

Abstract
Transposons permit permanent cellular genome engineering in vivo. However, transgene expression falls rapidly postdelivery due to a variety of mechanisms, including immune responses. We hypothesized that delaying initial transgene expression would improve long-term transgene expression by using an engineered piggyBac transposon system that can regulate expression. We found that a 2-part nonviral Tet-KRAB inducible expression system repressed expression of a luciferase reporter in vitro. However, we also observed nonspecific promoter-independent repression. Thus, to achieve temporary transgene repression after gene delivery in vivo, we utilized a nonintegrating version of the repressor plasmid while the gene of interest was delivered in an integrating piggyBac transposon vector. When we delivered the luciferase transposon and repressor to immunocompetent mice by hydrodynamic injection, initial luciferase expression was repressed by 2 orders of magnitude. When luciferase expression was followed long term in vivo, we found that expression was increased >200-fold compared to mice that received only the luciferase transposon and piggyBac transposase. We found that repression of early transgene expression could prevent the priming of luciferase-specific T cells in vivo. Therefore, transient transgene repression postgene delivery is an effective strategy for inhibiting the antitransgene immune response and improving long-term expression in vivo without using immunosuppression.—Doherty, J. E., Woodard, L. E., Bear, A. S., Foster, A. E., Wilson, M. H. An adaptable system for improving transposon-based gene expression in vivo via transient transgene repression.
Keywords: piggyBac, inducible expression, gene transfer
Long-term gene expression is necessary for successful correction of an inherited disease or genome engineering. The development of a host immune response to the transgene product can be major obstacle to long-term gene expression after gene delivery in vivo. A simple gene delivery system with improved long-term expression that can be adapted to any transgenes would be a significant advance.
Immune responses to gene delivery products have been described in a number of models of human disease (1, 2). Reporter genes, just like therapeutic genes, can be targeted by the immune system in vivo, providing a model for evaluating mechanisms affecting long-term transgene expression. A variety of reporters are targeted by the immune system, including lacZ (3, 4), fluorescent proteins (5, 6), and firefly luciferase (7, 8). Luciferase is a popular reporter gene due to the ability to noninvasively monitor its expression in vivo (9). However, a specific T-cell response to luciferase has been characterized after in vivo gene transfer (8), resulting in lack of persistent expression after lentivirus-mediated gene delivery in the lung (10). Therefore, luciferase represents a model transgene for evaluating long-term transgene expression in vivo after gene delivery.
The use of nonviral vector obviates the potential immune response to viral vector (11–14). We have previously demonstrated long-term transgene expression in mouse liver using the nonviral but integrating piggyBac transposon system (15). Improvements in the efficiency of transposon systems have increased their attractiveness for gene transfer as an alternative to viral vectors (16–18). Hydrodynamic tail vein injection, an accepted method of nonviral gene delivery to mouse liver in vivo, injures the liver in the process of delivery, leading to inflammation (19). We hypothesized that temporarily suppressing gene expression after gene delivery would improve long-term gene expression by mitigating immune response to transgene expressed in the liver during the time of liver injury. Methods of improving long-term transgene expression after gene delivery to the liver remain of interest as the liver continues to be a site of consideration for delivery of genes, even in human clinical trials (20).
An important part of implementing any gene transfer strategy is the ability to control the level of expression. Although this can be modulated by optimization of the vector dose, regulatable expression systems remain critical for controlling gene expression. Most current systems allow regulation of gene expression via control of transcription through administration of small molecules (21, 22). Systems include those using rapamycin (23), mifepristone (24), and ecdysone (a Drosophila hormone; ref. 25). The most widely used are those controlled by tetracycline (Tet; refs. 26, 27). In this study, we used a modified version of the Tet-regulated system that uses the Krüppel-associated box (KRAB) domain of human Kox1 to act as a transcriptional repressor (28, 29).
Plasmid-mediated transgene delivery results in a spike in transgene expression soon after gene delivery. There is a subsequent fall in transgene expression after initial delivery, resulting from lack of transgene integration, transgene silencing, and immune response to the transgene. Transposons can prolong transgene expression by promoting transgene integration in vivo. Though long-term expression is improved compared to nonintegrating plasmid DNA alone, a high initial level of transgene expression is required in order to achieve sufficient long-term stable expression (15). High levels of gene expression soon after delivery, in concert with nonspecific inflammation from gene delivery, likely contribute to the development of a specific immune response to the transgene. In this study, we tested whether we could improve long-term expression of a model reporter transgene delivered using hydrodynamic tail vein injection of the piggyBac transposon system by inhibiting early transgene expression in vivo with the Tet-KRAB inducible expression system. Luciferase is known to provoke an immune response after gene transfer (7, 8, 10), and the inflammation caused by tissue damage after hydrodynamic tail vein injection (30) likely exacerbates this. Our goal was to engineer a transgene expression system for improved long-term expression in vivo that could be easily adapted to other transgenes by simply swapping out the transgene in the regulatable transposon vector.
