

SUMMARY
Focused ion beam milling at cryogenic temperatures (cryo-FIB) is a valuable tool that can be used to thin vitreous biological specimens for subsequent imaging and analysis in a cryo-transmission electron microscope (cryo-TEM) in their frozen-hydrated state. This technique offers the potential benefit of eliminating the mechanical artifacts that are typically found with cryo-ultramicrotomy. However, due to the additional complexity in transferring samples in and out of the FIB, contamination and devitrification of the amorphous ice is commonly encountered. In order to address these problems, we have designed a new sample cryo-shuttle that specifically accepts Polara TEM cartridges directly in order to simplify the transfer process between the FIB and TEM. We used the quality of the ice in the sample as an indicator to test various parameters used the process, and demonstrated with successful milling of large mammalian cells. By comparing the results from larger HeLa cells to those from E. coli cells, we discuss some of the artifacts and challenges we have encountered using this technique.
Keywords: Cryo-TEM, Cryo-FIB, HeLa Cell, E. coli
INTRODUCTION
Cryo-transmission electron microscopy (cryo-TEM) is a very valuable analytical technique used in structural biology in order to determine 3D structures of protein and protein complexes in frozen-hydrated state. Another important extension of cryo-TEM with respect to determination of sub-cellular structure is cryo-electron tomography, which can be used to turn the 2D projections of TEM images taken over a range of angles into a 3D reconstruction of that volume (Baumeister, 2005). Cryo-TEM and tomography have been shown to be a reliable tool for determining the structure of small biological specimens such as cellular organelles and bacteria frozen at cryogenic temperatures in order to preserve their native state (Grünewald et al., 2003; Kurner et al., 2005; Murphy et al., 2006).
In larger mammalian cells, such as HeLa cells, the volume of the cell is very large and not necessarily flat, which means that the sample becomes too thick to be electron transparent in the TEM. When the thickness of the sample exceeds the mean free path of electrons, multiple inelastic scattering events begin to degrade the image, meaning only the leading edge of the cell can be imaged (Medalia et al., 2002). For example in the study of the interactions of the human immunodeffiency virus (HIV) with mammalian HeLa cells, the virus particle can only be visualized by cryo-TEM near the edge of the cell. For the particles that have traveled much farther towards the nucleus, the sample is too thick to image these interactions using TEM (Jun et al., 2011).
Thinning biological specimens for use in the TEM has traditionally been performed in one of two ways. In the first method, the specimens are stained and fixed in a polymer resin and then sectioned using a diamond knife at room temperature (McEwen and Marko, 2001; McIntosh, 2001). This method was shown to alter the cells and fine structure from their native state due to the additional chemical fixation and dehydration (Baumeister, 2002). Using frozen specimens in the TEM has been shown to allow for imaging of biological samples in their close-to-native state (Baumeister, 2002), which forms the basis of the second method used for thinning of biological specimens. In this method, the samples are rapidly frozen under high pressure in liquid nitrogen to preserve their structure and then sectioned with a diamond knife under cryogenic temperature (Hsieh et al., 2002; Al-Amoudi et al., 2003, 2005; Zhang et al., 2004). The cryosectioning method is currently the only way of making sections of 50 nm of cells as well as tissues and many reports showed its strength at molecular resolution (Al-Amoudi et al., 2007; Pierson et al., 2010). However, cryosectioning is technically very demanding and challenging and the yield of high resolution tomograms is low. Although it is a fast method capable of producing many thin sections on an EM grid, the reproducibility is poor. The main problem with cryosectioning is that the sample does not lie flat on the grid after cutting, which results in poor attachments to the supporting film, making collection and alignment of the tilt series very difficult (Hsieh et al., 2002, 2006). This method also leads to many mechanical cutting artifacts, including knife marks, crevasses in thicker samples and compression in thinner samples (Zhang et al., 2004; Al-Amoudi et al., 2005). At the molecular level, the knife marks and crevasses might be avoided, however, the compression of the sample (30–50%) is unavoidable (Zhang et al., 2004; Al-Amoudi et al., 2005; Marko et al., 2006).
Recently, it has been shown that a focused ion beam (FIB) can be used to thin biological samples as well (Marko et al., 2007; Hayles et al., 2010; Rigort et al., 2010). A dual-beam FIB/SEM system uses a beam of focused Ga+ ions to mill away parts of the sample leaving few defects and is commonly used in material science applications to make TEM samples of hard materials (Nastasi et al., 1996). It has been demonstrated that using a FIB to mill away ice at cryogenic temperatures does not cause the sample to devitrify when using a 10 pA FIB beam, with accelerating current of 30 keV (Marko et al., 2006). In fact, bacterial cells were successfully milled in a FIB for use in cryo-TEM tomography (Marko et al., 2007; Rigort et al., 2010; Strunk et al., 2011). Also, Hayles and Marko have demonstrated cryo-FIB preparation of cell pellets for use in cryo-TEM (Marko et al., 2005; Hayles et al., 2010). Yet, thinning of larger mammalian cells has not been demonstrated using a cryo-FIB. We have found that this technique can be applied to larger cells and demonstrate successful results on HeLa cells using a modified sample shuttle to reduce frost contamination. We characterized milling conditions that result in samples of adequate quality for further cryo-TEM analysis. We also show how different types of ice can indicate where in the freezing, milling, transfer and cryo-TEM process problems may occur leading to poor quality samples. Finally, we examined the differences in milling large mammalian cells versus small bacterial cells and the unique difficulties presented when milling these large cells as opposed to smaller cells.
MATERIALS AND METHODS
Cell Preparation
Wild type K12 E. coli cells were grown in LB broth (10 % tryptone, 5 % yeast extract and 10 % NaCl). Starter cultures were grown overnight at 34°C with 280 rpm shaking to an approximate optical density of 2.0 at 600 nm. Overnight cultures were diluted 1/50 into the same media and allowed to grow to an optical density of 0.5–0.6 at 600 nm. E. coli cells (3–5ul) were withdrawn directly from cultures, mixed with 15nm gold beads (2 μl), and placed on R2/2 Quantifoil grids (Micro Tools GmbH, Germany). The grids were manually blotted from the back side of the grid with a filter paper and plunge-frozen in liquid ethane using a home-made manual gravity plunger. The frozen grids were loaded onto modified Polara cartridges (FEI, Hillsboro, OR) with cell side facing up and stored in liquid nitrogen for future use.
HeLa cells were cultured at 37 °C with 5 % CO2 in DMEM with 4.5 g/L L-glutamine and glucose (Lonza Group Ltd, Basel, Switzerland) containing 10% heat inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen Corporation, Carlsbad, CA). Cultures at ~ 80 % confluence were routinely split 1:5 in 60 mm culture dishes. Cells were centrifuged at 1000× g and plated onto the gold R2/2 Quantifoil finder EM grids (Quantifoil Micro Tools GmbH, Jena, Germany) at a density of 2 × 104 cells/ml (total 2 ml culture) in glass-bottom culture dishes (MatTek Corporation, Ashland, MA). The gold EM grids were disinfected under UV light for 2 hours and coated with 50 μg/ml fibronectin (Sigma) before use. 15 nm gold beads (4μl) were applied to the cell culture on EM grids, blotted with a filter paper and plunged into liquid ethane for rapid freezing using an FEI Vitrobot Mark III (FEI, Hillsboro, OR). The frozen grids were loaded onto modified Polara cartridges with cell side facing up and stored in liquid nitrogen for future use.
FIB Milling
For FIB milling of the cells, an FEI Quanta 200 3D DualBeam FIB/SEM (FEI Corp., OR.) equipped with a Quorum PolarPrep 2000T Cryo Transfer Station and a Quorum PP7465 dual slusher system (Quorum Technologies Ltd, East Sussex, UK) was used. It is important in these experiments to keep the samples under liquid nitrogen at all times. If the samples come to a temperature above approximately −135°–140°C, the ice begins to devitrify (Hsieh et al., 2002).
