+TGA)RvPstI primers, and the fragments were inserted into pNT204, resulting in the plasmid pNT206. PpATG18 original promoter and ORF were amplified with PpATG18promoterFwBamHI and PpATG18ORF(-TAA)RvXhoI, and the PCR product was inserted into pNT206 or pNT205. The plasmids were designated pNT1801 and pNT1808, respectively. The PpATG18ORF was amplified using PpATG18ORF(-ATG)FwBamHI and PpATG18ORF(-TAA)RvXhoI and then introduced into pSY003 or pNT401, resulting in the pNT1811 and pNT1812 plasmids. pNT1801, pNT1808, and pNT1812 were subjected to site-directed mutagenesis by using PpATG18 ST387-391AFw, PpATG18 ST387-391ARv, PpATG18 ST387-391DFw, PpATG18 ST387-391DRv, PpATG18 ST491-495AFw, PpATG18 ST491-495ARv, PpATG18 ST491-495DFw, PpATG18 ST491-495DRv, PpATG18FTTGFw, and PpATG18FTTGRv. These resulting plasmids were designated pNT1802–1807, pNT1809, pNT1810, pNT1813, pNT1814, pNT1816, pNT1826, and pNT1827, respectively. To construct a more efficient expression vector, the KpnI–XhoI fragment including PpATG18ORF from pNT1812 was inserted into pGAPZB, resulting in pNT1815. For single mutation analysis, pNT1801 was subjected to site-directed mutagenesis by using PpATG18 S387AFw, PpATG18 S387ARv, PpATG18 S388AFw, PpATG18 S388ARv, PpATG18 T389AFw, PpATG18 T389ARv, PpATG18 T390AFw, PpATG18 T390ARv, PpATG18 S391AFw, PpATG18 S391ARv, PpATG18 S492AFw, PpATG18 S492ARv, PpATG18 S493AFw, PpATG18 S493ARv, PpATG18 T494AFw, PpATG18 T494ARv, PpATG18 S495AFw, and PpATG18 S495ARv. These resulting plasmids were designated pNT1817–1825.
For disruption of each null mutant, we used the primers (pDV−) listed in Table S2. The amplified PCR products were inserted into the disruption vectors.
Immunoblot analysis
To prepare samples for immunoblot analyses, harvested cells were suspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 1 mM PMSF, 1 mM EDTA, EDTA-free complete protease inhibitor cocktail [Roche], and phosphatase inhibitor PhosSTOP [Roche]) and then lysed using Multi-Beads Shocker (Yasui Kikai). Cell extracts were centrifuged at 800 g for 5 min at 4°C to remove cell debris, and dissolved in sample buffer as described previously (Tamura et al., 2010). Each sample (5 µg for PpAtg18-5×Flag detection or 15 µg for YFP-PpAtg8, PpPex12, or PpPgk1 detection) was electrophoresed on a 7.5–8.0% (PpAtg18-5×Flag) or 12% SDS-PAGE gel (YFP-PpAtg8, PpPex12, and PpPgk1). The gels were transferred to nitrocellulose (Flag, YFP, and Pgk1) or PVDF membranes (PpPex12) by semidry blotting (ATTO). The blots were incubated with anti-Flag (diluted 1:1,000), anti-GFP (diluted 1:1,000), anti-PpPex12 (diluted 1:3,000), or anti-Pgk1 (diluted 1:1,000) in TBS-T buffer for 1 h or overnight. Then, the membranes were washed three times with TBS-T buffer and incubated with anti–mouse-HRP (diluted 1:10,000) or anti–rabbit-HRP (diluted 1:10,000) for 1 h. Finally, bound secondary antibodies were detected using Western Lightning (PerkinElmer) and the signals were analyzed with a Light Capture system (ATTO).
λ-Phosphatase with or without heat inactivation was added to cell lysates and incubated at 30°C for 1 h. The samples were precipitated with acetone and analyzed by immunoblotting. For Phos-tag experiments, we followed the manufacturer’s instructions. An 8% SDS-PAGE was prepared using Phos-tag acrylamide (25 or 50 µM) and an equal volume of 10 mM MnCl2. After electrophoresis, the samples were transferred to a PVDF membrane and analyzed by immunoblotting.
