

Abstract
Free base octaethylporphyrin (OEP) was converted, in two steps (β,β′-dihydroxylation, oxidative diol cleavage with concomitant aldol condensation), to the corresponding oxypyriporphyrin. This conversion was previously described to be only applicable to the Ni(II) complex of OEP. Modified diol cleavage conditions made this reaction sequence now applicable to free base OEP. The single crystal structure of the resulting free base oxypyriporphyrin was determined, proving its near-perfect planarity. The reaction sequence can also be applied to oxypyriporphyrin itself, generating the unprecedented bacteriochlorin-type bis-oxypyriporphyrin as two separable isomers. The ground state (UV/Vis and fluorescence) and excited state (transient triplet-triplet absorption, triplet lifetimes, and triplet EPR) photophysical properties of all chromophores are described and contrasted against OEP, chlorins, and oxochlorins. The pyridone-modified porphyrins possess unique spectroscopic signatures that distinguish them from regular porphyrins or chlorins. The presence of the pyridone moiety alters the ESI+ collision-induced fragmentation properties of these oxypyriporphyrins only to a minor degree when compared to those of OEP or chlorins, attesting to their stability.
Keywords: porphyrins, porphyrinoids, octaethylporphyrin, chlorins, photophysics, tandem mass spectrometry
Introduction
The chemical and physical properties of porphyrinic compounds in nature are adjusted by the variation of the degree of saturation of the tetrapyrrolic macrocycle, as in chlorins (2,3-dihydroporphyrins) or bacteriochlorins (2,3,12,13-tetrahydroporphyrins), or by variation of the substituent pattern on the macrocycle periphery.[1] The latter may affect the porphyrinic π–system either directly through conjugation or indirectly through the tuning of the conformation of the macrocycle.[2]
Porphyrins and chlorins are attractive targets for use in a diverse range of technical and medicinal applications.[3] The majority of these applications requires the ability to tune their photophysical properties. While progress has been made in the understanding of the factors controlling the optical properties of porphyrins, much is left to be desired for the synthesis of porphyrins with designed physical and chemical properties. As a consequence, this area represents one of the foci in current porphyrin research.[4]
The search for porphyrins with tunable long wavelength absorbing and emitting properties led to the development of porphyrinic chromophores that have no natural precedent, including expanded porphyrins, porphyrin isomers, or porphyrin analogues that contain non-pyrrolic building blocks.[5–7] The bulk of these porphyrinoids are made by total synthesis, i.e., they are synthesized in one or more steps from mono-pyrrolic precursors.
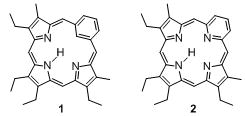
Examples of porphyrinoids containing non-pyrrolic building blocks are carbaporphyrins such as benziporphyrin 1[7,8] or pyriporphyrin 2.[9–12] However, the benzene and pyridine moieties, respectively, are not fully conjugated into a porphyrin-like aromatic system. In contrast, oxypyriporphyrin 3 contains a porphyrin-like π-aromatic system incorporating the pyridone moiety.[13–16] Lash and his group have optimized the 3+1 total synthesis approach toward oxybenziporphyrins and related macrocycles (Scheme 1).[6] A tripyrrane, such as 4 (often prepared in situ), is condensed with a bisaldehyde, such as 2-formylsalicylaldehyde, and the intermediate macrocycle is aromatized by means of an oxidation. This pathway proved to be extremely successful and led to the synthesis of a wide structural variety of pyrrole-modified porphyrins and carbaporphyrins.[6]
Scheme 1.
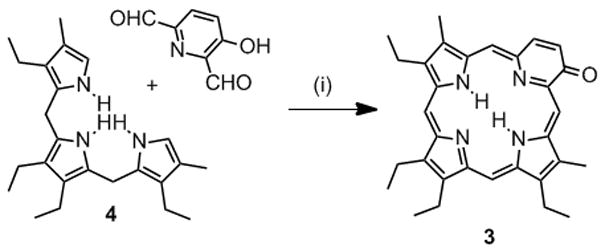
3+1 synthesis of oxypyriporphyrin 3.[15] Reaction Conditions: 1. [H+], 2. DDQ.
A Fe-complex of oxypyriporphyrin was prepared and apomyoglobin reconstituted with this heme analogue.[17,18] The holoenzyme did not exhibit a measurable affinity for oxygen and exhibited a number of other functional anomalies when compared to native myoglobin, but provided valuable insight into the structural ligand control of oxygen binding and provided a model compound for one other heme enzyme, guanylate cyclase.
An alternative approach toward pyrrole-modified porphyrins is the modification of a readily available porphyrin, such as meso-tetraphenylporphyrin (TPP) or octaethylporphyrin (OEP, 5, Scheme 2). The earliest example of a porphyrinoid resulting from an insertion reaction of a carbon into a pyrrolic unit of a porphyrin was reported by Callot and Schaeffer.[11,19] A Ni(II)-induced rearrangement of a N-substituted TPP resulted in the ring expansion of one of the pyrrole moieties to form azine-modified porphyrin 6. Crossley and King reported a series of oxidation reactions of 2-amino-substituted TPP that lead to the observation of a series of pyrrole-modified porphyrins, among them anhydride 7.[20]
Scheme 2.
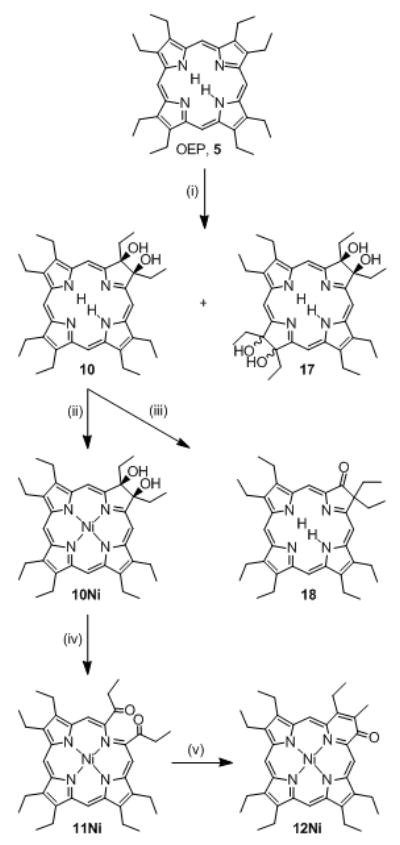
Bonnett’s synthesis of secochlorin 11Ni, pyriporphyrin 14Ni, and the literature-known pathway towards tetraol bacteriochlorin 17 and oxochlorin 18. Reaction Conditions: (i)[25] 1. OsO4/pyridine, 2. H2S, 3. chromatographic separation; (ii)[25] Ni(II) acetate, pyridine, Δ; (iii)[26] cat. HClO4, benzene, Δ; (iv)[27,28] Pb(IV) acetate, THF; (v)[27,28] base.
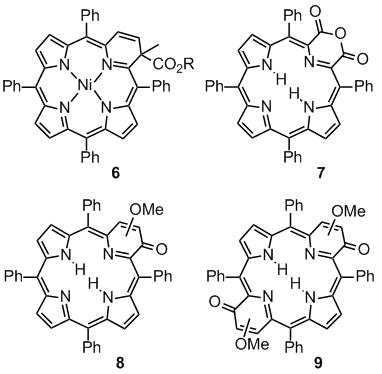
We reported the step-wise functionalization of TPP that leads to its conversion to, for instance, morpholino- or pyrazinochlorins and we demonstrated their unique properties as compared to, porphyrins and chlorins.[21] The one-step reaction of 2,3-dioxo-TPP and 2,3,12,13-tetraoxo-TPP[22] with diazomethane allowed the preparation of oxypyriporphyrin 8 and bis(oxypyri)porphyrin 9, each prepared as mixtures of inseparable regioisomers.[23] Compound 9 is a rare examples of a bis-pyrrole-modified system.[24] It is, to our knowledge, the only porphyrinoid system containing two six-membered rings known to date.
Bonnett and co-workers firstly described the step-wise functionalization of OEP to form an octaalkyloxypyriporphyrin (Scheme 2).[27,28] OEP was first converted to 2,3-dihydroxychlorin 10, followed by insertion of Ni(II) to form 10Ni and a Pb(IV)(acetate)4-induced diol cleavage reaction to form the Ni(II) complex of secochlorin 11Ni. Under base catalysis, this diketone underwent an intramolecular aldol condensation, resulting in the formation of the oxypyriporphyrin Ni(II) complex 12Ni.[27,28] Thus, one pyrrole unit in OEP was formally replaced by a six-membered pyridin-3-on moiety in two well controlled and efficient steps. The influence of this seminal paper on the development of strategies toward the conversion of porphyrins to other pyrrole-modified porphyrins cannot be underestimated and, as the forthcoming will reveal, this contribution is a direct extension of this work.
The potentially much more useful free base analogue to 12Ni could not be prepared, however.[27,28] Demetalation of 12Ni was unsuccessful and the oxidation of free base diol 10 resulted in a complex mixture from which no product could be isolated. Since the structurally very similar free base compound 3 and related compounds are stable when prepared by total synthesis,[15,16,29] the problem likely lies in the instability of the intermediate free base secochlorin 11. Whereas its Ni(II) complex 11Ni is stable and could be structurally characterized,[27,28] free base secochlorin 11 may not be stable enough under the reaction conditions employed to allow high-yielding conversions. This conclusion is mirrored by our own findings that Ni(II) imparts particular stability on TPP-derived secochlorins.[30,25]
To account for the limited stability of TPP-derived free base secochlorins and to circumvent the problem of removing Ni(II) from the final product, we devised two principle synthetic methodologies: one uses Ag(II) as a stabilizing but removable metal ion template,[31] the other is a two-step, one-pot approach in which the free base secochlorin, prepared in situ using mild diol cleavage conditions (NaIO4 heterogenized on silica gel), is trapped with an appropriate reagent.[32]
The latter synthetic methodology was successfully tested. Thus, we report here the step-wise conversion of OEP, 5, to free base pyrrole-modified porphyrin 12. Moreover, the mild β,β′-cleavage and ring-fusion conditions allowed the preparation of two isomeric bis(oxypyri)porphyrins 16, the first examples of their class. We further report the crystal structure of 12, the ground (UV/Vis and fluorescence) and excited state (triplet spectra, triplet life times and EPR spectra) photophysical properties of all novel macrocycles, and their collision-induced fragmentation mass spectra. We contrast the results obtained to their non-pyrrole-modified counterparts. This allows the derivation of some structure-physical properties relationships for these porphyrinoids.