For use on the cold stage of the Quorum PolarPrep, a specialized shutter was designed by the University of Pittsburgh School of Medicine Machine Shop as seen in figure 1(a–c). There are many advantages to using this shuttle design. Since the Polara TEM requires the use of cartridges in the loading mechanism instead of just electron microscopy grids, we are able to design the shuttle to accept the cartridge too. This means that the grid is only handled during the initial loading of the grid into the cartridge and reduces the contamination and damage problems usually associated with loading and unloading an EM grid. In other studies, a loading device was used that covered the samples and protected them from frost and contamination, however the TEM grids still needed to be loaded into the shuttle and then into the TEM, which increases the exposure of the samples to contaminants (Marko et al., 2007; Hayles et al., 2010; Rigort et al., 2010; Plitzko et al., 2011)
Figure 1.

Specialized parts created for FIB milling. (a) Shuttle designed to accept Polara TEM cartridges. (b) Polara cartridge inserted into specialized shuttle. (c) Shutter used to reduce contamination and frost protecting the Polara cartridge. (d) Comparison of the regular Polara cartridge (left) and specialized cartridge with channels to allow for lower milling angle. (e) SEM image of a mill, showing the x and y-axis in the SEM (white) and TEM stage (black), the red dashed arrow indicates milling direction. (f) TEM image of a mill, also showing the x and y axis of the SEM (white) and TEM stage (black), the TEM stage is at a 90° rotation to the FIB axis. The red dashed arrow indicates milling direction. The scale bars are (e) and (f) are 5μm and 1μm respectively.
The design of the shuttle also keeps the grids in the fixed orientation from FIB to the TEM. The axis from the FIB to the TEM is rotated ~90° clockwise and is shown in figure 1(e and f). This will allow for easier acquisition of the tomography tilt series. Another important advantage of using this shuttle design is that it includes a shutter mechanism that protects the grid from contamination during transfer into and out of the FIB as seen in figure 1(b and c), which eliminates the major problem typically encountered in cryo-FIB milling.
Another very important factor in cryo-FIB milling is the milling angle. The smaller the angle between the sample and the FIB beam, the thinner the sample and the more useable area for tomography (discussed more in depth in the following sections). The Polara cartridge itself was also modified slightly with small channels cut into the sides to allow for shallower milling angles and can be seen in figure 1(d) compared to the standard Polara cartridge.
The frozen EM grids are loaded into the Polara cartridges and then loaded onto the shuttle while immersed in liquid nitrogen. The shuttle is loaded into the polar prep transfer station and kept under liquid nitrogen. It is important to use coated Styrofoam cups in the transfer station because the holes in regular Styrofoam allow air to enter the liquid nitrogen and condense, causing contamination in the liquid nitrogen, which can then be transferred to the sample. Then the transfer station is pumped down until the liquid nitrogen is slushed and the shuttle is retracted into the transfer arm and sealed under vacuum to protect the sample during transfer to the FIB prep chamber.
The transfer arm is attached to the prep gate valve of the FIB and pumped down before insertion into the prep chamber. The sample is not in contact with liquid nitrogen for approximately 30 seconds while the valve is pumped down. The vacuum seal that was formed in the transfer station helps protect the sample from devitrification during this time. Inside the prep chamber is a small cryo stage kept at approximately −180° C. After the shuttle is successfully transferred, the FIB prep chamber is pumped down to 10−6 mbar and then the gate valve between the prep chamber and main FIB chamber is opened and the shuttle is transferred to the cold stage within the main chamber of the FIB.