Protein purification from P. pastoris
For purification of PpAtg18 variants, we used P. pastoris cells expressing GST-tagged proteins. Cells were cultivated in YPD media up to stationary growth and harvested by centrifugation at 2,000 g for 5 min. Cells were washed with 1× PBS buffer once, and then suspended in lysis buffer (PBS, 1 mM PMSF, 10 mM MgCl2, 2 µg/ml Pepstatin A, 10 µg/ml Leupeptin, 0.05% [wt/vol] Tween 20, and phosphatase inhibitor PhosSTOP). Next, cells were lysed by sonication and centrifuged at 6,000 g for 15 min to yield cell lysates. Cell lysates were incubated with GS 4B (GE Healthcare) at RT for 30 min. The column was washed with 1× PBS multiple times and the captured proteins were eluted with reduced glutathione (for PIP strips, PIP arrays, and liposome pull-downs), or cleaved with PreScission Protease (for surface plasmon resonance; GE Healthcare). In the phosphatase treatment experiment, we incubated the captured GST-PpAtg18 with λ-phosphatase for 2 h on the column. Then, to remove λ-phosphatase from the sample, the column was washed with 1× PBS buffer multiple times before exchanging the buffer with liposome binding assay buffer: LB buffer, 10 mM Hepes-KOH, pH 8.0, and 150 mM NaCl.
Lipid dot blot analysis
PIP strip and PIP array assays were performed according to the protocols of Echelon Bioscience, Inc. For the PIP strip, membranes were incubated with 0.5 µg/ml purified proteins for 1 h. For the PIP array, membranes were incubated with 1.0 µg/ml protein for 3 h. Anti-GST antibody diluted 1:5,000 was incubated for 1 h and anti–goat-HRP diluted 1:10,000 was incubated for 1 h. For both PIP strip and PIP array experiments, the signals were acquired using the same exposure time.
Liposome preparation
Lipids used in this study were purchased from Sigma-Aldrich: DOPC, 1,2-Dioleoyl-sn-glycero-3-phosphocholine; DSPE, 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine; DPPS, 1,2-Dipalmitoyl-sn-glycero-3-phospho-L-serine; PI(3)P, l-a-Phosphatidyl-D-myo-inositol 3-monophosphate dipalmitoyl; PI(4)P, l-a-Phosphatidyl-d-myo-inositol 4-monophosphate dipalmitoyl; PI(3,5)P2, l-a-Phosphatidyl-d-myo-inositol 3,5-bisphosphate dipalmitoyl; PI(4,5)P2, l-a-Phosphatidyl-d-myo-inositol 4,5-bisphosphate dipalmitoyl. For the liposome pull-down assay, 100:45:45:10 DOPC/DSPE/DPPS/PIs was mixed in lipid mixing buffer (14:5:1 chloroform/methanol/dissolved water) to give a final concentration of 1 mg/ml. Similarly, for surface plasmon resonance analysis, 97/3 DOPC:PIs was mixed. These mixed lipids were lyophilized, and then resuspended in an aliquot of LB buffer by multiple freeze/thaw steps and sonication. The lipid-containing mixture was passed through a Mini-Extruder set (Avanti Polar Lipids, Inc.) with a 100-nm pore size filter to make liposomes.
Liposome binding assay
The purified GST-PpAtg18 variants (1 µg) were incubated with 50 µl of liposomes in 200 µl of LB buffer containing 2.0 × 10−3% (wt/vol) Tween 20 (which is less than the critical micelle concentration [CMC], 1.0 × 10−2) at RT for 20 min. Then, the liposomes were pelleted at 16,000 g for 20 min and the pellets were washed twice with LB buffer. Next, the pellets were suspended in sample buffer and subjected to immunoblot analysis. The blots were incubated with anti-GST (1:5,000) for 1 h, followed by the secondary antibody anti–goat-HRP (1:10,000). The experiments were repeated at least three times and the bands were analyzed by densitometry using a CS analyzer (ATTO).
Surface plasmon resonance analysis
A Biacore2000 (GE Healthcare) with an L1 sensor chip was used. Liposomes were suspended in 10 mM Hepes-KOH, pH 7.5, 150 mM NaCl, and 1 mM MgCl2 and were loaded up to 6,000 RUs (resonance units). Before injection, 0.1% (wt/vol) BSA was applied at 20 µl/min for 2 min. Atg18, from which the N-terminal affinity tag had been cleaved, was loaded and dissociated at 80 µl/min. Injection and dissociation were performed for 90 s and 240 s, respectively. Data were analyzed by BIAevaluation software (GE Healthcare).
Microscopy analysis
A motorized inverted microscope (IX81; Olympus) equipped with a Uplan-Apochromat 100×/1.35 NA oil iris objective lens using a XF104-3 filter set (Omega Optics) was used for YFP, and a U-MRFPHQ unit (Olympus) was used for FM 4-64 as described previously (Tamura et al., 2010). Both YFP and FM 4-64 signals were acquired using a Plan-Fluor 100× lens (Carl Zeiss) with pinhole setting to 1.02 airy units for YFP acquisition. Image data were captured with charge-coupled device camera DP30BW (Olympus). These data were acquired and analyzed by using MetaMorph version 7.0 (Molecular Devices), and saved as TIFF files. The TIFF image files were optimized for their contrast on Photoshop CS3 (Adobe). The scale bars in all experimental microscopy figures are equal to 2 µm.
Morphometric analysis
The number of budded vacuoles from each cell (VSM) was counted after cells had been shifted from methanol to glucose medium. In each experiment, at least 200 cells were analyzed, and the experiment was repeated at least three times. Similarly, we counted budding vacuoles in cells grown on SD medium or under hyperosmotic conditions. When the cells were cultured with oxidants (1 mM H2O2, 15 mM tBOOH, or 4 mM diamide), the ratio of the number of cells possessing a single spherical vacuole was determined from the acquired images. At least 500 cells were counted for each time point, and the experiments were repeated at least three times. In all experiments, the vacuoles were stained with FM 4-64.
Sample preparation for mass spectrometry
The purified protein was applied to an anion-exchange column (HiTrap DEAE; GE Healthcare) equilibrated with 20 mM Tris-HCl, pH 8.0, and eluted with a linear gradient of 0–1 M NaCl. Purified protein (60 pmol) in 20 mM Tris-HCl, pH 8.5, was diluted to 200 µl with 100 mM ammonium bicarbonate, and 20 µl of acetonitrile was added. Each sample was treated with 10 µl of a reducing agent, 10 mM Tris(2-carboxyethyl)phosphine, at 37°C for 45 min, and alkylated with 10 µl of cysteine blocking reagent, 50 mM iodoacetamide, at 24°C for 1 h in the dark. Proteins were digested with 0.1 µg/µl trypsin (Promega) in 100 mM ammonium bicarbonate at 37°C overnight. The digestion reaction was stopped by addition of 250 µl of 1% trifluoroacetic acid. After vortexing, 200 fmol amounts of the resulting samples were directly subjected to liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis.
LC-MS/MS
LC-MS/MS analysis of the digested samples was performed with an RP-LC system (Paradigm MS4B; Michrom BioResources, Inc.) interfaced with a hybrid mass spectrometer (LTQ-Orbitrap; Thermo Fisher Scientific) using a nano-electrospray ionization device (AMR). Samples were loaded onto the trap cartridge and washed with mobile phase A (98% H2O, 2% acetonitrile, and 0.1% formic acid) for concentration and desalting. Subsequently, peptides were eluted over 20 min from the analytical column via the trap cartridge using a linear gradient of 5–45% mobile phase B (10% H2O, 90% acetonitrile, and 0.1% formic acid) at a flow rate of 1 µl/min. To survey the phosphorylated peptides in each sample, the mass spectrometer was operated in the data-dependent mode to automatically switch between one high-resolution MS scan (resolution, 60,000; scan range, m/z 400–1,600) by the Orbitrap hybrid mass spectrometer and up to three concurrent MS/MS scans in the LTQ for the three most intense peaks selected from each MS scan. For determination of the phosphorylation sites on the phosphorylated peptides, the mass spectrometer was operated in the targeted MS/MS scan mode to switch between high-resolution MS and high-resolution MS/MS scans (resolution, 60,000); both were acquired using the Orbitrap in profile mode. Mascot software (version 2.1.1; Matrix Science) was used for matching searches against the PpAtg18 sequence without an initial Met and with an artificial Gly-Pro-Leu-Gly-Ser sequence at the N terminus. Peptide mass tolerance was 20 ppm, fragment mass tolerance was 0.8 D, and trypsin specificity was applied with a maximum of two missed cleavages. Carbamidomethylation of Cys was allowed as a fixed modification, whereas oxidation of Met and phosphorylation of Ser and Thr were allowed as variable modifications. Phosphorylation sites were determined by manually comparing datasets of product ions acquired from Orbitrap MS/MS spectra, and theoretical fragment ions were estimated using the MS-product tool on the ProteinProspector web site (http://prospector.ucsf.edu/prospector/mshome.htm).
Online supplemental material
Fig. S1 shows identification of phosphorylated regions by LC-MS/MS. Fig. S2 shows purification of PpAtg18 proteins from P. pastoris. Fig.S3 shows the role of PpAtg18 in autophagic pathways. Table S1 lists yeast strains used in this study. Table S2 lists oligonucleotides used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201302067/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201302067.dv.
Supplementary Material
Acknowledgments
We thank Dr. Jun Hoseki, for critical reading of the manuscript and valuable comments, and Dr. Michael Thumm for discussions regarding the crystal structure of PROPPIN. We thank Dr. Subramani for the gift of the PpPex12 antibody and all members of Sakai laboratory.
This work was supported partly by a Core Research for Evolutional Science and Technology grant (2006–2010) and by an Advanced Low Carbon Technology Research and Development Program (2012–present) from the Japan Science and Technology Agency, and Grants-in-Aid for Scientific Research 18076002 (2006–2010) and 2238005 (2010–2012) from the Japan Society for the Promotion of Science.
Footnotes
Abbreviations used in this paper:
- AOX
- alcohol oxidase
- GS 4B
- Glutathione Sepharose 4B
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MIPA
- micropexophagy-specific membrane apparatus
- PAS
- preautophagosomal structure
- PI
- phosphoinositide
- PI(3)P
- phosphatidylinositol 3-monophosphate
- PI(3,5)P2
- phosphatidylinositol 3,5-bisphosphate
- PIP
- phosphatidylinositol phosphate
- VSM
- vacuolar sequestering membrane
References
- Asakura M., Ninomiya S., Sugimoto M., Oku M., Yamashita S., Okuno T., Sakai Y., Takano Y. 2009. Atg26-mediated pexophagy is required for host invasion by the plant pathogenic fungus Colletotrichum orbiculare. Plant Cell. 21:1291–1304 10.1105/tpc.108.060996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran S., Ragusa M.J., Boura E., Hurley J.H. 2012. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol. Cell. 47:339–348 10.1016/j.molcel.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera M., Ostrowicz C.W., Mari M., LaGrassa T.J., Reggiori F., Ungermann C. 2009. Vps41 phosphorylation and the Rab Ypt7 control the targeting of the HOPS complex to endosome-vacuole fusion sites. Mol. Biol. Cell. 20:1937–1948 10.1091/mbc.E08-09-0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera M., Langemeyer L., Mari M., Rethmeier R., Orban I., Perz A., Bröcker C., Griffith J., Klose D., Steinhoff H.J., et al. 2010. Phosphorylation of a membrane curvature-sensing motif switches function of the HOPS subunit Vps41 in membrane tethering. J. Cell Biol. 191:845–859 10.1083/jcb.201004092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443:651–657 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- Dove S.K., Cooke F.T., Douglas M.R., Sayers L.G., Parker P.J., Michell R.H. 1997. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 390:187–192 10.1038/36613 [DOI] [PubMed] [Google Scholar]
- Dove S.K., Piper R.C., McEwen R.K., Yu J.W., King M.C., Hughes D.C., Thuring J., Holmes A.B., Cooke F.T., Michell R.H., et al. 2004. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 23:1922–1933 10.1038/sj.emboj.7600203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove S.K., Dong K., Kobayashi T., Williams F.K., Michell R.H. 2009. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem. J. 419:1–13 10.1042/BJ20081950 [DOI] [PubMed] [Google Scholar]