Results and Discussion
Diol cleavage of dihydroxychlorin 10 – synthesis of oxypyriporphyrin 12
Confirming the report by Bonnett,[27,28] oxidation of 10 with Pb(IV) acetate does not allow the isolation of any product in reasonable yield. We surmised that the Pb(IV)-acetate/THF reaction conditions are too acidic for secochlorin 11. Thus, reaction of blue diolchlorin 10 with a slurry of NaIO4-impregnated silica gel in CHCl3 containing 1–10% Et3N results in the formation of a less polar red-purple product (Scheme 3). Its ESI+ mass spectrum suggests the composition for the expected bisketone 11 (m/z = 567.3679 for [11·H]+). The major fragment ion of this compound is [MH - H2O]+, perhaps indicating that an aldol condensation reaction of 11 to form 12 is taking place under the conditions of the ESI+ mass spectrometric measurement. However, the 1H NMR spectrum of the red product shows in the chemical shift range typical for meso-protons, instead of the two signals expected, four singlets (at 10.46, 9.94, 9.88, and 9.80 ppm, corresponding to 1H each), and two non-equivalent NH protons. The 13C NMR spectrum confirms the presence of a molecule lacking two-fold symmetry and indicates the presence of one carbonyl carbon (δ = 202.1 ppm in the 13C NMR) in conjunction with two significantly low-field-shifted pyrrolidine-like sp3-carbons (δ = 79.7 and 71.0 ppm). All together, the data suggests that diol cleavage of 10 to diketone 11 has taken place but that this diketone has reacted spontaneously to form aldol product 14. Commensurate with this assignment, the Soret band in the UV/Vis spectrum is significantly more red-shifted (λmax = 410 nm; Figure 1) compared to the spectrum for the starting diol chlorin 10, but altogether very much different from that described for oxypyriporphyrin 3.[15,16,29] This is likely an effect of the presence of the reduced ‘β,β′-bond’ in the dihydropyridone moiety. Compound 14 is not stable and significantly decomposes in solution within a day. The addition of base (up to 10 vol% DBU) or catalytic amounts of acid (TFA fumes) hastens its disappearance but leads cleanly to the appearance of a bright green product with an m/z ratio of 548.3533 in its ESI+ spectrum, corresponding to C36H45N4O + H+, the composition expected for the target free base aldol condensation product oxypyriporphyrin 12.
Scheme 3.
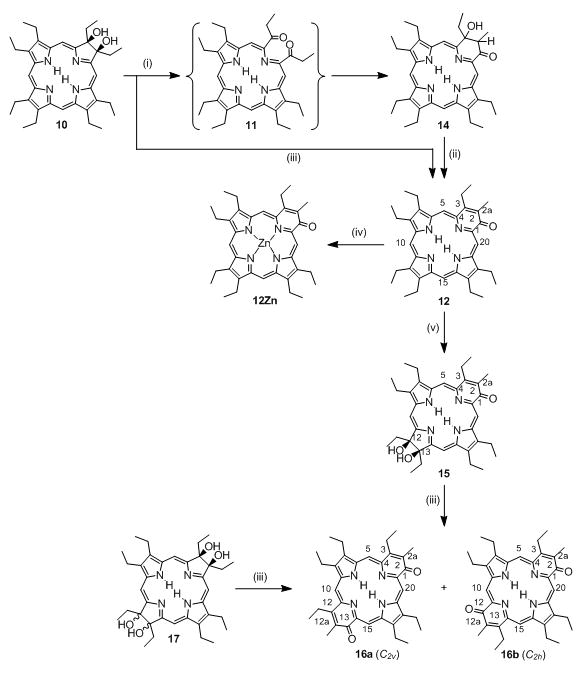
Syntheses of the free base chromophores 12, 14 - 18. Reaction Conditions: (i) 1. NaIO4/silica, Et3N, CHCl3, 2. chromatographic separation; (ii) cat. acid or base; (iii) 1. NaIO4/silica, DBU, CHCl3, 2. chromatographic separation; (iv) Zn(II) acetate, CHCl3/MeOH, Δ; (v) 1. OsO4/py, CHCl3 2. H2S, 3. chromatographic separation.
Figure 1.

Normalized UV/Vis spectra (CHCl3) of dihydroxychlorin 10 (solid trace), cleavage product 14 (dashed trace), and 12,13-dihydroxyoxypyriporphyrin 15 (dotted trace).
These findings suggested the use of DBU as base for a one-pot, two-step conversion of free base diolchlorin 10 to oxypyriporphyrin 12. Indeed, addition of free base diolchlorin 10 to a slurry of the silica gel/NaIO4 oxidant in CHCl3 in the presence of 5–10 vol% DBU leads, over the course of 2 to 12 h at ambient temperature, to a conversion of the initially blue solution to a green solution. TLC analysis revealed the formation of one major bright green compound of lesser polarity. This product can be, after chromatographic separation and crystallization, isolated in over 50% yield (at up to 1 × 10−4 mol scales) as a purple microcrystalline solid, and proved to be identical to oxypyriporphyrin 12 described above (Scheme 3). Of all the bases tested (pyrazole and imidazole – no conversions; pyridine and Et3N – slow conversion) we found DBU to be the best base to cleanly and rapidly lead to the formation of the product.
When compared to the spectrum of diol chlorin 10, the chromophore is blue-shifted and more porphyrin-like (Figure 4). As expected, the UV/Vis spectrum of the product is near-identical to that of oxypyriporphyrin 3.[15] A discussion of the UV/Vis spectra of this and the other novel compounds prepared herein is presented below. Also, we will detail the ESI+ collision-induced fragmentation spectra of 12 and other novel compounds below.
Figure 4.

The normalized absorption and fluorescence emission spectra and triplet life-times of 5, 12, 18, 10, 15, 17, 16a, and 16b (all at ambient temperature, degassed toluene). The triplet excited state lifetimes were calculated as mean of at least eight decay constants of the decay kinetics taken (440 nm for 5, 470 nm for 12, 470 nm for 18, 430 nm for 10, 460 nm for 15, 420 nm for 17, 520 nm for 16a and 510 nm for 16b). For the transient triplet-triplet absorption spectra, see SI Figure S16.
The 1H NMR spectrum of oxypyriporphyrin 12 shows the diagnostic peaks described for this compound class,[15,16,29] modulated by the particular substitution pattern present: next to the four low-field signals assigned to the non-equivalent meso-protons, two high-field signals assigned to the NH protons can be distinguished. One methyl group (s at 2.82 ppm, 3H) can also be differentiated; the remaining ethyl groups show significant overlaps. The combination of HMQC, HMBC and NOESY spectra allowed the unambiguous assignment of most peaks in the 1H and 13C NMR spectra. In summary, the spectroscopic properties of this compound unambiguously identify it as free base oxypyriporphyrin 12. It thus expands the number of known derivatives of this compound class,[15,16,29] but more importantly, it demonstrates the applicability of the porphyrin β,β′-bond cleavage and ring-fusion strategy toward the synthesis of free base OEP-derived pyrrole-modified porphyrins.
Standard Zn(II) insertion protocols generate the complex 12Zn. This derivative also possesses the spectroscopic properties one would expect when projecting the data reported by Lash for similar complexes,[15] including its peculiar solubility and solvochromic properties that point toward extensive aggregation behavior (see SI).[33]
Single crystal X-ray structure of oxypyriporphyrin 12
The spectroscopically derived connectivity of 12 could be confirmed by single crystal X-ray crystallography, the first example of a structural characterization of this class of pyrrole-modified porphyrins (Figure 2A). Somewhat surprising is the pronounced planarity of the oxypyriporphyrin chromophore (Figure 2B). Evidently, the expansion of a pyrrolic moiety by a carbonyl group is readily absorbed by the macrocycle without causing any major distortions. In comparison, the related free base pyriporphyrins, such as 2, are expected to be decidedly non-planar, with the mean plane of the pyridine moiety taking up a steep angle with respect to the mean plane of the remaining tripyrrolic moiety, a feature that is even preserved in their metal complexes.[9,34]
Figure 2.

View of the molecular structure of 12. Only one of the two whole molecule-disordered molecules is shown. A. View from an oblique angle. B. Side view along the N3-N1 axis, highlighting the degree of planarity of the macrocycle.
Figure 3 illustrates the bond length and inner core distance differences observed in 12 with respect to the metrics of OEP.[35] The differences in the pyridone moiety are likely the result of the fixed enone conjugation of this cross-conjugated moiety. The additive effects of many small changes in the bond distances and angles (not shown) lead to a significant tetragonal distortion of the inner core of 12. In OEP, the distances between opposing inner nitrogens are 4.052 (N-N) and 4.195 Å (NH-NH), respectively, i.e. the cavity is about 0.14 Å longer than wide, with the NH hydrogens positioned on the longer axis. In 12, the cavity is significantly longer (4.326 Å, N-N) and slightly narrower (4.155 Å, NH-NH), but with the NH protons lying on the shorter axis. This elongation was recently predicted for the Fe(III) complex of an oxypyriporphyrin based on EPR data and quantum chemical calculations.[18]
Figure 3.
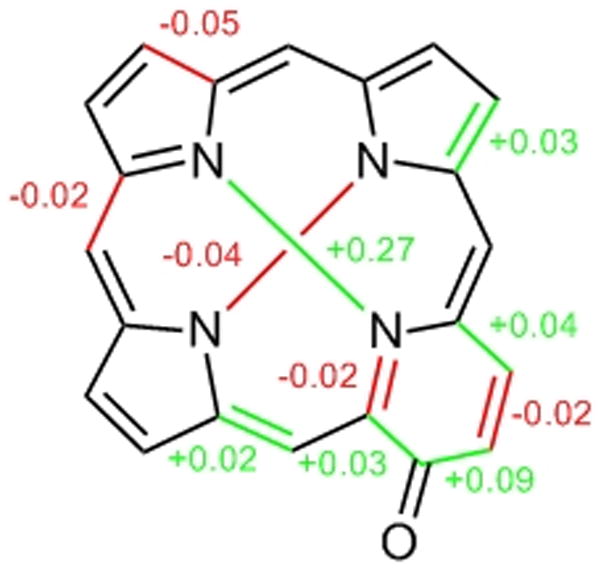
Differences in bond length (in Å) observed in 12 compared to 5.[31] Arbitrarily, only differences that are equal or larger than 0.02 Å, i.e. equal or larger than 2 × the average e.s.d. for C-C bond lengths determined for 12 (e.s.d. = 0.01 Å), are considered. Comparison of the distances in the pyridone moiety with those of a pyrrolic moiety in 5 carrying an imine-type nitrogen.
Dihydroxylation of oxypyriporphyrin 12
The ability to formally replace one pyrrolic building block of free base OEP by a pyridin-3-one moiety in two reasonably high yielding reaction steps allows us to test whether this reaction sequence can be performed twice on OEP, giving rise to novel doubly pyrrole-modified chromophores. Two reaction pathways are perceivable: Firstly, an additional dihydroxylation of oxypyriporphyrin 12, followed by diol cleavage/aldol reaction and, secondly, the onestep, double diol cleavage/aldol reaction on known tetraolbacteriochlorins 17 (Scheme 2). Both approaches proved successful and each has its own merits.
Reacting green 12 with stoichiometric amounts of OsO4 in CHCl3/pyridine leads, over the course of one week, to the formation of one major, forest-green, polar product. Quenching of the osmate ester with H2S, followed by chromatography and recrystallization, leads to the formation of a dark-green microcrystalline compound in acceptable yields (58%, 10−4 mol scale). High resolution ESI+ mass spectrometry confirms the composition of the product to be C36H47N4O3, i.e. the expected composition of the (protonated) diol adduct 15.
It has been well established that OsO4 adds to the double bond that leads to the smallest loss in resonance energy.[36] In porphyrins specifically, it attacks the double bond of largest olefinic character. Hence, at least two locations for the diol functionality are perceivable. Lash has shown that oxypyriporphyrins are aromatic, with an implied chlorin-like conjugation pathway.[15] Following the known directing effects of free base chlorins with respect to a second reduction or dihydroxylation,[37,38,39] we can expect the β,β′-double bond opposite to the oxypyridine moiety (C12-C13) to be the most likely dihydroxylation site, thus rendering the two β,β′-double bonds adjacent to the non-pyrrolic building block (either C7-C8 or C17-C18) as the less likely sites. On the other hand, we cannot exclude the enone functionality (C3-C2a double bond) in the pyridone moiety as a potential dihydroxylation site.[40]
Dihydroxylation of the pyridone moiety in 12 would generate a chromophore that is predicted to possess similar optical properties to 14. Inspection of the UV/Vis spectrum of the bright-green compound, however, shows a much red-shifted spectrum compared to that of 14 (Δλmax of the longest wavelength band = + 23 nm), with the longest wavelength absorbance also being the most intense of all Q-bands (Figures 1 and 4). These changes in the UV/Vis upon dihydroxylation of 12 are suggestive of a β,β′-bond dihydroxylation forming 12,13-dihydroxyoxypyrichlorin 15.
The diagnostic resonances in the 1H NMR of 15 for the pyridone moiety are preserved and new signals indicative for a dihydroxy-pyrrolidine moiety appeared. If hydroxylation of a β,β′-bond adjacent to the pyridone moiety would have taken place, it would have generated a mixture of two regioisomers (each present as a racemic mixture), but we could not find any indication for the formation of any regioisomers. Diol 15 is expected to be a simple racemic mixture. Taken all together, the NMR data support the formation of bacteriochlorin-like 15. As we will show below, definitive proof for this assignment is provided by the reaction products of 15.
Diol cleavage of dihydroxyoxypyrichlorin 15 – synthesis of bis(oxypyri)porphyrins 16
Reaction of dihydroxyoxypyriporphyrin 15 with silica/NaIO4 in CHCl3/10% DBU at ambient temperature leads, over the course of ~2 h, to the formation of two less polar compounds (Rf-16a = 0.43, Rf-16b = 0.34, silica-CHCl3/1% MeOH) in an 1:1 ratio. Their UV/Vis spectra are similar (Figure 4). Both possess identical compositions as determined by HR ESI+ mass spectrometry (M+H+ corresponding to C36H43N4O2). The 13C NMR spectra of 16a and 16b suggest the presence of two similar two-fold symmetric molecules: each possess only two signals assigned to meso-carbons (186.0, 152.4 and 185.6, 152.5 ppm, respectively), one methyl signal (2.74 and 2.72 ppm, respectively), and one carbonyl carbon (186.0 and 185.6 ppm, respectively). Both compounds also possess different IR spectra (Figures S28 and S29). These data are as expected for the two isomeric bis(oxypyri)porphyrins 16a and 16b (Scheme 3). The identity of each isomer can be unambiguously assigned. One diagnostic feature in their 1H NMR spectra are the number of NH signals. While the two NH groups in C2v-symmetric 16a are non-equivalent and show two signals (br s at −4.40 and −4.45 ppm, 1H each), both NH groups are symmetry-equivalent in the C2h-symmetric isomer 16b, and only one signal can be distinguished (br s at −3.80 ppm, 2H).
Some of the NMR-derived symmetry arguments brought forward in support of the bacteriochlorin-type connectivity of chromophores 16a/b can also be made for an isobacteriochlorin-like substitution pattern. However, such an isobacteriochlorin-type substitution pattern for 15 would have led to formation of three isomeric bis(oxypyri)porphyrins in an (idealized) 1:2:1 ratio. But most convincingly, the double oxidation of a genuine bacteriochlorin, tetraol 17, also produced 16a/b.
Diol cleavage of tetrahydroxybacteriochlorins 17 – alternate synthesis of bis(oxypyri)porphyrins 16
A one-pot, four-step conversion of tetraolbacteriochlorin 17 proved to be an alternate synthesis of bis(oxypyri)porphyrins 16a/b. Thus, subjecting tetraol 17 to the standard diol cleavage reaction conditions, the pink solution turns green within 2 h at room temperature. Once TLC analysis of the reaction mixture has indicated the consumption of the tetraolbacteriochlorin, three products can be identified: The mono-oxidized compound 15 – thus establishing the bacteriochlorin-type substitution pattern of this compound – and the two isomers of 16 in a ~1:1 ratio. Lengthening of the reaction time or addition of more oxidant converts the majority of 15 into 16a/b, isolable in a combined yield of about 50%. While this reaction is simpler than the stepwise approach, the preparation and purification of the tetraols 17 are more tedious than that of diol 10, largely canceling the advantages of the simplicity of the final step.
Both stereoisomers of 17 (resulting from the two possible relative orientations of the vic diols with respect to each other – syn and anti) form identical sets of bis(oxypyri)porphyrins 16a/b but for sake of simplicity, we generally used mixtures of the isomers of 17 in these reactions.
Absorption and fluorescence spectroscopy of singlet-states
We will be discussing the optical properties of the oxypyri-modified chromophores 12, 15, and 16a/b in comparison to those of the parent porphyrin 5, a typical chlorin, 10, and a bacteriochlorin, 17. To assess the influence of a carbonyl group, we also included known oxochlorin 18 in the comparison, made by a pinacol-pinacolone rearrangement of diol 10 (Scheme 2).[41] Focus in this comparison is the question in as far the pyridone-modified chromophores possess optical properties that are porphyrin- or chlorin-like or whether they are altogether unique.
Inspection of the UV/Vis spectra shown in Figure 4 reveals that the Q-bands in the UV/Vis spectrum of oxypyriporphyrin 12 are not at all chlorin-like. They are porphyrin-like with respect to their descending intensity with longer wavelengths, and the small intensity ratio of Q-bands to Soret band. This was noted before.[15,16,29] Likewise, bis-modified systems 16 are not chlorin or bacteriochlorin-like (cf. to 10 or 17). The Soret band of 12 is, compared to porphyrin 5 or chlorin 10, 25 and 32 nm red-shifted, respectively, and split. The latter feature is shared, albeit expressed to different degrees, by all pyridone-modified chromophores (12, 15, 16). Generalized, the effect of the presence of pyridone units merely results in a ~25 nm shift per unit of the Soret and side bands. Thus, removal of a β,β′-double bond in ‘porphyrin’ 12 to its 12,13-dihydroxyderivative 15 generates a chromophore that is equivalent to a 25 nm red-shifted 12,13-dihydroxychlorin, such as 10.
The absorption spectrum of oxochlorin 18 is chlorin-like. Therefore, the presence of a conjugated ketone alone cannot account for the unexpected behavior of the oxypyri-systems and, therefore, it must be the combination of the presence of the six-membered ring composed of sp2 carbons and the conjugated ketone. Removal of the pyridone C-C double bond, as in 14, results in a chlorin-type spectrum (cf. to Figure 1).
The high extinction coefficients for 12 (log ε for λSoret = 5.14), support the notion that 12 is also essentially planar in solution, excluding conformational effects as primarily responsible for the modulation of the optical spectra. Of note is also the observation that the two regioisomers of the bis-modified systems 16a/b have distinct, if similar, absorption and emission spectra.
The fluorescence emission spectra of the chromophores also show a signature feature for the presence of a pyridone moiety (in the absence of any other pyrrole modification), in that the second vibronic band possesses higher intensity than the (0-0) emission (Figure 4). This is not typically observed for porphyrins or chlorins. In the hybrid molecule 15, the (red-shifted) chlorin character prevails. A possible practical advantage of the high intensity second vibronic band in the pyridone-modified systems 12 and 16 are the large separations between suitable long wavelengths of excitation and the highest intensity emission wavelengths. Otherwise, the porphyrin-typical small Stoke’s shifts for the (0-0) band are observed in all systems. Moreover, the λmax-emission for 16a/b are significantly red-shifted compared to those of mono-pyridone 12 or even bacteriochlorin 17, though combined with similar, or even blue-shifted λmax-absorption. Thus, the pyridone moieties introduce unique optical features into the porphyrinoid chromophore.
Base properties
Monoprotonation of the inner imine-type nitrogens of a regular porphyrin such as OEP distorts it from planarity, exposing its second imine-type nitrogen and making the second protonation to occur more readily than the first (pKb1 < pKb2). Hence, the monoprotonated species is never observed and a spectrophotometric titration of OEP with TFA in CH2Cl2 exhibits sharp isosbestic points (Figure S17). In contrast, and confirming observations made by Lash,[15,16,29] the corresponding titrations of 12 or 16a/b indicate step-wise protonation events (Figures S18–23). Step-wise addition of 1 equiv TFA to a solution of 12 in CDCl3 shows the presence of only three NH protons (at −0.8, −1.2, and −2.7 ppm at ~0.1% TFA; see Figure S22). Aside from the high-field shift of a pair of methylene signals assigned to the 121,131-CH2 groups and a high-field shift of the meso-protons, except the one signal assigned to 20-CH, no other major changes are observed. This indicates that only relatively minor conformational changes have taken place upon monoprotonation. This is consistent with the assumption that the pyrrole moiety opposite of the pyridone moiety is protonated. Given that the pyridone nitrogen is in conjugation with the carbonyl group, its basicity is also expected to be greatly diminished.
Triplet lifetimes
The triplet lifetimes, determined by the observation of the dynamics of the transient triplet-triplet absorption profiles (Figure S16), show a clear trend (Figure 4). The most symmetric, rigid chromophore that does not carry a conjugated ketone functionality, porphyrin 5, has a lifetime of 530 μs.[42] The lifetime of the corresponding more flexible chlorin is about half this value (270 μs). The effects of the introduction of a second pyrrolidine moiety, as in bacteriochlorin 17, is additive and its presence once again about halves the lifetime (110 μs). A conjugated ketone moiety, as in oxochlorin 18, induces a lesser lifetime reduction (360 μs) compared to a pyrrolidine or a pyridone (12, 260 μs). Thus, both the introduction of a pyridone or pyrrolidine moiety have the same net effect. However, the effects of an introduction of a second pyridone (as in 16) or a pyrrolidine (as in 17) to a framework already incorporating a pyridone are not linear; instead, a much more dramatic reduction of the lifetimes is observed, ranging from 46 to 32 μs.
The rate constant of intersystem crossing (kisc) from the triplet state of π-systems to the ground singlet state is largely governed by non-radiative processes and can be expressed as:
| (Eq. 1) |
Here, i indicates triplet state spin sublevel, Hint is the perturbation operator responsible for driving the population from the triplet (T1) state to the isoenergetic vibrational level of the singlet ground state (S0ν), and ρ(E) is the density of final states into which isc proceeds. The energy gap rule is expected to hold for T1→S0 transitions. Thus, a direct relationship between the energy of T1 and its lifetime should be observed; the lower the T1 energy, the shorter a lifetime can be expected. The difficulty in rationalizing the observed triplet lifetimes is the lack of phosphorescence data as a measure of the T1 energy levels.
In general, the energy of the first excited triplet state is determined by the singlet-triplet splitting.[43] Assuming that this splitting is approximately the same for all molecules studied here, the order of the excited triplet state energies is expected to be the same as for their first excited singlet states. If this is correct, the shortest lifetimes for the first excited triplet state T1 are expected for bacteriochlorin 17 and 16a/b. While these chromophores indeed possess short lifetimes, chlorin-pyridone-hybrid 15 is also characterized by a very short lifetime τ without a concomitant very red-shifted λmax-absorption. Thus, molecular structure also plays an important role in driving the T1→S0 transition. Ketones possessing nπ* T1 states accelerate isc due to strong spin-orbit coupling with the low-lying ππ* states.[43,44] This rationalizes why the chromophores undergoing the fastest T1→S0 crossing are substituted by (multiple) ketone groups (like 16a/b). It is important to note, however, that the presence of a ketone alone (as in oxochlorin 18) is not sufficient to induce fast relaxation. Thus, the combination of π-extended chromophore, likely larger conformational flexibility of the bis-pyrrole modified compounds, and the presence of a (conjugated) ketone are required to rationalize the observed triplet lifetime trends. Again, this clearly differentiates the electronic structures of pyridone-modified systems from those of traditional porphyrins and chlorins.
Triplet-state EPR spectroscopy
A triplet state is, by definition, characterized by three-fold degeneracy due to three possible alignments of two unpaired electron spins. In absence of an external magnetic field, the spin-spin interactions in organic molecules have anisotropic character, leading to quantization along the principal magnetic axes system. The separation of the triplet Tx, Ty, Tz sublevels in absence of an external magnetic field is termed zero field splitting. Applying an external magnetic field provides the experimental possibility of switching the spin coupling from the internal molecular frame to a new frame quantized along the external magnetic field. Increase of the external magnetic field will increase the energy separation between triplet sublevels. EPR spectroscopy at liquid He temperatures allows the measurement of the transitions between magnetic triplet sublevels. The spin system can be described by a spin Hamiltonian. Proper treatment of this Hamiltonian allows the extraction of a number of factors that characterize the EPR spectrum and that allow conclusions to be made regarding the structural features of the chromophore.[45–47]
Among these measurables are the zero field splitting parameters E and D. The D-parameter defines the overall spatial extent of the triplet spin distribution. The E parameter describes the degree of in-planar asymmetry of the triplet state. The quantity 3|E|/|D|, termed rhombicity, lies in the range between 0 and 1. For E = 0, the molecules posses a fully symmetrical (axial) distribution of the triplet spin, typical for molecules with 3-fold or higher axial symmetry. 3|E|/|D| = 1 is characteristic for molecules with lower, orthorhombic symmetry.[45,48] Furthermore, the average inter-electron distance can be estimated, providing an estimate of the size of the triplet orbital.
The photoexcited triplet EPR spectra for oxypyriporphyrin 12 and its diol derivative 15 in comparison to porphyrin 5, diol chlorin 10, and oxochlorin 18 are shown in Figure 5. The D and E parameters and related values are collected in Table 1.
Figure 5.
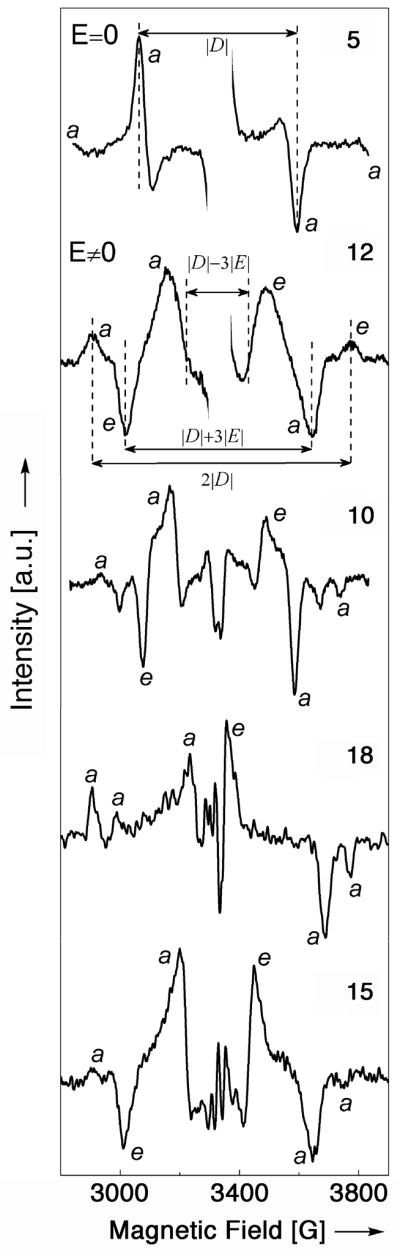
X-band EPR spectra of the light-induced triplet states, presented as light-minus-dark difference plots of (5), (10), (12), (15) and (18). The letters a and e correspond to signals associated with absorption and emission.
Table 1.
Room temperature triplet state EPR polarization data (white light excitation sources) of the molecules indicated.
| Molecule | |D|a [10−4 cm−1] | |E|b [10−4 cm−1] |
|
d [Å] | Polarization pattern | |
|---|---|---|---|---|---|---|
| 5e | 495 ± 1 | 0 | 0 | 2.72 | aa/aa | |
| 12e | 397 ± 10 | 66 ± 1 | 0.50 | 2.93 | aea/eae | |
| 10e | 370 ± 7 | 69 ± 1 | 0.55 | 3.00 | aea/eaa | |
| 15f | 393 ± 1 | 66 ± 1 | 0.50 | 2.94 | aea/eaa | |
| 18e | 399 ± 3 | 88 ± 1 | 0.66 | 2.92 | aaa/eaa |
zero field splitting parameters;
rhombocity;
mean inter-electron distance;
solvent toluene:tyridine 10:1 (v/v); room temperature;
solvent tyridine
The line shape of the EPR spectrum of randomly oriented molecules in an excited triplet state is represented by a first derivative spectrum, generally possessing six resonances, two for every x, y, z canonical Cartesian axis of the external applied magnetic field (field parallel to principal molecular axis). The absolute magnitude of D and E parameters can be directly obtained from the spectral traces, assuming 2|D| for the splitting between the outmost lines, |D| + 3|E| for splitting between intermediate lines and |D| − 3|E| for splitting between the innermost lines (Figure 5). For |E| ≈ 0 the intermediate and innermost resonances degenerate, and only four lines are observable in the EPR spectrum.[49]
The EPR spectrum of the parent porphyrins OEP, 5, has four resonance signals (two of them are weak and appear at the fringes of the spectrum).[49] This line shape is characteristic for triplet state spin system with |E| ≈ 0, and a |D| parameter value of 0.0495 cm−1, the largest number for all measured molecules. OEP possesses low orthorhombic (D2h) symmetry due to the presence of the inner NH hydrogens. However, a value of E ≈ 0 is clear evidence that their influence on the electron distribution in the π-chromophore is negligible. The average inter-electron distance between interacting spins is 2.72 Å.
The compounds 10, 12, 15, and 18 of lesser symmetry all possess six resonance lines. Their D-parameter values are notably smaller and range from 0.0370 to 0.0399 cm−1. Their lower symmetries are also evident in their non-zero E-parameters. The rhombocity varies from 0.50 to 0.66. The average inter-electron distance between spins is slightly larger compared to OEP, 5, and range from 2.92 – 3.00 Å. This indicates that the triplet state electron density is less condensed for molecules with modified pyrrole rings. It suggests also that the triplet state has expanded to include ring substitutents possessing free electron pairs, such as the ketone functionalities.
Another characteristic property of EPR triplet state spectra is their polarization pattern. At low temperatures, the T1 lifetime is shorter than the spin lattice relaxation between two spin alignment patterns of each triplet state Tx, Ty, Tz sublevel. Thus, a non-Boltzman occupation of the triplet manifold results. Observed transitions may be absorptive (a) or emissive (e). Interpretation of a particular polarization pattern observed can furnish information about intersystem crossing, spin dynamics and relaxation.[48,50,51]
Porphyrin 5 possesses an aa/aa polarization pattern, molecule 12 aea/eae, 10 aea/eaa, 18 aaa/eaa and 15 aea/eaa. Only oxypyriporphyrin 12 possesses the polarization pattern typical for free-base porphyrin macrocycles.[51,52] However, on the basis of structure and optical spectroscopic properties it possesses neither porphyrin nor chlorin symmetry. The other molecules have different and unique polarization patterns, reflecting their unique population of spin sublevels in the intersystem crossing process from S1 state. Again, this clearly differentiates these chromophores from regular porphyrins and chlorins.
Tandem ESI(+) mass spectrometry
We, and others, reported previously ESI mass spectrometry investigations of porphyrins that revealed that their fragmentation patterns were diagnostic for the presence of certain functional groups and, in some cases, the observed fragmentation patterns were reflecting their solution state chemistry.[53],[54] Lash already noted a small peak in the EI mass spectrum of 3 corresponding to the loss of CO.[29] Is this presumed ring contraction reaction of the oxypyriporphyrins a common and general fragmentation pattern for oxypyriporphyrins under ESI(+) conditions, and might this be an indication of the relative stability of the oxypyriporphyrins compared to porphyrins? To answer these and other questions, we investigated the collision-induced fragmentation spectra of the pyridone-derived porphyrinoids 12, 15, and 16a and 16b, and compared them to those obtained for OEP, 5, diol chlorin 10, and oxochlorin 18.
In general, the ESI(+) spectra of all chromophores investigated show primarily the MH+ signal, and, as commonly observed under ESI conditions, no or only minor fragmentations are observed. However, the collision-induced fragmentation spectra (MS2 spectra) of the MH+ ions show rich fragmentation patterns (Figure 6).
Figure 6.

Collision-induced fragmentation spectra (ESI+, 100% CH3CN) of 5, 10, 12, 15, 16a, and 18. Except for the spectrum of 15, all spectra were recorded using a Q-TOF mass spectrometer providing high accuracy data, allowing a direct conclusion on the compositions of the fragment ions. Composition differentials listed are based on [M+H]+.
The porphyrin OEP, 5, shows the consecutive loss of at least five single carbon fragments (largely CH4, and CH3 units), all of which are solely attributed to the degradation of the β-ethyl groups. Diol chlorin 10 shows a similar pattern as 5, modified by the loss of H2O and, eventually, both oxygens. Of interest is that the peak corresponding to the loss of water (m/z = 551.3866) is, against expectations, and irrespective of the collision energy used, always of very low abundance.
Oxochlorin 18 possesses the same composition as this fragment ion at m/z = 551.3866; in fact, 18 was prepared by dehydration of 10. But do both species possess the same structure? Indeed, the MS3 spectra of both peaks are identical (not shown). Hence, an explanation for the low abundance of the [M - H2O + H]+ peak in the MS2 spectrum of 10 likely lies in the fact that the loss of water implies that a pinacol-pinacolone-type rearrangement has to take place that, evidently, is not a facile reaction in the absence of a solvent matrix. The result also demonstrates that any exocyclic loss of water is of even higher energy.
The MS2 spectrum of 18 shows no loss of CO (or any other oxygen containing species) but instead again the loss of a number of single carbon fragments, with the most abundant fragment being that corresponding to [M - C3H8 + H]+ at m/z = 507.3178. The loss of C3H8 (44 amu) is observed in all compounds investigated, safe for the diol derivatives 10 and 15, and in all cases it a very prominent fragment. We speculate that this is due to the degradation of two adjacent β-ethyl groups (I) to form a β,β′-fused cyclopropane moiety (II) (Scheme 4). This ring fusion is surprising but appears to us to be more reasonable than the alternative diradical structure III. Aside from a report on the formation of a cyclopropane fused to a chlorin,[55] this ring fusion has not been observed in porphyrins.
Scheme 4.
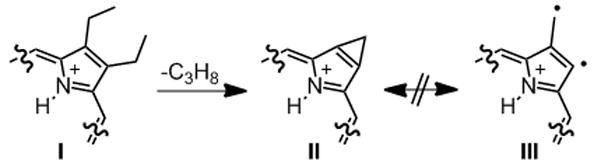
Proposed rationalization for the prominent C3H8 fragment from β,β′-diethyl-substituted porphyrins.
The MS2 spectrum of oxypyriporphyrin 12 shows the now familiar one carbon fragment fragmentation pattern typical for β-octaethyl porphyrinoids. Only one of the major fragments can be associated with the loss of oxygen (in the form of CO), following the loss of C3H8. Hence, the CO loss is only a minor fragmentation pathway for 12 under tandem ESI(+) mass spectrometry conditions, a finding also confirmed by the MS2 spectra of 15 – most similar to that of diol 10 – and of bisoxypyriporphyrin 16a – very similar to that of the mono-oxypyriporphyrin 12. The spectrum of 16b (not shown) is identical to that of 16a. Overall the results attest to the porphyrin-like stability of the planar and fully conjugated oxypyriporphyrins.
Conclusion
We have shown that a formal replacement of a pyrrolic building block in free base OEP by a pyridin-3-one moiety via step-wise derivatization is possible. The synthetic methodology used – dihydroxylation, followed by diol cleavage and aldol condensation of the resulting secochlorin in situ – produces a oxypyriporphyrin that is complementary to the compounds prepared by total syntheses.[15,16,29] We thus fill the gap between the total syntheses of the free base forms of these chromophores by Lash and the conversion of the Ni(II) complex of OEP to yield the [oxypyriporphyrinato]Ni(II) complexes featured by Bonnett.[27,28]
Surprising in its extent, the near-perfect planarity of free base oxypyriporphyrin 12 was shown by single crystal diffractometry. Compared to OEP, a small tetragonal distortion of the central cavity was noted. The doubly-modified systems, hitherto not accessible through total synthesis approaches, are readily accessible in regioisomerically pure forms. The photophysical properties of oxypyriporphyrins are characteristically different from both porphyrin or chlorins, whereby we surmise the combination of a π-conjugated ketone group and peripheral β,β′-like double bond are responsible for the observed effects. The expansion of the porphyrinic chromophore by a six-membered ring may also influence conformational flexibility. Collision-induced fragmentation mass spectra have demonstrated the minute effect that the presence of the pyridone moiety has on the gas-phase fragmentation properties of this class of chromophores. We interpret this fining as an indication of their porphyrin-like stability.
In summary, the results shed further light on the structure-physical properties and structure-chemical properties relationships of pyrrole-modified porphyrins. The straightforward syntheses of oxypyriporphyrin derivatives, combined with their stability and unique optical properties, invites their further study.
Experimental Section
General
1H NMR and 13C NMR spectra, recorded on Bruker instruments, were referenced to residual solvent peaks. All peak assignments listed were based on HMQC, NOESY, and HMBC spectra. IR spectra were recorded on a JASCO FT-IR-410 using a diffuse reflectance attachment (all IR spectra are shown in the SI). Elemental analyses were provided by Numega Resonance Labs Inc., San Diego, CA, U.S.A. The analytical TLC plates were aluminium backed Silicycle ultra pure silica gel 60, 250 mm; preparative TLC plates (20 × 20 cm, silica gel on glass) and the flash column silica gel (standard grade, 60Å, 32–63 μm) used were provided by Sorbent Technologies, Atlanta, GA, U.S.A.
Reagents
All reagents were used as received. Octaethylporphyrin[56] was provided by D. Dolphin, University of British Columbia, Canada. 2,3-Dihydroxy-octaethylchlorin (10), 2,3,12,13-tetrahydroxyoctaethylbacteriochlorin (17), 2-oxo-3,3,7,8,12,13,17,18-octaethylporphyrin (18)[25], and the silica-bound NaIO4[57] were prepared as described previously. In a variation of the literature procedures, we found it advantageous to chromatograph the osmate esters of 10 and 17 (silica - CHCl3 with a gradient of 1 to 3% MeOH), instead of the diol or tetraols, respectively. Once the respective esters were separated, they were treated with H2S to liberate the target compounds.
Mass Spectrometry
Mass spectral analysis were performed on a quadrupole time-of-flight (Q-TOF) mass spectrometer (QSTAR Elite) and a 4000 QTrap hybrid triple quadrupole linear ion trap mass spectrometer (both Applied Biosystems/MDS Sciex, Foster City, CA, USA), both equipped with an ESI sources. The data acquisition was under the control of the Analyst QS software (Foster City, CA). All samples were dissolved in spectral grade CH3CN to achieve a final concentration of 10–20 μM. For either instrument, samples were infused into the ESI source at a flow rate of 10 μL·min−1 (either using from a build-in or Harvard Apparatus syringe pump). Typical source conditions for the Q-Star were: Capillary voltage, 5500 V; declustering potential, 80 V; focusing potential, 280 V; declustering potential 2, 15 V; resolution 15,000 (full-width half-maximum). Ultra high pure N2 was used as the nebulizer, curtain and collision gas. For the CID experiments, the precursor ion was selected using the quadrupole analyzer and the product ions were analyzed using the TOF analyzer. Collision energies between 50–65 V were used. For QTrap mass spectral analysis, the turboionspray ion source conditions were optimized and set as: Curtain gas, 10; collision gas, high; ionspray voltage, 5500 V; ion source gas 1, 12; declustering potential, 30 V. Nebulizer and collision gas was N2. The most abundant product ions in the MS/MS (MS2) were also subjected to MS/MS/MS (MS3) experiments.
UV/Vis and Fluorescence Spectroscopy
Absorption spectra were recorded using a Varian Cary 50 UV/Vis spectrophotometer in toluene at ambient temperature. The fluorescence spectra were recorded in the same solvent on a Cary Eclipse instrument with λexcitation = λSoret.
Transient Triplet-Triplet Absorption Spectra and Triplet Lifetime Measurements
Samples were dissolved to OD = 1.5 at their respective λSoret in either toluene (Fisher HPLC grade) or 2-methyltetrahydrofuran (2-MTHF, Sigma-Aldrich 99+%) in a 1.00 cm cuvette fitted with a vacuum adapter. Every sample was degassed by means of at least 6 freeze-pump-thaw cycles. Samples were kept under vacuum during the measurements. The experiments were performed at room temperature with excitation from a Quanta-Ray Pro-230/MOPO-710 Nd:YAG laser system having a pulse repetition rate of 9.8 Hz and an output energy of ~9 mJ. The laser beam was focused onto the sample at a right angle to the measuring beam which consisted of an Oriel 150 W Xe arc lamp filtered by a 40% transmitting neutral density filter. The light transmitted through the sample was passed through an Instrument SA model LH290 1200 g/mm monochromator and focused onto a photodiode detector. The output was then amplified using a home-built amplifier and fed to a Tektronix digital oscilloscope model TDS 620A for signal averaging. Each transient profile consists of an average of 2000 scans (spectra are shown in Figure S16). The excitation wavelengths were as follow: 623 nm for 5, 605 nm for 12, 646 for 18, 645 nm for 10, 675 nm for 15, 500 nm for 17, 646 nm for 16a and 650 nm for 16b.
EPR Spectroscopy
Approximately 1 mg of each sample was dissolved in 1 mL of the solvents listed in Table 1 (toluene - Fisher HPLC grade; pyridine - J.T. Baker 99.9%). All samples were degassed before the experiments by subjecting them to at least 6 freeze-pump-thaw cycles. The EPR experiments were performed at ~20 K using a Bruker EMX X-band spectrometer equipped with ESR 900 continuous helium cryostat and Oxford ICT4 temperature controller. The samples were excited with white light from a 1000 W Xe arc lamp (Kratos LH151N/1S) filtered by a 5 cm layer of water and focused onto the cavity by a set of lenses. For all the spectra, the following instrument settings were used: microwave frequency, 9.4 GHz; modulation frequency, 100 kHz; center field, 3337 G; sweep width, 1000 G; time constant, 328 ms; conversion time, 328 ms; field resolution, 2048 points; number of scans, 8. The light minus dark difference spectra were obtained by subtraction, using Galactic DataMax 2.2 software.
3,7,8,12,13,17,18-Heptaethyl-2a-methyl-2-oxo-2a-homoporphyrin (12)
To a solution of 2,3,7,8,12,13,17,18-octaethyl-2,3-dihydroxychlorin 10 (52 mg, 9.0 × 10−5 mol) in CHCl3 (20 mL) and DBU (1 mL) was added silica-bound NaIO4 (250 mg). The reaction mixture was stirred for 12 h at ambient temperature and monitored by TLC and UV/Vis until most to all of the starting material was consumed (Rf of 10 (silica-CHCl3/3 % MeOH) = 0.31). If necessary, more oxidant was added. Upon consumption of the starting material, the mixture was filtered to remove the silica, washed repeatedly with water and dried over Na2SO4. The solvent was removed in vacuo and the residue was purified by preparative TLC (500 μm silica, CHCl3/1% MeOH) and recrystallized by slow solvent exchange of CH2Cl2 to EtOH to provide 12 as a purple solid (28 mg, 0.05 mmol, 55 %). The reaction is amenable to 10-fold scaling. Rf (silica-CHCl3/3 % MeOH) = 0.50; 1H NMR (500 MHz, [D1]CHCl3, 25 °C, TMS); δ = 11.03 (s, 1H, 20-CH), 9.78 (s, 1H, 5-CH), 9.62 (s, 1H, 15-CH), 9.56 (s, 1H, 10-CH), 4.13 (m, 4H, 131,181-CH2), 4.01 (m, 2H, 171-CH2), 3.90 (m, 2H, 71-CH2), 3.81 (m, 2H, 121-CH2), 3.75 (m, 2H, 81-CH2), 3.71 (m, 2H, 31-CH2), 2.82 (s, 3H, 2a1-CH3), 1.83 (m, 21H, 31,71,81,121,131,171,181-CH3), −3.63 (s, 1H, exchangeable with D2O, NH,), −3.84 ppm (s, 1H, exchangeable with D2O, NH,); 13C NMR (125 MHz, [D1]CHCl3, 25 °C, TMS): δ = 185.9 (2-C=O), 155.0 (11-C), 154.3 (6-C), 152.8 (3-C), 144.8 (12-C), 144.6 (13-C), 143.1 (18-C), 141.7 (14-C), 141.2 (7-C), 138.4 (16,17-C), 138.1, 137.9 (1-C, 4-C), 137.5 (C8), 137.1 (2a-C), 134.0 (19-C), 132.8 (9-C), 103.5 (20-CH), 103.2 (5-CH), 96.2 (10-CH), 95.9 (15-CH), 25.9 (31-CH2), 19.7, 19.6, 19.5, 19.4 (71,81,121,131,171,181-CH2), 18.5, 18.4, 18.2, 18.1 (71,81,121,131,171,181-CH3), 16.0 (31-CH3), 12.4 ppm (2a1-CH3); UV/Vis (CH2Cl2): λmax (log ε) = 339 (sh), 424 (5.14), 443 (4.82), 547 (3.80), 587 (4.36), 609 (4.21), 662 nm (3.05); UV/Vis [CHCl3-5% TFA, λmax (log ε)]: 434 (5.29), 546 (3.3), 587 (4.04), 638 (4.24); MS (ESI, cone voltage 30 eV, 100% CH3CN): m/z: 549.4 [M+H]+; HRMS (ESI+, 100% CH3CN): m/z calcd for C36H44N4O: 549.3593, found 549.3589; elemental analysis calcd (%) for C36H44N4O: C, 78.79; H, 8.08; N, 10.21; O, 2.92; found: C, 78.33; H, 8.05; N, 10.00; O, 3.20.