The dual beam contains both a FIB and SEM column that are focused at the same point on the sample but 52 apart as shown in figure 2(a). After the shuttle has been loaded onto the FIB cold stage, the shutter is opened by unscrewing the transfer rod and using the end of the rod to flip open the shutter. The shutter is kept closed until the sample is all the way into the FIB and then closed before it is removed from the cold stage in the same fashion. The sample is then tilted to approximately 10° with respect to the axis of the FIB column. It is important that this angle is as small as possible because the smaller this milling angle, the larger the available viewing area in the TEM, as shown in figure 2(b). Also, a smaller angle will produce a large thin sample area, which is ideal for tomography. However, the sample must still be viewable in the FIB, so that the milling area can be selected and to ensure that the beam is not blocked by the sample.
Figure 2.
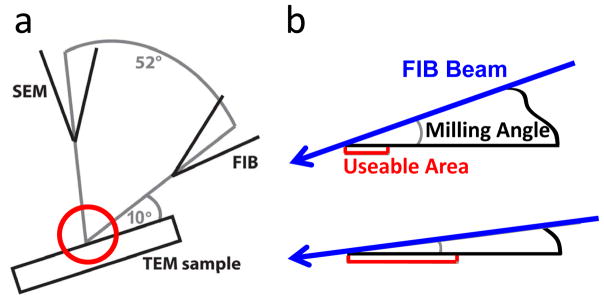
Schematic of FIB milling. (a) Setup of the dual beam system. (b) Magnification of the red circle indicated in figure (a). The blue arrows indicate the FIB beam and the red area indicates the useable area for TEM imaging. The smaller the milling angle, the larger the useable area, in our experiments, this angle was ~9–11°.
After the sample has been properly situated with respect to the FIB and the SEM, milling begins. A FIB milling current of 10–30 pA was used depending on the specimen thickness and an accelerating voltage of 30 kV. Thinner samples milled much faster (approximately 30 seconds to a minute) and therefore only needed a current of 10 pA. The larger specimens, like HeLa cells, required a higher current of 30 pA and a longer time of about 20 minutes per mill due to their increased thickness. The milled area was approximately 1 × 5 μm, which allows for a focusing and tracking region in the TEM. As stated above, the milling angle was approximately 9–11° to ensure a thin sample. The SEM images were obtained using an accelerating voltage of 10 kV and 0.45 nA current.
TEM characterization
After cryo-FIB processing, EM cartridges were stored in liquid nitrogen before they were examined by cryo-EM with an FEI Polara G3 (FEI Corp., OR.) transmission electron microscope equipped with a field emission gun and a Gatan 4K × 4K CCD camera (Gatan, Inc., Warrendale, PA). The FIB-milled regions were first located in low magnification (170 x) EM projection images. Low dose (20 e−/Å2) projection images of the identified regions of interest were recorded on a CCD camera at a nominal magnification of 50,000x and under-focus values ranging 2–4 μm.
RESULTS AND DISCUSSION
Characterizing Ice Quality
The quality of the ice is very important in the final condition of the samples. The ice needs to stay amorphous and vitreous throughout the milling process and TEM analysis in order for the samples to be viable for TEM and tomography. In cryogenic samples, crystalline ice can disrupt the fine structure of the cells. In our experiments, we observed three different types of crystallized “bad ice”. Each type can indicate where in the process the sample came above temperature. Electron diffraction patterns would further identify the types of ice observed more clearly, however that data was not collected initially. Alternatively, we analyzed these ice forms by their resemblance to previously characterized ones by electron diffraction (Dubochet et al., 1988; Marko et al., 2006). After fixing the problems at each step that led to these crystalline ice formations, we no longer observe these characteristic types of ice.
The first type of bad ice we observed was granular ice. It is characterized by small grains of ice as seen in figure 3(a). This happens when the sample comes close to the devitrification temperature but does not rise above it significantly or for a significant amount of time. This type of bad ice can be the most difficult to diagnose. Many times the sample was only in the devitrification zone for a few seconds before this ice formed. This happens either when the sample temperature was not monitored closely enough, allowing the temperature to rise towards the devitrification temperature for a short time, or due to slightly heating of the sample during milling.
Figure 3.