MATERIALS AND METHODS
Plasmids
The hyperactive transposase plasmids (pCMV-m7pB and pCMV-HA-m7pB; refs. 16, 17) and luciferase transposon pTCAGluc (15) have been previously described. The repressor constructs were cloned from the lentiviral vector pLCVT-rtTR-KRAB (plasmid 11643; Addgene, Cambridge, MA, USA; ref. 28). The rtTR-KRAB-2SM2 open reading frame (ORF) and the woodchuck hepatitis post-transcriptional regulatory element (WPRE) were excised from the donor plasmid using KpnI and MscI and cloned into the recipient plasmids KpnI/blunt to create the plasmids pT-CMV-rtTR-KRAB-2SM2 and pCMV-rtTR-KRAB-2SM2, with the repressor construct in a transposon and in a nonintegrating plasmid vector, respectively.
To clone a liver-specific luciferase reporter, the expression cassette was excised from the adenoviral vector HD-Ad-N6-FVIII (31) by AscI digest and cloned into the piggyBac transposon vector Zeo-pT-MCS (32). The firefly luciferase ORF was excised from pTCAGluc and bluntly cloned into the vectors, creating pT-phosphoenolpyruvate carboxykinase (PEPCK)-luc. To insert a Tet-responsive element (TRE) into the vector, the TRE was amplified by PCR from pLCVT-rtTR-KRAB-2SM2 with primers 5′-CCCAACGAAGACAAGATCTC-3′ and 5′-CTGCTGCCGCGGCAGTGGGTTCCCTAGTTAG-3′. The resulting product was directionally cloned upstream of the promoter in the plasmid pT-PEPCK-luc PspXI/SacII. To add a second TRE, the TRE was excised and bluntly cloned into pT-TRE-PEPCK-luc between the promoter and ORF in the same orientation as the first TRE, yielding the plasmid pT-TRE-PEPCK-TRE-luc.
The qPCR control plasmid pItk was created by TaqBead (GE Healthcare, Waukesha, WI, USA) PCR amplification of FVB mouse genomic DNA. The 246-bp PCR product was gel-purified and TOPO-cloned into the vector pCR2.1 (Invitrogen, Grand Island, NY, USA). The desired mouse Itk sequence represents a product spanning intron 13 to exon 14 of the mouse Itk genomic DNA sequence (chromosome 11: 46,177,282 to 46,181,527; Ensembl release 67). Plasmid pCR2.1-ZeoExc was created so that the number of copies of excised transposon backbone plasmid could be quantified by qPCR. The excision fragment was obtained by PCR with TaqProRed from a liver sample isolated from a mouse that had received pCMV-HA-m7pB and pT-TRE-PEPCK-TRE-Luc. The primers were those described below for qPCR of excision copy number. The fragment was gel purified and cloned with the pCR2.1-TOPO cloning kit. All plasmids were verified by DNA sequencing.
Cell culture
All cell culture experiments were conducted using HeLa cells [American Type Culture Collection (ATCC), Manassas, VA, USA], which were plated in 6-well plates 24 h prior to transfection; for experiments using doxycycline, cells were split into medium containing 1 μg/ml doxycycline. Cells were transfected using FuGene 6 (Roche Applied Science, Indianapolis, IN, USA). All transfections consisted of 200 ng of luciferase plasmid and the indicated ratio of repressor plasmid, and inert (pUC19; Invitrogen) plasmid was added to all transfections to make a total DNA content of 1 μg. Each transfection was performed in triplicate.
Luciferase expression in vitro
Luciferase expression was assayed 48 h after transfection in all cell culture experiments. The medium was aspirated, and the cells were washed with phosphate-buffered saline (PBS). Luciferase expression in cell culture was quantified using the Steady-Glo Luciferase Assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions. Each transfection was assayed in duplicate on a FLUOstar Omega plate reader (BMG Labtech, Ortenberg, Germany).
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. FVB/N mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and were 8–10 wk old at the time of injection. Mice were treated with 0.2 mg/ml doxycycline in the drinking water for 7 d prior to injection, and doxycycline treatment was continued for 2 wk after injection.
Plasmid DNA was delivered by hydrodynamic tail vein injection as described previously (33). Briefly, the indicated amounts of each plasmid were combined with 100 μl/g gram body weight of TransIT QR hydrodynamic injection solution (Mirus Bio, Madison, WI, USA), which was then injected via the tail vein in <10 s. All plasmid DNA was prepared with an endotoxin-free Maxiprep kit (Qiagen, Valencia, CA, USA).
In vivo imaging of luciferase expression was conducted using a Xenogen IVIS imaging system (Caliper Life Sciences, Hopkinton, MA, USA). Mice were imaged 5 min after intraperitoneal injection of 100 μg luciferin substrate (Caliper Life Sciences) in PBS as per the manufacturer's instructions.
Liver sample collection
Mice were euthanized by ketamine/xylazine injection, and their livers were dissected into 1-cm2 pieces. Half of the liver was minced with a straight razor into a fine pulp and placed into a 100-μm strainer (BD Falcon, Franklin Lakes, NJ, USA) with 10 ml of PBS. The cells in the strainers were washed twice with 5 ml PBS and recovered by centrifugation at 500 g for 20 min. