

Abstract
The endoplasmic reticulum transmembrane protein, Unc93b1, is essential for trafficking of endosomal TLRs from the endoplasmic reticulum to endosomes. A genetic defect in the human UNC93B1 gene is associated with immunodeficiency. However, systemic lupus erythematosus (SLE) patients express increased levels of the UNC93B1 protein in B cells. Because SLE in patients and certain mouse models exhibits a sex bias and increased serum levels of type I interferons in patients are associated with the disease activity, we investigated whether the female sex hormone estrogen (E2) or type I interferon signaling could up-regulate the expression of the murine Unc93b1 gene. We found that steady-state levels of Unc93b1 mRNA and protein were measurably higher in immune cells (CD3+, B220+, CD11b+ and CD11c+) isolated from C57BL/6 (B6) females than age-matched males. Moreover, treatment of CD11b+ and B220+ cells with E2 or interferons (IFN-α, IFN-β or IFN-γ) significantly increased the levels of Unc93b1 mRNA and protein. Accordingly, a deficiency of estrogen receptor-α or STAT1 expression in immune cells decreased the expression levels of the Unc93b1 protein. Interestingly, levels of Unc93b1 protein were appreciably higher in B6.Nba2 lupus-prone female mice compared with age-matched B6 females. Furthermore, increased expression of the interferon- and E2-inducible p202 protein in a murine macrophage cell line (RAW264.7) increased the levels of the Unc93b1 protein, whereas knockdown of p202 expression reduced the levels. To our knowledge, our observations demonstrate for the first time that activation of interferon and estrogen signaling in immune cells up-regulates the expression of murine Unc93b1.
Keywords: estrogen, IFN, innate immune response, p202, SLE, TLRs, Unc93b1
Introduction
Sensing pathogen-derived nucleic acids by endosomal TLRs in innate immune cells is critical for host defense against pathogens (1, 2). Upon endocytosis of pathogen-associated nucleic acids, TLR9 binds to CpG-rich unmethylated double-stranded DNA (dsDNA), whereas TLR7 binds to single-stranded RNA (ssRNA). Moreover, activation of TLR9 and TLR7 by nucleic acids induces innate immune responses that result in production of pro-inflammatory cytokines, including type I interferons (3, 4). Because mammalian nucleic acids also share certain features that are recognized by both TLR9 and TLR7, self-nucleic acids under some conditions enter the endosomes (1, 5), resulting in the activation of TLR7- and TLR9-induced innate immune responses (2–4). Aberrant activation of TLR9- and TLR7-mediated innate immune responses is associated with the development and progression of autoimmune diseases, including systemic lupus erythematosus (SLE) (3–5).
One of the ways the innate immune system prevents the recognition of self-nucleic acids is through tight regulation of intracellular trafficking of TLRs (6, 7). Upon sensing viral infection or activation of TLR signaling, the endoplasmic reticulum pool of the TLR7 and TLR9 is mobilized to traffic from the endoplasmic reticulum to endosomal vesicles where it allows recognition of virus-derived nucleic acids (7). At a steady state, Unc93b1, a multi-transmembrane protein in the endoplasmic reticulum, regulates trafficking of endosomal TLRs (such as TLR3, TLR7, TLR8 and TLR9) in humans and mice (8–11). Thus, the endosomal localization of both TLR7 and TLR9 prevents accessibility to self-derived nucleic acids. Additionally, activation of TLR7 and TLR9 by proteolytic cleavage by endosomal proteases in the acidified ‘signaling endosomes’ further prevents exposure to self-nucleic acids (12, 13). In the signaling endosomes, the activated TLR9 and TLR7 through recruitment of the adaptor protein MyD88 and IL-1 receptor-associated kinase-4 initiate signaling, which results in activation of NF-κB transcription factor and transcriptional activation of genes that encode pro-inflammatory cytokines, including type I interferons (2, 14, 15).
Unc93b1 protein is conserved between human and mouse (90% amino acid identity) (2, 13). The proteins are predicted to be multi-transmembrane proteins. Accordingly, the GFP fusion protein of human UNC93B localizes to the endosomal compartment (8–10). A point mutation (H412R) in the endoplasmic-reticulum localized murine Unc93b1 protein abolishes its interactions with TLR3, TLR7 and TLR9, resulting in the loss of signaling via these TLRs (8).
A study has noted that expression levels of UNC93B mRNA in PBMCs isolated from patients with active SLE were significantly higher than those of healthy controls (16). Moreover, the expression levels of UNC93B protein in B cells (CD20+) isolated from patients with active SLE were also higher than healthy controls. Of note, the higher expression levels of UNC93B protein in B cells from SLE patients correlated significantly with higher levels of anti-dsDNA antibody (16). Consistent with these observations, a study has noted that Unc93b1 function is required for optimal production of auto-antibodies in the B6-Faslpr and BXSB lupus-prone mice (17). These observations suggest that Unc93b1 protein function contributes to the development of autoimmunity, including SLE.