[3,7,8,12,13,17,18-Heptaethyl-2a-methyl-2-oxo-2a-homoporphyrinato]zinc(II) (12Zn)
Zn(II) acetate (40 mg) was added to a solution of 12 (50 mg, 9.0 × 10−5 mol) in CHCl3/10% MeOH (20 mL). The reaction mixture was heated to reflux until the starting material was exhausted, as monitored by TLC and UV/Vis (~30 min). The solvent was removed in vacuo, the residue dissolved in CHCl3, washed several times with water, and dried over Na2SO4. Crystallization by solvent exchange from CHCl3 to EtOH on the rotary evaporator, followed by filtration, provided 12Zn as a purple solid in 67% yield (35 mg, 6.0 × 10−5 mol). Rf (silica-CHCl3/5% MeOH) = 0.50; 1H NMR (400 MHz, 1:1 [D6]DMSO:[D4]MeOH, 25 °C, TMS): δ = 10.78 (s, 1H, meso-H), 10.09 (s, 1H, meso-H), 9.82 (s, 2H, meso-H), 3.99 (m, 14H, CH2CH3), 2.80 (s, 3H, CH3), 1.91, 1.81 ppm (m, 21H, CH2CH3); 13C NMR (100 MHz, [D1]CHCl3, 25 °C, TMS): δ = 183.9, 152.6, 149.8, 149.6, 148.8, 148.7, 146.9, 145.8, 144.4, 144.2, 143.4, 143.3, 142.7 142.2, 140.3, 136.7, 135.7, 103.0, 102.6, 97.8, 97.7, 31.2, 26.3, 19.7, 19.5, 19.4, 19.2, 19.1, 19.0, 16.8, 13.0 ppm; UV/Vis (CHCl3): λmax (log ε) = 434 (4.64), 453 (4.36), 578 (3.45), 627 nm (3.83); MS (ESI+, cone voltage 30 eV, 100% CH3CN): m/z 611.1 [M+H]+; HRMS (EI): m/z calcd for C36H42N4O: 549.3593, found 549.3589.
3,7,8,12,13,17,18-Heptaethyl-2a-hydro-3-hydroxy-2a-methyl-2-oxo-2a-homo-porphyrin (14)
Rf (silica-CHCl3) 0.21; 1H NMR (400 MHz, [D1]CHCl3, 25 °C, TMS): δ = 10.46 (s, 1H), 9.94 (s, 1H), 9.88 (s, 1H), 9.80 (s, 1H), 4.08 (m, 8H), 3.90 (m, 4H), 3.63 (m, 4H), 2.97 (m, 3H), 1.84 (m, 21H), −3.59, −3.79 ppm (s, 2H, NH): 13C NMR (100 MHz, [D1]CHCl3, 25 °C, TMS): δ = 202.1, 154.1, 153.1, 151.3, 144.5, 143.5, 139.81, 139.4, 138.3, 137.0, 135.7, 135.3, 134.3, 132.5, 98.8, 98.0, 97.9, 96.6, 79.7, 70.6, 51.6, 45.7, 42.2, 32.3, 19.7, 19.6, 19.6, 19.5, 19.1, 18.6, 18.5, 18.4, 18.3, 18.2, 18.1, 13.7, 12.3, 11.2, 8.5, 8.1 ppm; UV/Vis (CH2Cl2): λmax (rel. int.) = 410 (1.00), 493 (0.05), 512 (0.05), 555 (0.08), 589 (0.05), 645 nm (0.12); MS (ESI+, cone voltage 30 eV, 100% CH3CN): m/z: 567.6 [M+1]+; HRMS (EI): m/e calcd for C36H46N4O2: 567.3699, found 567.3679.
3,7,8,12,13,17,18-Heptaethyl-12,13-dihydroxy-2a-methyl-2-oxo-2a-homochlorin (15)
In a single neck round bottom flask, oxypyriporphyrin 12 (55 mg, 9.8 × 10−5 mol) is dissolved in a 2:3 mixture (15 mL) of CHCl3 (EtOH-stabilized) and pyridine (distilled from KMnO4). To this are added 25 mg OsO4 (1.25 mL of a stock solution of 0.1 g OsO4 in 5 mL pyridine). CAUTION: Fume hood and eye protection! The flask is stoppered and protected from light (aluminum foil) and stirred for 7 d at ambient temperature. The osmate ester of 15 forms as the only polar, green product. After this time, gaseous H2S is bubbled through the solution for 1 min. The mixture is stirred for 30 min and TLC control should confirm the conversion of the new compound into a slightly more polar product of the same color and UV/Vis spectrum. The mixture is then filtered through a pad of Celite (diatomaceous earth) and is evaporated to dryness on the rotary evaporator. Preparative plate or column chromatography (silica-CH2Cl2), followed by recrystallization by slow solvent exchange from CHCl3 to EtOH on the rotary evaporator yielded 15 as a green powder (40 mg, 68% yield). Alternatively, the compound can be isolated in varying yields as an intermediate product during the MnO4–-oxidation of tetraolbacteriochlorin 17, see below. Rf (silica-CHCl3/1 % MeOH) = 0.25; 1H NMR (400 MHz, [D1]CHCl3, 25 °C, TMS): δ = 9.22 (s, 1H, 5-H), 8.94 (s, 1H, 20-H), 8.71 (s, 1H, 10-H), 8.32 (s, 1H, 15-H), 5.52 (br s, 1H, exchangeable with D2O, OH), 5.31 (br s, 1H, exchangeable with D2O, OH), 3.84 (m, 4H, 71,81-CH2), 3.53 (m, 2H, 31-CH2), 3.28 (m, 1H, 171-CH2), 3.01 (m, 1H, 171-CH2), 2.73 (m, 2H, 181-CH2), 2.58 (s, 3H, 2a1-CH3), 2.47 (4H, m, 121,131-CH2), 1.78 (t, 3J = 8 Hz, 3H, 71-CH3), 1.76 (t, 3J = 8 Hz, 3H, 81-CH3), 1.62 (t, 3J = 7 Hz, 3H, 31-CH3), 1.37 (t, 3J = 7 Hz, 3H, 171-CH3), 1.13 (t, 3J = 7 Hz, 3H, 181-CH3), 1.07 (t, 3J = 7 Hz, 6H, 121,131-CH3) −2.55, −3.23 ppm (s, 2H, exchangeable with D2O, NH); 13C NMR (125 MHz, [D1]CHCl3, 25 °C, TMS): δ = 181.5 (2-C=O), 151.2 (3-C), 143.6, 142.4, 141.4, 139.1, 135.2, 135.0, 134.5 (1,4,6,7,8,9,11,14,16,17,18,19-C), 135.6 (2a-C), 105.5 (5,20-C), 92.2 (10-C), 92.0 (15-C), 85.1, 84.9 (12,13-C), 28.4, 28.2 (121,131-CH2), 25.6 (31-CH2), 19.7, 19.2, 18.0, 17.9, 17.6, 17.2, 16.0 (31,71,81,171,181-CH3, 71,81,171,181-CH2), 12.4 (2a1-CH3), 8.50, 8.46 ppm (121,131-CH3); UV/Vis (CHCl3): λmax (log ε) = 402 (3.86), 426 (4.57), 449 (4.03), 541 (4.12), 575 (sh), 587 (3.48), 613 (3.46), 668 nm (3.52); MS (ESI+, 30 V cone voltage, 100% CH3CN): m/z: 583.2 [M+H]+; HRMS (ESI+, 30 V cone voltage, 100% CH3CN): m/z calcd for C36H47N4O3: 583.3643, found 583.3644.
3,7,8,12,17,18-Hexaethyl-2a,13a-dimethyl-2,13-dioxo-2a,12a-homoporphyrin (16a) and 3,7,8,13,17,18-hexaethyl-2a,12a-dimethyl-2,12-dioxo-2a,12a-homo-porphyrin (16b)
To a solution of 2,3,7,8,12,13,17,18-octaethyl-2,3,12,13-tetrahydroxybacteriochlorin (17, isomerically pure or mixture of the two possible stereoisomers) (50 mg, 8.3 × 10−5 mol) in CHCl3 (50 mL) and DBU (1.0 mL) were added the silica-bound NaIO4 (250 mg). The reaction mixture was stirred for ~2 h until all the starting material was consumed (monitored by TLC – Rf of 17 (silica-CHCl3/5% MeOH) = 0.15 – and UV/Vis spectroscopy – reduction of the band at λ = 709 nm). The mixture was filtered to remove the silica gel and washed with water, 1% aq HCl, water, and dried over Na2SO4. The solvent was removed in vacuo. The residue was separated and purified by preparative TLC (500 μm silica, CH2Cl2). The two major dark-green bands were isolated and recrystallized by slow solvent exchange from CH2Cl2 to MeOH to provide the two isomeric bis(oxypyri)porphyrins 16a and 16b (each 11 mg, 2.1 × 10−5 mol, 25%) as purple solids. An additional product, appearing in varying yields, could be identified as 15. Alternatively, dihydroxy-oxypyriporphyrin 15 could be, using the identical methodology, oxidized to provide 16a and 16b in 35% yields each.