Characterizing ice quality. (a) Granular ice, characterized by small grains of ice and occurs when the sample comes close to the devitrification temperature but does not rise above it significantly or for a significant amount of time. This ice is similar to that observed by Marco at temperatures of −155° to 145°C (Marco et. al., 2006). (b) Devitrified ice, characterized by large ice crystals that run into each other and can be seen to disrupt the structure of the target cells and occurs when the sample has been above the devitrification temperature for a significant amount of time, usually a few minutes, mainly observed this ice when there was a problem with the loading process and is similar to that observed by Marco at temperatures of −135° to −115°C (Marco et. al., 2006). (c) Directional ice appears to have grown in a directional manner and is caused by incomplete initial freezing, as evidenced by the directional freezing patterns. This ice indicates that the samples were not frozen quickly enough to give the ice an amorphous structure during the plunge freezing process. (d) Good ice appears to be completely gray and smooth. There is no evidence of any type of structure or crystal and is the ideal ice for TEM characterization and tomography because there are no ice crystals disrupting the structure of the cells. The scales bars are all (a–d) 100 nm.
The second type of bad ice is completely devitrified ice. This occurs when the sample has been above the devitrification temperature for a significant amount of time, usually a few minutes. These samples are characterized by large ice crystals that run into each other and can be seen to disrupt the structure of the target cells as shown in figure 3(b) and correspond to a rise in temperature between −135° to −115°C and most likely indicates cubic ice (Marko et al., 2006). We mainly observed this ice when there was a problem with the loading process.
Another possible type of ice that is caused by problems in the initial sample freezing step is directional ice. The ice appears to have grown in a directional manner as shown in figure 3(c). This ice is caused by incomplete initial freezing, as evidenced by the directional freezing patterns. This ice indicates that the samples were not frozen quickly enough to give the ice an amorphous structure during the plunge freezing process. It should be mentioned that in plunge freezing where the sample in deposited onto the grid and then plunged into liquid ethane, the entire sample grid does not always completely vitrify. One advantage of using our wedge shaped milling method (figure 2) is that the area imaged in the mill is closest to the grid surface, and therefore the areas that are most likely to be vitreously frozen are imaged. In much larger cells, a high pressure freezing technique can be used to rapidly freeze the entire sample (Hayles et al., 2010; Marko et al., 2010).
Good amorphous ice after FIB milling is shown in figure 3(d). The ice appears to be completely gray and smooth. There is no evidence of any type of structure or crystalline diffraction pattern (data not shown). This is the ideal ice for TEM characterization and tomography because there are no ice crystals disrupting the structure of the cells. We found that in order to keep the ice vitrified, it was important to keep the FIB stage as cold as possible during the entire process. For our system, we were able to keep the FIB stage below −183°C. Keeping the samples as cold as possible (significantly below the devitrification temperature) gives us a temperature buffer in which the sample can be transported between different apparatus without too much of an increase in temperature.
Milling Artifacts
Even at the low milling currents and times used (10 pA and 30 seconds), we found that there were still some artifacts left from milling as seen in figure 4(b). The artifacts appear as small gray spots and larger gray “blobs” on the specimen, which appear to have a regularity and pattern. In figures 4(b and c) it can also be seen that the artifacts are only present in milled areas (figure 4(b)) and not present in unmilled sections (figure 4(c)). The artifacts are not present in all samples, and the presence of the artifacts does not seem to correlate with milling currents, or any other milling parameters. In general the quality of the milled regions of the E. coli cells was very good.
Figure 4.
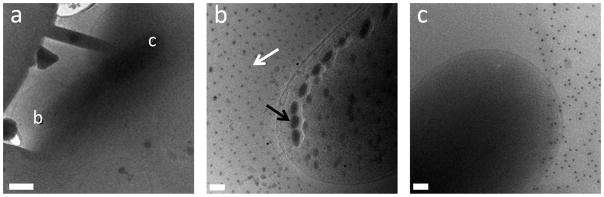
Surface contamination resulted from deposition of milled material. (a) Overview of milled area (b) E. coli cell located in the milled region. (c) E. coli cell located outside the milling area, no artifacts are observed. The solid black dots are gold fiducial markers. The white arrow indicates the artifacts that show regularity and the black arrow indicates the “blob-like” artifacts. The scale bars are (a) 1 μm, (b and c) 100 nm.