SLE is a complex polygenic autoimmune disease (18, 19). The disease in patients and mouse models is characterized by increased levels of pathogenic auto-antibodies against nuclear antigen and DNA, increased serum levels of IFN-α and increased expression of the interferon-inducible genes (the ‘interferon-signature’) (18). Additionally, SLE in patients and certain mouse models also exhibits a sex bias (develops at a female-to-male ratio of 9:1) (20, 21). Studies have indicated that epistatic interactions among the New Zealand Black (NZB)-derived autoimmunity 2 (Nba2) interval genes on chromosome 1 contribute to the modification of lupus susceptibility in the B6.Nba2 congenic (congenic for the Nba2 interval on C57BL/6 mice) mice (22–28). The interval harbors candidate lupus susceptibility genes that encode the Fcγ receptors, SLAM/CD2-family proteins and interferon-inducible p200-family proteins (24). Generation of sub-congenic lines, including the B6.Nba2-C line (which harbors only genes that encode for the p200-family proteins) (28) and mouse strains that are deficient in the expression of either Fcgr2b (encoding the inhibitory FcγRIIB receptor) (26) or Aim2 (encoding the Aim2 protein from the p200 family) (25) gene has revealed that epistatic interactions among the Nba2 interval genes contribute to the regulation of the interferon responses. These studies indicated that mouse sex and the sex hormone estrogen-dependent increase in the levels of the p202 protein (a lupus susceptibility modifier protein in the p200 family) in immune cells are associated with increased expression of type I interferons (and activation of an interferon response) in the B6.Nba2 (same as B6.Nba2-ABC) female mice compared with age-matched C57BL/6 and B6.Nba2-C female mice (21, 25, 28, 29).
Because patients with active SLE express increased levels of Unc93b1 (16), we explored whether the expression of Unc93b1 in murine immune cells is up-regulated by activation of type I interferon response or estrogen signaling. Here we report that steady-state levels of Unc93b1 in immune cells depend on the sex of mice and that activation of interferon or estrogen signaling in immune cells up-regulates the expression of Unc93b1. Moreover, we identify p202 protein from the p200 family of proteins as one of regulators of the Unc93b1 expression.
Methods
Mice
C57BL/6 (B6) male and age-matched female mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The Stat1-deficient and the corresponding wild-type male and age-matched female mice were purchased from the Taconic Farms (Germantown, NY, USA). The B6.Nba2 mice (originally purchased from The Jackson Laboratory) were bred at the Laboratory Animal Medical Services (LAMS) facilities at the University of Cincinnati. Mice were housed in specific pathogen-free LAMS facilities. The Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati approved the protocol for the studies described here.
Isolation of splenocytes, purification of cells, cell culture and treatments
Total splenocytes were isolated from mice as described previously (25). Freshly isolated cells were cultured in RPMI 1640 medium that was supplemented with 10% fetal bovine serum (Invitrogen, Grand Island, NY, USA) and antibiotics. When indicated, splenic CD3+ or B220+ cells and bone marrow-derived Cd11b+ or Cd11c+ cells were purified using cell purification kits (kits purchased from Miltenyi Biotech, Auburn, CA, USA). The purified (90–95% pure) cells were used immediately for experiments described. Normally, total splenic cells from two or more age- and gender-matched mice were pooled to purify the above indicated immune cells. Cells were either left untreated or treated with the universal IFN-α (1000U ml−1; from PBL Biomedical Laboratories, Piscataway, NJ, USA) or murine IFN-γ (10ng ml−1; from R&D Systems, Minneapolis, MN, USA) for the time period indicated. For the treatment of immune cells with sex hormone estrogen (E2) or dihydrotestosterone (DHT), cells were cultured in phenol red-free RPMI 1640 medium (Invitrogen), which was supplemented with 10% charcoal-stripped fetal bovine serum (Invitrogen).
Murine macrophage cell lines RAW264.7 and J774.A1 (purchased from the American Type Culture Collection) were maintained as suggested by the supplier. Generation of a stable RAW264.7 cell line that over-expresses the p202 protein and a control cell line (transfected with a vector alone) has been described (25). Similarly, the generation of a stable cell line that expresses reduced levels of p202 protein and a control cell line (infected with a control lentivirus) has been described (25). For experiments, cells of stable lines were incubated without using any antibiotics for 2 days. When indicated, cells were treated with either a control TLR9 ligand or a stimulatory murine-specific TLR9 ligand (ODN-1668; from InvivoGen, San Diego, CA, USA) at a final concentration of 5nM for 6h.
Nucleofections
Plasmids to express p202 [pCMV-202; (25)] have been described. RAW264.7 cells (2–4×106) from sub-confluent cultures were nucleofected with the purified (endotoxin free) plasmid DNA. As a control, we used pCMV plasmid. For nucleofections, we used the Nucleofector-II device (Amaxa Biosystems, Germany), the kit-V and the program D-032. After nucleofections, cells were incubated for ~24h before harvesting for total RNA or proteins. Nucleofections resulted in ~50–60% cell survival.
Preparation of RNA and RT–PCR
Total RNA was prepared from the indicated cells (splenocytes, purified immune cells or macrophage cell lines) using TRIzol (Invitrogen, Carlsbad, CA, USA) method. RNA (0.5–2 µg) was used to generate cDNA using the Superscript one-step RT–PCR system (from Invitrogen). Semi-quantitative PCR was performed using a pair of primers specific to the Unc93b1 (primers—forward: 5′-AGCTGCCCTTCAAACAC-3′; reverse: 5′-CCTGCCTCT CTTTGTCTTC-3′) gene. The conditions for the regular PCR were the same as described previously (25).
The quantitative real-time TaqMan PCRs were performed using the 7300 Real-Time PCR System (from Applied Biosystems, Foster City, CA, USA) as described previously (25). We used the TaqMan gene expression assays for the reactions. The TaqMan assays for the Unc93b1 (Assay Id# Mm00457643_m1), Tlr3 (Assay Id# Mm00628112_m1), Tlr9 (Assay Id# Mm00446193_m1), the endogenous Actb control (cat # 4352933E) and β2-microglobulin (Assay Id# Mm00437762_m1) were purchased from Applied Biosystems and used as suggested by the supplier.