- Efe J.A., Botelho R.J., Emr S.D. 2007. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol. Biol. Cell. 18:4232–4244 10.1091/mbc.E07-04-0301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré J.-C., Manjithaya R., Mathewson R.D., Subramani S. 2008. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell. 14:365–376 10.1016/j.devcel.2007.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J., Stromhaug P.E., George M.D., Habibzadegah-Tari P., Bevan A., Dunn W.A., Jr, Klionsky D.J. 2001. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol. Biol. Cell. 12:3821–3838 10.1091/mbc.12.12.3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi K., Yurimoto H., Oku M., Sakai Y. 2011. Yeast methylotrophy and autophagy in a methanol-oscillating environment on growing Arabidopsis thaliana leaves. PLoS ONE. 6:e25257 10.1371/journal.pone.0025257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Suzuki K., Ohsumi Y. 2012. Autophagosome formation can be achieved in the absence of Atg18 by expressing engineered PAS-targeted Atg2. FEBS Lett. 586:2473–2478 10.1016/j.febslet.2012.06.008 [DOI] [PubMed] [Google Scholar]
- Krick R., Tolstrup J., Appelles A., Henke S., Thumm M. 2006. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett. 580:4632–4638 10.1016/j.febslet.2006.07.041 [DOI] [PubMed] [Google Scholar]
- Krick R., Busse R.A., Scacioc A., Stephan M., Janshoff A., Thumm M., Kühnel K. 2012. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc. Natl. Acad. Sci. USA. 109:E2042–E2049 10.1073/pnas.1205128109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrassa T.J., Ungermann C. 2005. The vacuolar kinase Yck3 maintains organelle fragmentation by regulating the HOPS tethering complex. J. Cell Biol. 168:401–414 10.1083/jcb.200407141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9:99–111 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- Li S.C., Kane P.M. 2009. The yeast lysosome-like vacuole: endpoint and crossroads. Biochim. Biophys. Acta. 1793:650–663 10.1016/j.bbamcr.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjithaya R., Nazarko T.Y., Farré J.C., Subramani S. 2010. Molecular mechanism and physiological role of pexophagy. FEBS Lett. 584:1367–1373 10.1016/j.febslet.2010.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Ohsumi Y. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- Obara K., Sekito T., Niimi K., Ohsumi Y. 2008. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 283:23972–23980 10.1074/jbc.M803180200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M., Sakai Y. 2010. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 277:3289–3294 10.1111/j.1742-4658.2010.07741.x [DOI] [PubMed] [Google Scholar]
- Powell D.J., Hajduch E., Kular G., Hundal H.S. 2003. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol. Cell. Biol. 23:7794–7808 10.1128/MCB.23.21.7794-7808.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieter E., Vinke F., Bakula D., Cebollero E., Ungermann C., Proikas-Cezanne T., Reggiori F. 2013. Atg18 function in autophagy is regulated by specific sites within its β-propeller. J. Cell Sci. 126:593–604 10.1242/jcs.115725 [DOI] [PubMed] [Google Scholar]
- Suzuki K., Kubota Y., Sekito T., Ohsumi Y. 2007. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 12:209–218 10.1111/j.1365-2443.2007.01050.x [DOI] [PubMed] [Google Scholar]
- Tamura N., Oku M., Sakai Y. 2010. Atg8 regulates vacuolar membrane dynamics in a lipidation-independent manner in Pichia pastoris. J. Cell Sci. 123:4107–4116 10.1242/jcs.070045 [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Kobayashi T., Yamamoto H., Hoshida H., Akada R., Inagaki F., Ohsumi Y., Noda N.N. 2012. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J. Biol. Chem. 287:31681–31690 10.1074/jbc.M112.397570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman L.S. 2006. Organelles on the move: insights from yeast vacuole inheritance. Nat. Rev. Mol. Cell Biol. 7:243–252 10.1038/nrm1892 [DOI] [PubMed] [Google Scholar]
- Yamashita S., Yurimoto H., Murakami D., Yoshikawa M., Oku M., Sakai Y. 2009. Lag-phase autophagy in the methylotrophic yeast Pichia pastoris. Genes Cells. 14:861–870 10.1111/j.1365-2443.2009.01316.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.