16a
Rf (silica-CHCl3/1 % MeOH) = 0.34; 1H NMR (500 MHz, [D1]CHCl3, 25 °C, TMS): δ = 10.44 (s, 2H, 15,20-H), 9.41 (s, 2H, 5,10-H), 3.99 (q, 3J = 8 Hz, 4H, 171,181-CH2), 3.73 (q, 3J = 8 Hz, 4H, 71,81-CH2), 3.68 (q, 3J = 8 Hz, 4H, 31,121-CH2), 2.72 (s, 6H, 2a1,12a1-CH3), 1.78 (t, 3J = 8 Hz, 6H, 171,181-CH3), 1.74 (t, 3J = 8 Hz, 6H, 31,121-CH3), 1.66 (t, 3J = 8 Hz, 6H, 71,81-CH3), −4.35 (s, 1H, NH), −4.40 ppm (s, 1H, NH); 13C NMR (125 MHz, [D1]CHCl3, 25 °C, TMS): δ = 185.6 (2,13-C=O), 152.5 (3,12-C), 142.7 (1,14-C), 142.3 (17,18-C), 140.3 (7,8-C), 139.5 (4,11-C), 137.4 (2a,12a-C), 136.4 (16,19-C), 134.6 (6,9-C), 103.2 (5,10-CH), 102.4 (15,20-CH), 25.7 (31,121-CH2), 19.6 (71,81,171,181-CH2) 18.0 (171,181-CH3), 17.8 (71,81-CH3), 15.6 (31,121-CH3), 12.2 ppm (2a1,12a1-CH3); UV/Vis (CHCl3): λmax (log ε) = 408 (sh), 435 (5.19), 465 (4.80), 578 (3.82), 624 (4.58), 652 (4.51), 708 nm (3.39); MS (ESI+, cone voltage 30 eV, 100% CH3CN): m/z: 563.3 [M+H]+, HRMS (ESI+) m/z calcd for C36H43N4O2: 563.3381, found 563.3366.
16b
Rf (silica-CHCl3/1% MeOH) = 0.43; 1H NMR (400 MHz, [D1]CHCl3, 25 °C, TMS): δ = 10.55 (s, 2H, 10,20-H), 9.72 (s, 2H, 5,15-H), 3.99 (q, 3J = 8 Hz, 4H, 81,181-CH2), 3.92 (q, 3J = 8 Hz, 4H, 71,171-CH2), 3.81 (q, 3J = 8 Hz, 4H, 31,131-CH2), 2.74 (s, 6H, 2a1,12a1-CH3), 1.83 (t, 3J = 8 Hz, 6H, 31,131-CH3), 1.79 (m, 12H, 71,81,171,181-CH3), −3.80 ppm (s, 2H, NH); 13C NMR (100 MHz, [D1]CHCl3, 25 °C, TMS): δ = 186.0 (2,12-C=O), 152.4 (3,13-C), 142.3 142.1, 140.7, 140.4, 135.8, 135.6 (1,4,6,9,11,14,16,19-C), 137.6 (2a-C, 12a-C), 103.2, 102.9 (5,10,15,20-CH), 25.7 (31,131-CH2), 19.7, 19.6 (71,81,171,181-CH2), 18.3, 17.8 (71,81,171,181-CH3), 15.7 (31,131-CH3), 12.2 ppm (2a1,12a1-CH3); UV/Vis (CHCl3): λmax (log ε) = 435 (5.05), 464 (4.83), 578 (3.71), 625 (4.40), 655 (4.32), 710 nm (3.48); MS (ESI+, cone voltage 30 eV, 100% CH3CN): m/z: 563.1 [M+H]+; HRMS (ESI+): m/z calcd for C36H43N4O2: 563.3381, found 563.3359.
X-ray Crystallography
Single crystals (black plates) of 12 were grown upon slow evaporation of MeOH/CH2Cl2 solutions to dryness.
Diffraction data of a 0.34 × 0.33 × 0.04 mm crystal were collected on a Bruker AXS SMART APEX CCD diffractometer with an Apex2 software upgrade at 100(2) K using monochromatic Mo Kα radiation (λ = 0.71073 Å) using the ω scan technique. The data were collected using SMART, and the data integration and unit cell determination was made using Apex2; the sadabs multi-scan absorption correction as embedded in Apex2 was applied.[58] The structure was solved by direct methods and refined by full matrix least squares against F2 against all reflections using SHELXTL.[58] A full-matrix refinement method on the least-squares on F2 was applied.[58] Non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed in calculated positions and were isotropically refined with a displacement parameter of 1.2 times that of the adjacent carbon atom. The refinement converged satisfactorily. Crystal structure and refinement data for 12 are summarized in Table 2.
Table 2.
Crystal data and refinement details for 12.
| 12 | |
|---|---|
| empirical formula | C36H44N4O |
| Mr | 548.75 |
| crystal size [mm] | 0.55 × 0.44 × 0.08 |
| crystal system | triclinic |
| space group | P-1 (No. 2) |
| a [Å] | 9.221(3) |
| b [Å] | 13.466(4) |
| c [Å] | 13.493(4) |
| α [°] | 71.062(4) |
| β [°] | 73.391(4) |
| γ [°] | 76.189(4) |
| V [Å3] | 1498.9(7) |
| Z | 2 |
| ρcalcd. [g cm−3] | 1.216 |
| F(000) | 592.5 |
| μ [mm−1] | 0.074 |
| Tmax/Tmin | 0.759/0.994 |
| hkl range | ±12, ±17, −14 – 17 |
| θ range [°] | 2.3355 – 29.232 |
| measured refl. | 12464 |
| unique refl. [Rint] | 7156 |
| observed refl. I>2σ(I) | 2989 |
| refined parameters | 655 |
| restraints | 292 |
| goodness-of-fit | 1.025 |
| R1 (I>2σ(I)) | 0.0827 |
| wR2 (all data) | 0.1876 |
| residual electron density [e Å−3] | −0.220/0.313 |
The molecule exhibits whole molecule disorder with an occupancy rate for the major moiety of 0.567(2). Equivalent disordered sections of the molecule were restrained to have similar geometries and a global restraint (DELU restraint as described in SHELXTL)[58] was applied to ensure that anisotropic displacement parameters of spatially nearby atoms are similar. For the atom pairs C36 and C31b, C31 and C36b, C24 and C24b, C25 and C25b, C35 and C30b, and C30 and C35b the anisotropic displacement parameters were constrained to be identical.[59] Atoms C24 and C25 were restrained to be close to isotropic.
Supplementary Material
Acknowledgments
We thank Professor David Dolphin, University of British Columbia, Vancouver, Canada, for a generous gift of octaethylporphyrin and Pedro Daddario for editorial assistance. This work was supported by the US National Science Foundation under Grant Numbers CHEM-0517782 and CCMI-0730826 (to CB). NLM acknowledges an NSF REU fellowship. This work is supported in the laboratory of HAF by the National Institute of Health (GM-30353) and the University of Connecticut Research Foundation. The diffractometer was funded by NSF grant 0087210, by Ohio Board of Regents grant CAP-491, and by YSU.
Footnotes
CCDC-718691 (12) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Scheer H. Chlorophylls. CRC; Boca Raton: 1991. [Google Scholar]
- 2.(a) Jentzen W, Ma JG, Shelnutt JA. Biophys J. 1998;74:753. doi: 10.1016/S0006-3495(98)74000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma JG, Laberge M, Song XZ, Jentzen W, Jia SL, Zhang J, Vanderkooi JM, Shelnutt JA. Biochemistry. 1998;37:118. doi: 10.1021/bi972375b. [DOI] [PubMed] [Google Scholar]; (c) Shelnutt JA, Song XZ, Ma JG, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31. [Google Scholar]
- 3.(a) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. 1–10. Academic Press; San Diego: 2000. [Google Scholar]; (b) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. 11–20. Academic Press; San Diego: 2003. [Google Scholar]
- 4.For recent examples, see: Muthiah C, Bhaumik J, Lindsey JS. J Org Chem. 2007;72:5839. doi: 10.1021/jo0707885.Ptaszek M, McDowell BE, Taniguchi M, Kim HJ, Lindsey JS. Tetrahedron. 2007;63:3826. doi: 10.1016/j.tet.2007.02.038.Taniguchi M, Ptaszek M, McDowell BE, Lindsey JS. Tetrahedron. 2007;63:3840. doi: 10.1016/j.tet.2007.02.076.Taniguchi M, Ptaszek M, McDowell BE, Boyle PD, Lindsey JS. Tetrahedron. 2007;63:3850. doi: 10.1016/j.tet.2007.02.040.O’Neal WG, Jacobi PA. J Am Chem Soc. 2008;130:1102. doi: 10.1021/ja0780075.Borbas KE, Ruzié C, Lindsey JS. Org Lett. 2008;10:1931. doi: 10.1021/ol800436u.Muthiah C, Ptaszek M, Nguyen TM, Flack KM, Lindsey JS. J Org Chem. 2007;72:7736. doi: 10.1021/jo701500d.Ruzié C, Krayer M, Balasubramanian T, Lindsey JS. J Org Chem. 2008;73:5806. doi: 10.1021/jo800736c. and references therein.