Milling E. coli cells
The E. coli cells were initially used to determine the feasibility of our technique and as a baseline to compare milling of smaller cells, which has been performed before (Marko et al., 2007; Rigort et al., 2010), to the milling of larger HeLa cells, which is the motivation for this research. In the E. coli samples, it was very easy to find and target specific sites for milling since the E. coli are readily apparent in the SEM and easily distinguished from the ice as seen in figure 5(a). This makes the milling of E. coli cells relatively accurate and quickly yields a large number of viable sites for later TEM study.
Figure 5.

Successful milling of E. coli cell. (a) SEM image taken from the FIB. The white arrow indicates the mill of interest. (b) Medium magnification TEM image of the E. coli cell. The white arrow shows the area of interest. (c) High magnification TEM image of the E. coli cell. The scale bars are (a) 5 μm, (b) 1 μm, and (c) 100 nm.
The E. coli samples are much thinner than those used for HeLa cells simply because the sizes of the E. coli cells are much smaller then HeLa cells. Since the samples are much thinner, we were able to mill them using a very low current ion beam of 10 pA. Typically, these samples were milled for only 30 seconds to 1 minute when using a beam current of 10 pA. We are able to make out the double walled structure of the E. coli cell in the TEM images as seen in figure 5(c). Despite the few artifacts that are present in some of the milled samples, this is a viable technique for thinning small bacterial cells for tomography.
Milling HeLa cells
After determining optimal conditions for sample transfer and milling using the E. coli cells, we applied these techniques to the HeLa cell samples. There is a much greater degree of difficulty in working with these larger cells making it hard to achieve a good yield of viable sites for TEM and tomography. Due to their large size, the HeLa cell samples require a much longer milling time compared to the E. coli samples tested previously. The amount of time required was often not feasible to do in a single FIB session when using the same beam current as applied to E. coli cells. In order to alleviate this problem, we tested different milling current applied to HeLa cells and corresponding time required for producing a thin cellular region. Although a higher current gives shorter time, the sample also endures higher risk of radiation damaged by ion beam interaction and heating which leads to devitrification. The optimal conditions were established with an increased ion current from 10 pA to 30 pA. The time was still much longer then required for the E. coli samples, but was reduced to only 20–30 minutes. This balanced milling current and milling time did not affect the quality of the ice or the samples.
Another challenge that arose in the milling of the HeLa cells was finding the appropriate location for milling in the SEM due to the lack of contrast with respect to the surrounding ice. The ice around the cells is so thick that it appears uniformly bright unless using a very high accelerating voltage in the SEM. This limits the ability to specifically target certain cells, or parts of cells, requiring milling of many adjacent sites in order to have any chance of finding the area of interest. In figure 6(a) it can be seen that there are quite a few mills in the vicinity of the area of interest (that contains the parts of the cell we are observing in the TEM micrograph). These surrounding mills were not found to have any HeLa cell present. This significantly lowers the success rate of the HeLa cell samples. Using a correlative imaging method such as the one established by us and others (Plitzko et al., 2009; Jun et al., 2011), we can further localize the target cells in SEM through light microscopy images of the same sample, thus greatly enhance the success rate.
Figure 6.

Successful milling of HeLa cell. (a) SEM image taken from the FIB. The white arrow indicates the mill of interest. (b) Medium magnification TEM image of the HeLa cell. The images for figures (c–e) are indicated in white. (c) High magnification image from the TEM, the white dashed line indicates the cell membrane of a HeLa cell. (d) The black arrows indicate vesicles. (f) The white arrows indicate filaments and microtubules. The scale bars are (a) 5 μm, (b) 2 μm, and (c–e) 100 nm.