Immunoblotting
Immunoblotting was performed using total cell lysates (containing approximately equal amounts of proteins) that were prepared from the indicated cells (splenocytes, purified immune cells or macrophage cell lines) as described previously (25). Antibodies to murine Unc93b1 protein were purchased from Abnova (cat# PAB13029; Walnut, CA, USA). Antibodies to detect murine TLR3 (cat # sc-28999) and TLR9 (cat # sc-47723) proteins were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies to actin (cat # 5125) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Reporter assays
Sub-confluent cultures of cells (in a six-well plate) were transfected with the reporter plasmids ISRE-luc (1.8 μg; the reporter plasmid purchased from Clontech, Palo Alto, CA, USA) and pRL-TK (0.2 μg) using the FuGENE 6 (Roche Applied Science, Indianapolis, IN, USA) transfection reagent as suggested by the supplier. Cells were harvested between 40 and 45h after transfections, and the firefly and Renilla dual luciferase activities were determined.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software 5 (GraphPad Software Inc., La Jolla, CA, USA). A Student’s t-test and one-way analysis of variance were used for comparison among all different groups. All values are expressed as mean ± SD. P < 0.05 was considered statistically significant.
Results
Steady-state levels of Unc93b1 protein depend on the sex of mice and treatment of immune cells with estrogen or type I interferon increases the levels
Expression of TLR7 and TLR9 has been reported in B cells, plasmacytoid dendritic cells (pDCs), DCs and macrophages (30). Therefore, to explore whether sex-dependent factors and/or type I interferons could up-regulate the expression of Unc93b1, we compared steady-state levels of Unc93b1 mRNA and protein in immune cells (CD3+, B220+, CD11b+ and CD11c+) isolated from C57BL/6 males and age-matched females. As shown in Fig. 1A, we were able to detect the expression of the Unc93b1 mRNA in all cell types analyzed. However, steady-state levels were appreciably higher in CD11b+ cells. Of interest, the levels of the Unc93b1 mRNA were higher in all cell types isolated from females than age-matched males and the levels were significantly higher in CD11b+ cells. Correspondingly, constitutive levels of Unc93b1 protein were appreciably (2.4- to 5.8-fold) higher in extracts from cells isolated from female mice than age-matched males (Fig. 1B). These observations encouraged us to test whether treatment of cells with the female sex hormone estrogen (E2), male hormone DHT or IFN-α could regulate the expression of Unc93b1. As shown in Fig. 1C, treatment of Cd11b+ cells from male or female mice with IFN-α, E2 or DHT differentially increased the levels of Unc93b1 protein. The increase by E2 treatment was evident in cells from both male and female mice. Similarly, treatment of cells with IFN-α increased the levels of Unc93b1 protein in cells from male and female mice. Together, these observations revealed that the constitutive and induced (induced by IFN-α or E2) expression levels of Unc93b1 depend on factors such as sex of the mice and activation of estrogen or type I interferon signaling.
Fig. 1.

Steady-state levels of Unc93b1 mRNA and protein depend on the sex of mice and treatment of immune cells with estrogen or type I interferon increases the levels. (A) Total RNA from the indicated purified immune cells isolated from either C57BL/6 male (n = 4) or age-matched female (n = 4) mice was subjected to quantitative real-time PCR using the TaqMan assays specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units (one unit being the ratio of the Unc93b1 mRNA to β2-microglobulin mRNA). The ratio of mRNA levels in CD3+ cells from male mice is indicated as 1. The error bars represent the standard deviation (*P < 0.05). (B) Total cell extracts from the indicated purified immune cells isolated from either C57BL/6 males (M; n = 4) or age-matched females (F; n = 4) were subjected to immunoblotting using antibodies specific to the indicated proteins. FC, fold change in the Unc93b1 protein levels is indicated. (C) Purified CD11b+ cells isolated from C57BL/6 male (n = 6) or age-matched female (n = 6) mice were either left untreated (control) or treated with IFN-α (1000U ml−1), 17β-estradiol (E2; 10nM) or DHT (10nM) as described in Methods for 18h. After the treatment, total cell extracts containing approximately equal amounts of proteins were subjected to immunoblotting using antibodies specific to the indicated proteins. FC, fold change in the Unc93b1 protein levels is indicated.
Estrogen signaling contributes to increases in Unc93b1 levels
Our preliminary experiments indicated that treatment of CD11b+ and B220+ cells with the female hormone estrogen (E2) at a 1 or 10nM concentration reproducibly increased the levels of Unc93b1 mRNA and protein (data not shown). Therefore, to begin to investigate the molecular mechanisms through which E2 up-regulates the expression of Unc93b1 in immune cells, we examined whether E2-mediated induction of Unc93b1 expression is estrogen receptor-α (ERα) dependent. As shown in Fig. 2A, treatment of CD11b+ cells (isolated from B6 females) with 10nM E2, but not DHT (10nM), significantly increased the levels of Unc93B1 mRNA. Furthermore, steady-state levels of Unc93b1 mRNA were relatively lower in splenic cells isolated from the ERα-deficient (NZB × NZW)F1 than age-matched wild-type females (Fig. 2B). However, the difference in the mRNA levels was not statistically significant. Next, we examined the time course of the induction of Unc93b1 by E2 treatment of cells. As shown in Fig. 2C, treatment of CD11b+ cells (isolated from B6 females) with E2 increased the levels of Unc93b1 mRNA ~2-fold after 6h of treatment. Further increase in the treatment length did not result in significant increases in levels of Unc93b1 mRNA. Accordingly, we noted that treatment of cells with E2 for 6 or 12h also increased the levels of the Unc93b1 protein and further treatment for 24h did not increase the levels further. Together, these observations revealed that activation of estrogen signaling in CD11b+ cells contributes to increases in Unc93b1 protein levels.
Fig. 2.

Estrogen signaling contributes to increases in Unc93b1 mRNA and protein levels. (A) Purified CD11b+ cells isolated from C57BL/6 female mice (n = 4) were either left untreated or treated with 17β-estradiol (E2; 10nM) or DHT (10nM) as described in Methods for 18h. Total RNA was subjected to quantitative real-time PCR using the TaqMan assay specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units (one unit being the ratio of the Unc93b1 mRNA to β2-microglobulin mRNA). The ratio of mRNA levels in untreated cells is indicated as 1. The error bars represent the standard deviation (**P < 0.005). (B) Total RNA that was isolated from splenic cells derived from the wild-type (NZB × NZW)F1 or age-matched ERα-deficient (NZB × NZW)F1 females was analyzed by quantitative real-time PCR using the TaqMan assay specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units (one unit being the ratio of the Unc93b1 mRNA to β2-microglobulin mRNA). The ratio of mRNA levels in one of the wild-type females is indicated as 1. The error bars represent the standard deviation. NS, not significant. (C) Purified CD11b+ cells from C57BL/6 female mice (n = 4) were either left untreated or treated with 17β-estradiol (E2; 10nM) for the indicated time (h). Total RNA was subjected to quantitative real-time PCR using the TaqMan assay specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units (one unit being the ratio of the Unc93b1 mRNA to β2-microglobulin mRNA). The ratio of mRNA levels in untreated cells is indicated as 1. The error bars represent the standard deviation (*P < 0.05; **P < 0.005). (D) Purified CD11b+ cells from C57BL/6 female mice (n = 4) were either left untreated or treated with 17β-estradiol (E2; 10nM) for the indicated time (h). After the treatment, total cell extracts containing approximately equal amounts of proteins were subjected to immunoblotting using antibodies specific to the indicated proteins. FC, fold change in the Unc93b1 protein levels is indicated.
Activation of interferon signaling up-regulates Unc93b1 expression
Our above observations (Fig. 1C) that treatment of Cd11b+ cells with type I interferon increased the levels of Unc93b1 protein levels prompted us to identify the potential molecular mechanisms. Therefore, we treated B220+ cells with type I interferons (IFN-α or IFN-β) or type II interferon (IFN-γ) for 6h and analyzed the steady-state levels of Unc93b1 mRNA by semi-quantitative (Fig. 3A) and quantitative real-time (Fig. 3B) PCR. We found that the treatment increased the levels of the Unc93b1 mRNA ~2- to 2.5-fold. Correspondingly, we also noted increases in levels of Unc93b1 protein after treatment of cells with type I or II interferons (Fig. 3C). Because activation of type I and II interferons signaling requires the expression of STAT1 (30, 31), an interferon-activatable transcription factor, we also compared steady-state levels of Unc93b1 mRNA and protein between STAT1-deficient and age-matched wild-type mice. As shown in Fig. 3D, levels of the Unc93b1 protein were measurably lower in STAT1-deficient male and female mice compared with gender-matched wild-type mice. Together, these observations indicated that basal levels of Unc93b1 protein in immune cells are increased by interferon treatment of cells and a deficiency of STAT1 transcription factor in immune cells decreases the basal levels of the Unc93b1 protein.
Fig. 3.

Activation of interferon signaling up-regulates Unc93b1 expression. (A) Purified B220+ cells isolated from C57BL/6 female mice (n = 4) were either left untreated or treated with IFN-α (1000U ml−1), IFN-β (1000U ml−1) or IFN-γ (10ng ml−1) for 6h. Total RNA was subjected to semi-quantitative PCR using a pair of primers that were specific to the murine Unc93b1 gene. (B) RNA samples that were prepared in panel (A) were also subjected to quantitative real-time PCR using the TaqMan assay specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units (one unit being the ratio of the Unc93b1 mRNA to β2-microglobulin mRNA). The ratio of mRNA levels in untreated cells is indicated as 1. The error bars represent the standard deviation (*P < 0.05; **P < 0.005). (C) Purified B220+ cells isolated from C57BL/6 female mice (n = 4) were either left untreated or treated with IFN-α (1000U ml−1), IFN-β (1000U ml−1) or IFN-γ (10ng ml−1) for 6h. Total cell extracts containing approximately equal amounts of proteins were subjected to immunoblotting using antibodies specific to the indicated proteins. FC, fold change in levels of Unc93b1 protein is indicated. (D) Total cell extracts from splenic cells derived from the wild-type or STAT1-deficient male or age-matched female mice (n = 2) were analyzed by immunoblotting using antibodies specific to the indicated proteins. FC, fold change in levels of Unc93b1 protein is indicated. (E) Total RNA from splenic cells derived from the wild-type or STAT1-deficient male or age-matched female mice (n = 2) was analyzed by quantitative real-time PCR using the TaqMan assay specific to the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the wild-type males is indicated as 1. The error bars represent the standard deviation (NS, not significant; *P < 0.05).
Lupus-prone B6.Nba2 female mice express increased levels of Unc93b1
Studies have indicated that the development of lupus-like disease in the B6.Nba2 [same as B6.Nba2-ABC strain; ref. (28)] congenic mice depends on sex (22, 32) and type I IFN signaling (33). Accordingly, studies have revealed that immune cells from the B6.Nba2-ABC female mice express increased levels of type I interferon (IFN-β) (and exhibit an activation of the interferon response) compared with the age-matched C57BL/6 and B6.Nba2-C female mice (25, 28, 29). Therefore, our above observations that activation of estrogen or type I interferon signaling in immune cells up-regulated the expression of Unc93b1 encouraged us to compare Unc93b1 levels between the lupus-prone B6.Nba2-ABC and age-matched non-lupus-prone B6 and B6.Nba2-C females (28). As shown in Fig. 4A, steady-state levels of Unc93b1 mRNA were significantly higher in the B6.Nba2-ABC females than in the non-lupus-prone B6 females. The levels of mRNA in B6.Nba2-C sub-congenic females, which express reduced levels of type I interferons (25), were also lower than the B6.Nba2-ABC mice. Although the difference in the Unc93b1 mRNA levels between the two strains was not statistically significant, we observed appreciably higher levels of the Unc93b1 protein in extracts from B6.Nba2-ABC females than from age-matched B6 or B6.Nba2-C females (Fig. 4B, compare lane 3 with lane 1 or 2). Together, these observations revealed that the lupus-prone B6.Nba2-ABC female mice express increased levels of Unc93b1 protein compared with age- and gender-matched non-lupus-prone B6 and B6.Nba2-C mice.
Fig. 4.

Lupus-prone B6.Nba2 female mice express increased levels of Unc93b1. (A) Total RNA isolated from splenic cells derived from age-matched C57BL/6J (B6), B6.Nba2-C (C) or B6.Nba2-ABC (ABC) female mice (n = 3 for each strain) was subjected to quantitative real-time PCR using the TaqMan assay specific to the Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the B6 females is indicated as 1. The error bars represent the standard deviation (**P < 0.005; ***P < 0.001; NS, not significant). (B) Total cell extracts from splenic cells isolated from age-matched C57BL/6J (B6), B6.Nba2-C (C) or B6.Nba2-ABC (ABC) female mice (n = 3) were subjected to immunoblotting using antibodies specific to the indicated proteins. FC, fold change in levels of Unc93b1 protein is indicated.
p202 lupus susceptibility modifier protein up-regulates Unc93b1 expression
Increased levels of p202, an estrogen and interferon-inducible protein (21), in B6.Nba2 mice are associated with the development of lupus-like disease (21, 22, 24). Therefore, the above observations that activation of estrogen or interferon signaling increased the levels of Unc93b1 protein in immune cells and lupus-prone B6.Nba2 females expressed increased levels of Unc93b1 protein (compared with age-matched C57BL/6 and B6.Nba2-C females) promoted us to explore whether the p202 protein could up-regulate the expression of Unc93b1 protein. Therefore, we knocked down p202 expression in J774.A1 cells (Fig. 5A and B). Interestingly, the knockdown reduced basal levels of Unc93b1 mRNA and protein by 50–60%. Moreover, the knockdown of p202 expression in cells (which reduced basal levels of the STAT1 transcription factor and increased the basal levels of the Aim2 protein) also decreased the levels of TLR3 and TLR9 (Fig. 5C). Furthermore, nucleofection of RAW264.7 cells with the pCMV-202 expression vector, but not control vector pCMV, increased the levels of Unc93b1 mRNA (Fig. 5C) and protein (Fig. 5D). Moreover, consistent with our above observations that knockdown of p202 expression in J774.A1 cells reduced steady-state levels of TLR3 and TLR9 proteins (Fig. 5C), over-expression of the p202 protein in RAW264.7 cells compared with pCMV transfected cells significantly increased the steady-state levels of mRNAs encoding the TLR3 and TLR9 (Fig. 5F). Together, these observations suggested that increased levels of the p202 protein in immune cells contribute to increased expression levels of the Unc93b1 protein and certain TLRs (such as TLR3 and TLR9).
Fig. 5.

p202 protein up-regulates Unc93b1 expression. (A) Total RNA isolated from J774.A1 cells that were stably infected with either control lentivirus (control) or the virus encoding shIfi202 was subjected to quantitative real-time PCR using TaqMan assay for the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the control cells is indicated as 1. The error bars represent the standard deviation (*P < 0.05). (B) Total cell extracts from cells described in panel (A) were subjected to immunoblotting using the antibodies indicated. (C) Total cell extracts from cells described in panel (B) were subjected to immunoblotting using antibodies specific to the indicated proteins. (D) Total RNA isolated from RAW264.7 cells that were nucleofected with either an empty pCMV vector or the pCMV-202 plasmid (encoding the p202 protein) was subjected to qPCR using TaqMan assay for the Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the pCMV nucleofected cells is indicated as 1. The error bars represent the standard deviation (**P < 0.005). (E) Total cell extracts from cells described in panel (C) were subjected to immunoblotting using the antibodies indicated. (F) Total RNA as indicated in panel (D) was subjected to qPCR using TaqMan assay for the Tlr3 and Tlr9 genes. The ratio of the TLR3 and TLR9 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the pCMV nucleofected cells is indicated as 1. The error bars represent the standard deviation (**P < 0.005; ***P < 0.001).
Estrogen or interferon-mediated up-regulation of Unc93b1 expression depends on the expression of p202 protein
Our above observations that knockdown of p202 expression in J774.A1 cells reduced levels of Unc93b1 mRNA and protein, whereas over-expression of the p202 protein in RAW264.7 cells increased the levels of Unc93b1 mRNA and protein prompted us to investigate whether estrogen or type I interferon-mediated up-regulation of Unc93b1 expression depends on p202 expression. As shown in Fig. 6A, treatment of control J774.A1 cells with either IFN-α or E2 significantly increased the levels of Unc93b1 mRNA. However, treatment of cells that expressed reduced levels of p202 protein with IFN-α or E2 did not significantly increase the levels of Unc93b1 mRNA. Accordingly, the levels of Unc93b1 protein were ~5-fold higher in control cells after treatment with either IFN-α or estrogen. In contrast, the treatment did not appreciably increase the levels of Unc93b1 protein in cells that expressed reduced levels of the p202 protein. These observations indicated that estrogen and interferon-mediated up-regulation of Unc93b1 expression in J774.A1 cells depends on the expression of p202 protein.
Fig. 6.

Estrogen or IFN-α-mediated up-regulation of Unc93b1 expression depends on p202 protein expression. (A) Murine J774.A1 cells that were stably infected with either control lentivirus or the virus encoding shIfi202 were either left untreated or treated with IFN-α (IFN, 1000U ml−1) or estrogen (E2, 10nM) for 14h. After the treatments, total RNA was isolated and was subjected to quantitative real-time PCR using TaqMan assay for the murine Unc93b1 gene. The ratio of the Unc93b1 mRNA levels to β2-microglobulin mRNA was calculated in units. The ratio of mRNA levels in the control cells is indicated as 1. The error bars represent the standard deviation (**P < 0.01; ***P < 0.001; NS, not significant). (B) Murine J774.A1 cells that were stably infected with either control lentivirus or the virus encoding shIfi202 were either left untreated or treated with IFN-α (IFN, 1000U ml−1) or estrogen (E2, 10nM) for 14h. After the treatments, total cell lysates were analyzed by immunoblotting for the indicated proteins. FC, fold change in the Unc93b1 protein levels is indicated.
Reduced levels of p202 protein decrease TLR9-mediated stimulation of interferon signaling
Knockdown of p202 expression in J774.A1 cells reduced the levels of Unc93b1 and TLR9 proteins (Fig. 5C). Therefore, we investigate whether the knockdown of p202 expression in cells also decreases the Unc93b1–TLR9 axis-mediated stimulation of interferon signaling. As shown in Fig. 7, knockdown of p202 expression in J774.A1 cells significantly (40%) reduced the basal activity of an interferon-responsive reporter (the ISRE-luc). Interestingly, treatment of control cells with a stimulatory TLR9 ligand stimulated the activity of the reporter compared with cells that were treated with a control ligand. However, knockdown of p202 expression in cells significantly (~50%) decreased the extent of stimulation. These observations suggested that the expression of p202 protein in J774.A1 cells contributes to increased expression of Unc93b1 and TLR9. Additionally, the p202 protein increases the Unc93b1–TLR9 axis-mediated stimulation of interferon signaling.
Fig. 7.

Reduced levels of p202 protein decrease TLR9-mediated stimulation of interferon signaling. J774.A1 cells that were stably infected with either control lentivirus or the virus encoding shIfi202 were transfected with the reporter plasmid ISRE-luc (1.8 μg) along with a second reporter pRL-TK (0.2 μg) using the FuGene 6 transfection agent. Twenty-four hours after transfections, cells were treated with either a control TLR9 ligand or a stimulatory mouse-specific TLR9 ligand (ODN-1668; 5nM) for 6h. At the end of the treatment, cells were lysed and the cell lysates were analyzed for dual luciferase activity. The ratio between the firefly luciferase and the Renilla luciferase in control cells is indicated as 1. The error bars represent the standard deviation. *P < 0.05; **P < 0.005.
Discussion
Increased levels of IFN-α in most SLE patients and certain mouse models, which exhibit a sex bias in lupus, are part of a ‘feed-forward loop’ (30, 34). The loop is activated by innate immune responses that are initiated by nucleic acids sensing TLRs (for example, TLR7 and TLR9) upon detection of immune complexes that contain auto-antibodies in complexes with nuclear antigens and DNA (3, 4). Given that increased production of IFN-α is associated with increased risk to develop SLE and increased serum levels of IFN-α are associated with the disease activity (4, 30, 34), an improved understanding of the molecular mechanisms that contribute to the ‘feed-forward loop’ of IFN-α production is important. Therefore, our observations that (i) steady-state levels of Unc93b1 protein depend on the sex of mice and treatment of immune cells with estrogen or type I interferons increases the levels (Fig. 1); (ii) activation of estrogen signaling contributes to increases in Unc93b1 levels (Fig. 2); (iii) activation of interferon signaling up-regulates Unc93b1 expression (Fig. 3); (iv) lupus-prone B6.Nba2-ABC female mice express increased levels of Unc93b1 compared with non-lupus-prone B6 and B6.Nba2-C females (Fig. 4) and (v) the interferon and estrogen-inducible p202 protein up-regulates the expression of Unc93b1 and certain TLRs (Fig. 5) are significant. Moreover, to the best of our knowledge, our observations for the first time provide evidence for a sex-dependent regulation of the Unc93b1 protein levels. Given that nucleic acids sensing TLR7 and TLR9 play a critical role in the development and progression of SLE (5–7), our observations provide a molecular basis for sex bias in murine SLE (Fig. 8).
Fig. 8.