- 5.(a) Asat A, Dolphin D. Chem Rev. 1997;97:2267. doi: 10.1021/cr950078b. [DOI] [PubMed] [Google Scholar]; (b) Sessler JL, Weghorn S. Expanded, Contracted & Isomeric Porphyrins. Pergamon; New York: 1997. [Google Scholar]; (c) Sessler JL, Gebauer A, Weghorn SJ. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 2. 2000. p. 55. [Google Scholar]; (d) Maeda H, Furuta H. Pure Appl Chem. 2006;78:29. [Google Scholar]; (e) Srinivasan A, Furuta H. Acc Chem Res. 2005;38:10. doi: 10.1021/ar0302686. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lash TD. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 2. Academic Press; San Diego: 2000. p. 125. [Google Scholar]; (b) Lash TD. J Porphyrins Phthalocyanines. 2001;5:267. [Google Scholar]; (c) Lash TD. Eur J Org Chem. 2007:5461. [Google Scholar]
- 7.Lash TD. Synlett. 2000:279. [Google Scholar]
- 8.Berlin K, Breitmaier E. Angew Chem, Int Ed Engl. 1994;33:1246. [Google Scholar]
- 9.Mysliborsky R, Latos-Grazynski L, Szterenberg L. Eur J Org Chem. 2006:3046. [Google Scholar]
- 10.Berlin K, Breitmaier E. Angew Chem, Int Ed Engl. 1994;33:219. [Google Scholar]
- 11.Callot HJ, Schaeffer E. Tetrahedron. 1978;34:2295. [Google Scholar]
- 12.Lash TD, Pokharel K, Serling JM, Yant VR, Ferrence GM. Org Lett. 2007;9:2863. doi: 10.1021/ol071052x. [DOI] [PubMed] [Google Scholar]
- 13.Schönemeier T, Breitmaier E. Synthesis. 1997:273. [Google Scholar]
- 14.(a) Lash TD. Angew Chem; Int Ed Engl. 1995;34:2533. [Google Scholar]; (b) Lash TD, Chaney ST. Chem—Eur J. 1996;2:944. [Google Scholar]
- 15.Lash TD, Chaney ST, Richter DT. J Org Chem. 1998;63:9076. [Google Scholar]
- 16.Liu D, Ferrence GM, Lash TD. J Org Chem. 2004;69:6079. doi: 10.1021/jo040180l. [DOI] [PubMed] [Google Scholar]
- 17.Eguchi H, Ohgo Y, Ikezaki A, Neya S, Nakamura M. Chem Lett. 2008;37:768. [Google Scholar]
- 18.Neya S, Suzuki M, Ode H, Hoshino T, Furutani Yuji, Kandori H, Hori H, Imai K, Komatsu T. Inorg Chem. 2008;47:10771. doi: 10.1021/ic801406x. [DOI] [PubMed] [Google Scholar]
- 19.Callot HJ. Dalton Trans. 2008:6346. doi: 10.1039/b807066k. [DOI] [PubMed] [Google Scholar]
- 20.Crossley MJ, King LG. J Chem Soc, Chem Commun. 1984:920. [Google Scholar]
- 21.(a) Campbell CJ, Rusling JF, Brückner C. J Am Chem Soc. 2000;122:6679. [Google Scholar]; (b) Daniell HW, Brückner C. Angew Chem, Int Ed. 2004;43:1688. doi: 10.1002/anie.200352970. [DOI] [PubMed] [Google Scholar]; (c) McCarthy JR, Hyland MA, Brückner C. Org Biomol Chem. 2004;2:1484. doi: 10.1039/b401629g. [DOI] [PubMed] [Google Scholar]; (d) Lara KK, Rinaldo CK, Brückner C. Tetrahedron. 2005;61:2529. [Google Scholar]; (e) Perez MJ, McCarthy JR, Brückner C, Weissleder R. Nano Lett. 2005;5:2552. doi: 10.1021/nl0519229. [DOI] [PubMed] [Google Scholar]
- 22.(a) Crossley MJ, Burn PL, Langford SJ, Pyke SM, Stark AG. J Chem Soc, Chem Commun. 1991:1567. [Google Scholar]; (b) Crossley MJ, Burn PL, Chew SS, Cuttance FB, Newsom IA. J Chem Soc, Chem Commun. 1991:1564. [Google Scholar]; (c) Starnes SD, Rudkevich DM, Rebek J., Jr J Am Chem Soc. 2001;123:4659. doi: 10.1021/ja010038r. [DOI] [PubMed] [Google Scholar]; (d) Daniell HW, Williams SC, Jenkins HA, Brückner C. Tetrahedron Lett. 2003;44:4045. [Google Scholar]
- 23.Kozyrev AN, Alderfer JL, Dougherty TJ, Pandey RK. Angew Chem; Int Ed Engl. 1999;38:126. [Google Scholar]
- 24.One other example of a bis-pyrrole-modified porphyrinoid is a bis-carbaporphyrin: Graham SR, Colby DA, Lash TD. Angew Chem Int Ed Engl. 2002;41:1371. doi: 10.1002/1521-3773(20020415)41:8<1371::aid-anie1371>3.0.co;2-q.
- 25.Brückner C, Rettig SJ, Dolphin D. J Org Chem. 1998;63:2094. [Google Scholar]
- 26.(a) Bonnett R, Dimsdale MJ, Stephenson GF. J Chem Soc C. 1969:564. [Google Scholar]; (b) Chang CG, Sotiriou C. J Heterocyclic Chem. 1985;22:1739. [Google Scholar]; (c) Adams KR, the late Berenbaum MC, Bonnett R, Nizhnik AN, Salgado A, Asunción Vallés M. J Chem Soc, Perkin Trans. 1992;1:1465. [Google Scholar]
- 27.Adams KR, Bonnett R, Burke PJ, Salgado A, Vallés MA. J Chem Soc, Chem Commun. 1993:1860. [Google Scholar]
- 28.Adams KR, Bonnett R, Burke PJ, Salgado A, Vallés MA. J Chem Soc, Perkin. 1997;1:1769. [Google Scholar]
- 29.Lash TD, Chaney ST. Chem Eur J. 1996;2:944. [Google Scholar]
- 30.(a) Brückner C, Sternberg ED, MacAlpine JK, Rettig SJ, Dolphin D. J Am Chem Soc. 1999;121:2609. [Google Scholar]; (b) Brückner C, Hyland MA, Sternberg ED, MacAlpine J, Rettig SJ, Patrick BO, Dolphin D. Inorg Chim Acta. 2005;358:2943. [Google Scholar]
- 31.McCarthy JR, Melfi PJ, Capetta SH, Brückner C. Tetrahedron. 2003;59:9137. [Google Scholar]
- 32.McCarthy JR, Jenkins HA, Brückner C. Org Lett. 2003;5:19. doi: 10.1021/ol027072a. [DOI] [PubMed] [Google Scholar]
- 33.Ketone-functionalized zinc chlorins have been designed to form Lewis-acid (zinc center)-Lewis base (ketone functionality) coordination aggregates, thus this behavior does not surprise. See, e.g.: Balaban TS. Acc Chem Res. 2005;38:612. doi: 10.1021/ar040211z.
- 34.Note the difference of the conformation of 2 compared to the carbaporphyrin isomers of pyriporphyrins, see, e.g. ref. 12.
- 35.CCSD code OETPOR10. Lauher JW, Ibers JA. J Am Chem Soc. 1973;95:5148. doi: 10.1021/ja00797a009.
- 36.Kolb HC, Van Nieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]
- 37.Whitlock HW, Jr, Hanamer R, Oester MY, Bower BK. J Am Chem Soc. 1969;91:7485. [Google Scholar]
- 38.Chang CG, Sotiriou C, Weishih W. J Chem Soc, Chem Comm. 1986:1213. [Google Scholar]
- 39.a) Pandey RK, Shiau FY, Isaac M, Ramaprasad S, Dougherty TJ, Smith KM. Tetrahedron Lett. 1992;33:7815. [Google Scholar]; b) Smith KM, Goff DA. J Am Chem Soc. 1985;107:4954. [Google Scholar]; c) Brückner C, Dolphin D. Tetrahedron Lett. 1995;36:9425. [Google Scholar]
- 40.Meunier I, Pandey RK, Walker MM, Senge MO, Dougherty MTJ, Smith KM. Bioorg Med Chem Lett. 1992;2:1575. [Google Scholar]
- 41.Pandey RK, Isaac M, MacDonald I, Medforth CJ, Senge MO, Dougherty TJ, Smith KM. J Org Chem. 1997;62:1463. [Google Scholar]
- 42.Knyukshto V, Zenkevich E, Sagun E, Shulga A, Bachilo S. Chem Phys Lett. 1998;297:97. [Google Scholar]
- 43.Turro NJ. Modern Molecular Photochemistry. University Science Books; California: 1991. [Google Scholar]
- 44.Brückner C, McCarthy JR, Daniell HW, Pendon ZD, Ilagan RP, Francis TM, Ren L, Birge RR, Frank HA. Chem Phys. 2003;294:285. [Google Scholar]
- 45.Carrington A, McLachlan AD. Introduction to Magnetic Resonance. Harper and Row; New York: 1967. [Google Scholar]
- 46.Weil JA, Bolton JR, Wertz JE. Electron Paramagnetic Resonance. Wiley and Sons; New York: 1994. [Google Scholar]
- 47.Welter W. Magnetic Atoms and Molecules. Dover Publications; New York: 1989. [Google Scholar]
- 48.Angiolillo PJ, Lin VSY, Vanderkooi JM, Therien MJ. J Am Chem Soc. 1995;117:12514. [Google Scholar]
- 49.Hamacher V, Wrachtrap J, von Maltzan B, Plato M, Pöbius K. Appl Magnetic Resonance. 1993;4:297. [Google Scholar]
- 50.Kleibeuker JF, Schaafsma TJ. Chem Phys Lett. 1974;29:116. [Google Scholar]
- 51.Shediac R, Gray MHB, Uyeda HT, Johnson RC, Hupp JT, Angiolillo PJ, Therien MJ. J Am Chem Soc. 2000;122:7017. [Google Scholar]
- 52.Ponte Goncalves AM, Burgner RP. J Chem Phys. 1976;65:1221. [Google Scholar]
- 53.(a) McCarthy JR, Melfi PJ, Capetta SH, Brückner C. Tetrahedron. 2003;59:9137. [Google Scholar]; (b) Lau KSF, Sadilek M, Khalil GE, Gouterman M, Brückner C. J Am Soc Mass Spectrom. 2005;16:1915. doi: 10.1016/j.jasms.2005.08.001. [DOI] [PubMed] [Google Scholar]; (c) Lau KSF, Sadilek M, Khalil GE, Gouterman M, Brückner C. J Am Soc Mass Spectrom. 2006;17:1306. doi: 10.1016/j.jasms.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 54.(a) Berkel GJV, McLuckey SA, Glish GL. Anal Chem. 1991;63:1098. doi: 10.1021/ac00018a014. [DOI] [PubMed] [Google Scholar]; (b) Domingues MRM, Domingues P, Reis A, Ferrer-Correira AJ, Tomé JPC, Tomé AC, Neves MGPMS, Cavaleiro JAS. J Mass Spectrom. 2004;39:158. doi: 10.1002/jms.561. [DOI] [PubMed] [Google Scholar]; (c) Domingues MRM, Marques MGOS, Domingues P, Graça Neves M, Cavaleiro JAS, Ferrer-Correia AJ. Am Soc Mass Spectrom. 2001;12:381. doi: 10.1016/s1044-0305(01)00207-0. [DOI] [PubMed] [Google Scholar]; (d) Izquerido RA, Barros CM, Santana-Marques MG, Correia AJF, Silva AMG, Tome AC, Silva A, Neves MGPMS, Cavaleiro JAS. Rapid Commun Mass Spectrom. 2004;18:2601. doi: 10.1002/rcm.1663. [DOI] [PubMed] [Google Scholar]; (e) Quirke JME. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 7. Academic Press; San Diego: 2000. p. 371. [Google Scholar]; (f) Silva EMP, Domingues MRM, Barros C, Faustino MAF, Tomé JPC, Neves MGPMS, Tomé AC, Santana-Marques MG, Cavaleiro JAS, Ferrer-Correia AJ. J Mass Spectrom. 2005;40:117. doi: 10.1002/jms.789. [DOI] [PubMed] [Google Scholar]; (g) Van Berkel GJ, McLuckey SA, Glish GL. Anal Chem. 1991;63:1098. doi: 10.1021/ac00018a014. [DOI] [PubMed] [Google Scholar]
- 55.Callot HJ, Johnson AW. J Chem Soc; Chem Commun. 1969:749. [Google Scholar]
- 56.Sessler JL, Mozaffari A, Johnson MR. Org Syn. 1992;70:68. [Google Scholar]
- 57.Zhong YL, Shing TKM. J Org Chem. 1997;62:2622. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- 58.(a) Bruker Advanced X-ray Solutions SMART for WNT/2000 (Version 5.628) Bruker AXS Inc; Madison, WI, USA: 1997–2002. [Google Scholar]; (b) Bruker Advanced X-ray Solutions SAINT (Version 6.45) Bruker AXS Inc; Madison, WI, USA: 1997–2003. [Google Scholar]; (c) Bruker Advanced X-ray Solutions SHELXTL (Version 6.10) Bruker AXS Inc; Madison, WI, USA: 2000. [Google Scholar]
- 59.Please note that the numbering system used for 12 in the diffractometry study differs from that used in the NMR studies, see SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.