Even though it takes more time and samples to successfully make a HeLa cell sample, it is possible to achieve a good sample for TEM. We are able to visualize in the milled samples: vesicles (figure 6(d), black arrows), actin filaments (figure 6(e), white arrows) and protein complexes (figure 6(c), black circles).
CONCLUSIONS
There are a few main differences in the milling procedures between small cell samples, such as E. coli and larger mammalian cells, such as HeLa cells. It is possible to mill both and create samples that are viable for cryo-TEM and tomography. However, the larger mammalian cells require more time and have a lower success rate. We were able to show that the extra time and milling currents do not affect our ability to image the cellular features of interest in the TEM. In comparison to established cryosectioning technique, we are able to create fast and reproducible mills for TEM cells using cryo-FIB, 30–60 seconds for E. coli cells, and 20–30 minutes for HeLa. We are also able to create multiple milling sites within each sample, typically 10 sites from each grid square in E. coli samples, which greatly raises the number of viable sites for tomography. Cryo-FIB is less technically demanding than cryosectioning, and does not require highly skilled experts to carry out the milling process.
More work needs to be done to increase the yield of the HeLa cell TEM samples and to reduce the time needed to achieve these results. However, the samples we have produced using these methods should be adequate for further cryo-TEM tomography analysis.
Acknowledgments
The authors thank Trevor Clark at the Materials Research Institute, Pennsylvania State University for help with cryo-FIB operation, Travis Wheeler and the machine shop at the Department of Cell Biology and Physiology for construction of the cryo-FIB shuttle, Drs. Gongpu Zhao, Sangmi Jun and Xin Meng for operation of the cryo-TEM. This work was supported by the National Institutes of Health (RR024424 and GM085043).
References
- Al-Amoudi A, Díez DC, Betts MJ, Frangakis AS. The molecular architecture of cadherins in native epidermal desmosomes. Nature. 2007;450:832–837. doi: 10.1038/nature05994. [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A, Dubochet J, Gnaegi H, Lüthi W, Studer D. An oscillating cryo-knife reduces cutting-induced deformation of vitreous ultrathin sections. J Microsc. 2003;212:26–33. doi: 10.1046/j.1365-2818.2003.01244.x. [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A, Studer D, Dubochet J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J Struct Biol. 2005;150:109–121. doi: 10.1016/j.jsb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Baumeister W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Curr Opin Struct Biol. 2002;12:679–684. doi: 10.1016/s0959-440x(02)00378-0. [DOI] [PubMed] [Google Scholar]
- Baumeister W. From proteomic inventory to architecture. FEBS Letters. 2005;579:933–937. doi: 10.1016/j.febslet.2004.10.102. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Grünewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC. Three-Dimensional Structure of Herpes Simplex Virus from Cryo-Electron Tomography. Science. 2003;302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- Hayles MF, de Winter DAM, Schneijdenberg CTWM, Meeldijk JD, Luecken U, Persoon H, de Water J, de Jong F, Humbel BM, Verkleij AJ. The making of frozen-hydrated, vitreous lamellas from cells for cryo-electron microscopy. J Struct Biol. 2010;172:180–190. doi: 10.1016/j.jsb.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Hsieh CE, Leith A, Mannella CA, Frank J, Marko M. Towards high-resolution three-dimensional imaging of native mammalian tissue: electron tomography of frozen-hydrated rat liver sections. J Struct Biol. 2006;153:1–13. doi: 10.1016/j.jsb.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Hsieh CE, Marko M, Frank J, Mannella CA. Electron tomographic analysis of frozen-hydrated tissue sections. J of Struct Biol. 2002;138:63–73. doi: 10.1016/s1047-8477(02)00034-5. [DOI] [PubMed] [Google Scholar]