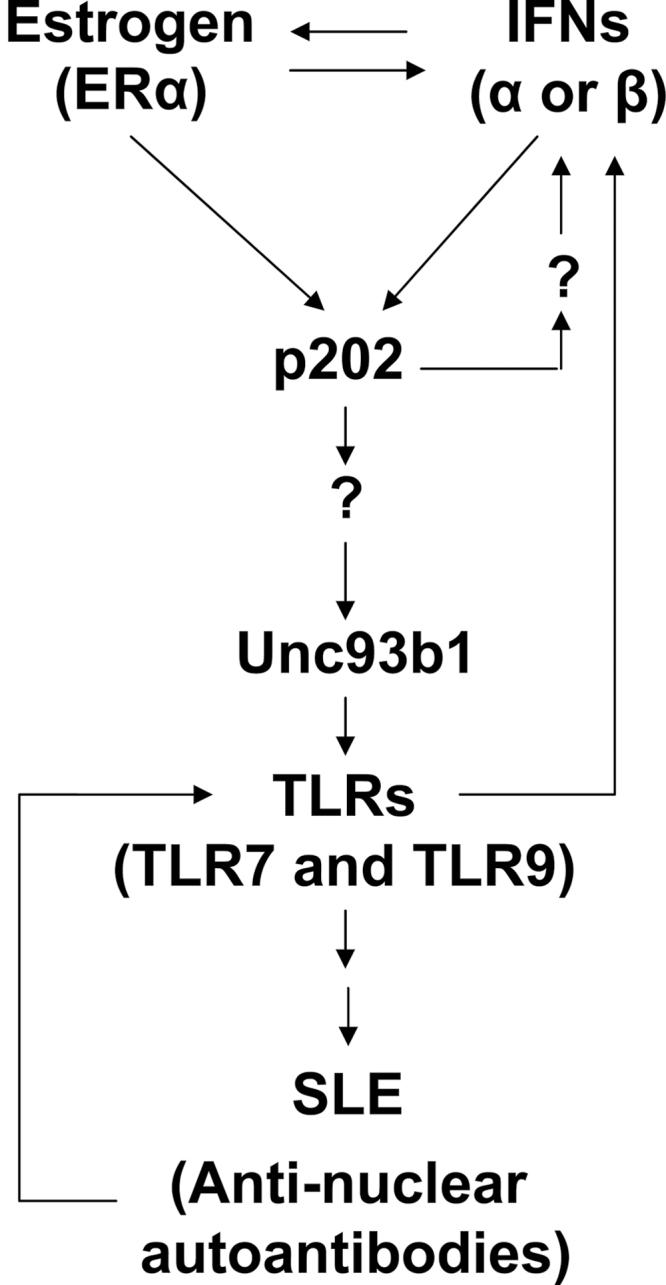
Proposed model for the up-regulation of murine Unc93b1 expression by activation of interferon and estrogen signaling in immune cells.
Expression of nucleic acids sensing TLRs has been reported in a variety of cell types (30). These cell types include myeloid DCs (mDCs), monocytes, macrophages, B cells and pDCs (30). Given that nucleic acids sensing TLRs such as TLR3, TLR7, TLR8 and TLR9 bind to Unc93b1 transmembrane protein and Unc93b1 transports these TLRs to signaling endosomes, our observations that expression of Unc93b1 protein is detectable in purified CD3+ cells (Fig. 1) raise the possibility that these cells also express certain nucleic acids sensing TLRs. Therefore, further work is in progress to test this possibility.
Increased levels of p202 protein in lupus-prone strains of female mice are associated with the development of antinuclear auto-antibodies, reduced expression of the inhibitory receptor FcγRIIB and reduced levels of the Aim2 protein (22–28). Given that the p202 protein can activate an interferon response in immune cells (27), our above observations raised the possibility that estrogen-dependent increased levels of p202 protein in immune cells in female mice contribute to increased expression of the Unc93b1 gene. Consistent with the above possibility, our observations revealed that estrogen signaling-mediated up-regulation of Un93b1 expression in J774.A1 cells was dependent upon the expression of p202 protein (Fig. 6).
The regulatory region of the murine Unc93b1 gene remains uncharacterized. Therefore, we have analyzed the 5′-regulatory region of the Unc93b1 gene for the presence of potential cis elements through which the p202 protein could activate the transcription of the Unc93b1 gene. Our analysis revealed that the regulatory region of the Unc93b1 gene contains potential DNA-binding consensus sequences for the interferon-activatable transcription factors such as ISGF3 and members of the interferon regulated factors. Additionally, the regulatory region also includes potential DNA-binding sites for ERα. Given that reduced expression of p202 protein in J774.A1 cells inhibited the IFN-α- or estrogen-mediated up-regulation of Unc93b1 expression (Fig. 6), our observations suggested that the p202 protein regulates the expression of Unc93b1. Therefore, further work will be needed to identify the molecular mechanisms.
In summary, our observations will serve as a basis to identify the molecular mechanisms through which levels of the murine Unc93b1 protein are regulated by the p202 protein, a lupus susceptibility modifier. Additionally, these studies will serve as a basis to investigate whether levels of the UNC93B1 protein are up-regulated by activation of interferon or estrogen signaling in immune cells in SLE patients.
Funding
National Institutes of Health (R01 AI066261 and R56 AI089775 to D.C.).
Acknowledgements
We thank Dr Bruce Beutler for suggesting certain experiments. Additionally, we also thank Drs Karen Gould and Lauren Erickson for providing reagents and helpful discussions.
References
- 1. Marshak-Rothstein A., Rifkin I. R. 2007. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 25:419 [DOI] [PubMed] [Google Scholar]
- 2. Saitoh S., Miyake K. 2009. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol. Rev. 227:32 [DOI] [PubMed] [Google Scholar]
- 3. Theofilopoulos A. N., Kono D. H., Beutler B., Baccala R. 2011. Intracellular nucleic acid sensors and autoimmunity. J. Interferon Cytokine Res. 31:867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niewold T. B. 2011. Interferon alpha as a primary pathogenic factor in human lupus. J. Interferon Cytokine Res. 31:887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rifkin I. R., Leadbetter E. A., Busconi L., Viglianti G., Marshak-Rothstein A. 2005. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 204:27 [DOI] [PubMed] [Google Scholar]
- 6. Kawai T., Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373 [DOI] [PubMed] [Google Scholar]
- 7. Lee C. C., Avalos A. M., Ploegh H. L. 2012. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 12:168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tabeta K., Hoebe K., Janssen E. M., et al. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 7:156 [DOI] [PubMed] [Google Scholar]
- 9. Brinkmann M. M., Spooner E., Hoebe K., Beutler B., Ploegh H. L., Kim Y. M. 2007. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 177:265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim Y. M., Brinkmann M. M., Paquet M. E., Ploegh H. L. 2008. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 452:234 [DOI] [PubMed] [Google Scholar]
- 11. Itoh H., Tatematsu M., Watanabe A., et al. 2011. UNC93B1 physically associates with human TLR8 and regulates TLR8-mediated signaling. PLoS One. 6:e28500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carter R. W., Tough D. F. 2006. ‘3d’ effects on global immunity. Nat. Immunol. 7:127 [DOI] [PubMed] [Google Scholar]
- 13. Conley M. E. 2007. Immunodeficiency: UNC-93B gets a toll call. Trends Immunol. 28:99 [DOI] [PubMed] [Google Scholar]
- 14. Fukui R., Saitoh S., Matsumoto F., et al. 2009. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J. Exp. Med. 206:1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fukui R., Saitoh S., Kanno A., et al. 2011. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 35:69 [DOI] [PubMed] [Google Scholar]
- 16. Nakano S., Morimoto S., Suzuki S., Watanabe T., Amano H., Takasaki Y. 2010. Up-regulation of the endoplasmic reticulum transmembrane protein UNC93B in the B cells of patients with active systemic lupus erythematosus. Rheumatology (Oxford). 49:876 [DOI] [PubMed] [Google Scholar]
- 17. Kono D. H., Haraldsson M. K., Lawson B. R., et al. 2009. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc. Natl Acad. Sci. USA. 106:12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsokos G. C. 2011. Systemic lupus erythematosus. N. Engl. J. Med. 365:2110 [DOI] [PubMed] [Google Scholar]
- 19. Liu Z., Davidson A. 2012. Taming lupus—a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 18:871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pennell L. M., Galligan C. L., Fish E. N. 2012. Sex affects immunity. J. Autoimmun. 38:J282 [DOI] [PubMed] [Google Scholar]
- 21. Choubey D., Panchanathan R., Duan X., Liu H., Liu H. 2011. Emerging roles for the interferon-inducible p200-family proteins in sex bias in systemic lupus erythematosus. J. Interferon Cytokine Res. 31:893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rozzo S. J., Allard J. D., Choubey D., et al. 2001. Evidence for an interferon-inducible gene, Ifi202, in the susceptibility to systemic lupus. Immunity. 15:435 [DOI] [PubMed] [Google Scholar]
- 23. Choubey D., Kotzin B. L. 2002. Interferon-inducible p202 in the susceptibility to systemic lupus. Front. Biosci. 7:e252 [DOI] [PubMed] [Google Scholar]
- 24. Choubey D., Panchanathan R. 2008. Interferon-inducible Ifi200-family genes in systemic lupus erythematosus. Immunol. Lett. 119:32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Panchanathan R., Duan X., Shen H., et al. 2010. Aim2-deficiency stimulates the expression of interferon-inducible Ifi202, a lupus susceptibility murine gene within the Nba2 autoimmune susceptibility locus. J. Immunol. 185:7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boross P., Arandhara V. L., Martin-Ramirez J., et al. 2011. The inhibiting Fc receptor for IgG, FcγRIIB, is a modifier of autoimmune susceptibility. J. Immunol. 187:1304 [DOI] [PubMed] [Google Scholar]
- 27. Panchanathan R., Shen H., Duan X., et al. 2011. Aim2-deficiency in mice suppresses the expression of the inhibitory Fcγ receptor (FcγRIIB) through the induction of the IFN-inducible p202, a lupus susceptibility protein. J. Immunol. 186:6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jørgensen T. N., Alfaro J., Enriquez H. L., et al. 2010. Development of murine lupus involves the combined genetic contribution of the SLAM and FcgammaR intervals within the Nba2 autoimmune susceptibility locus. J. Immunol. 184:775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Panchanathan R., Shen H., Bupp M. G., Gould K. A., Choubey D. 2009. Female and male sex hormones differentially regulate expression of Ifi202, an interferon-inducible lupus susceptibility gene within the Nba2 interval. J. Immunol. 183:7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hall J. C., Rosen A. 2010. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat. Rev. Rheumatol. 6:40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meraz M. A., White J. M., Sheehan K. C., et al. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 84:431 [DOI] [PubMed] [Google Scholar]
- 32. Gubbels M. R., Jørgensen T. N., Metzger T. E., et al. 2005. Effects of MHC and gender on lupus-like autoimmunity in Nba2 congenic mice. J. Immunol. 175:6190 [DOI] [PubMed] [Google Scholar]
- 33. Jørgensen T. N., Roper E., Thurman J. M., Marrack P., Kotzin B. L. 2007. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 × NZW)F1 mice. Genes Immun. 8:653 [DOI] [PubMed] [Google Scholar]
- 34. Theofilopoulos A. N., Baccala R., Beutler B., Kono D. H. 2005. Type I interferon (α/β) in immunity and autoimmunity. Annu. Rev. Immunol. 23:307 [DOI] [PubMed] [Google Scholar]