- Jun S, Ke D, Debiec K, Zhao G, Meng X, Ambrose Z, Gibson GA, Watkins SC, Zhang P. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure. 2011;19:1573–1581. doi: 10.1016/j.str.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurner J, Frangakis AS, Baumeister W. Cryo-Electron Tomography Reveals the Cytoskeletal Structure of Spiroplasma melliferum. Science. 2005;307:436–438. doi: 10.1126/science.1104031. [DOI] [PubMed] [Google Scholar]
- Marko M, Hsieh C, Moberlychan W, Mannella CA, Frank J. Focused ion beam milling of vitreous water: prospects for an alternative to cryo-ultramicrotomy of frozen-hydrated biological samples. J Microsc. 2006;222:42–47. doi: 10.1111/j.1365-2818.2006.01567.x. [DOI] [PubMed] [Google Scholar]
- Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Meth. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]
- Marko M, Hsieh C, Vetter D, Salmon N, Mannella C. Cryo-FIB Preparation for Cryo-TEM Tomography. Microsc Microanal. 2010;16:178–179. [Google Scholar]
- Marko M, Hsieh C-E, MoberlyChan WJ, Mannella CA, Frank J. Feasibility of Focused Ion Beam Milling for Preparation of TEM Specimens of Biological Material Embedded in Vitreous Ice. Microsc Microanal. 2005;11 [Google Scholar]
- McEwen BF, Marko M. The Emergence of Electron Tomography as an Important Tool for Investigating Cellular Ultrastructure. J Histochem Cytochem. 2001;49:553–563. doi: 10.1177/002215540104900502. [DOI] [PubMed] [Google Scholar]
- McIntosh JR. Electron Microscopy of Cells. J Cell Biol. 2001;153:F25–F32. doi: 10.1083/jcb.153.6.f25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalia O, Weber I, Frangakis AS, Nicastro D, Gerisch G, Baumeister W. Macromolecular Architecture in Eukaryotic Cells Visualized by Cryoelectron Tomography. Science. 2002;298:1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- Murphy GE, Leadbetter JR, Jensen GJ. In situ structure of the complete Treponema primitia flagellar motor. Nature. 2006;442:1062–1064. doi: 10.1038/nature05015. [DOI] [PubMed] [Google Scholar]
- Nastasi M, Mayer J, Hirvonen JK. Ion-Solid Interactions: Fundamentals and Applications. Cambridge University Press; 1996. [Google Scholar]
- Pierson J, Fernández JJ, Bos E, Amini S, Gnaegi H, Vos M, Bel B, Adolfsen F, Carrascosa JL, Peters PJ. Improving the technique of vitreous cryo-sectioning for cryo-electron tomography: electrostatic charging for section attachment and implementation of an anti-contamination glove box. J Struct Biol. 2010;169:219–225. doi: 10.1016/j.jsb.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Plitzko J, Rigort A, Baeuerlein F, Laugks T, Villa E, Baumeister W. Hybrid Imaging-Novel Approaches and Recent Advances in Correlative Microscopy. Microsc Microanal. 2011;17:964–965. [Google Scholar]
- Plitzko JM, Rigort A, Leis A. Correlative cryo-light microscopy and cryo-electron tomography: from cellular territories to molecular landscapes. Curr Opin Biotechnol. 2009;20:83–89. doi: 10.1016/j.copbio.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Rigort A, Bäuerlein FJB, Leis A, Gruska M, Hoffmann C, Laugks T, Böhm U, Eibauer M, Gnaegi H, Baumeister W, et al. Micromachining tools and correlative approaches for cellular cryo-electron tomography. J Struct Biol. 2010;172:169–179. doi: 10.1016/j.jsb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Strunk K, Clark T, Gray J, Zhang P. 3D Imaging of Biological Cells Using a CryoFIB/SEM and a CryoTEM. Microsc Microanal. 2011;17:942–943. [Google Scholar]
- Zhang P, Bos E, Heymann J, Gnaegi H, Kessel M, Peters PJ, Subramaniam S. Direct visualization of receptor arrays in frozen-hydrated sections and plunge-frozen specimens of E. coli engineered to overproduce the chemotaxis receptor Tsr. J Microsc. 2004;216:76–83. doi: 10.1111/j.0022-2720.2004.01395.x. [DOI] [PubMed] [Google Scholar]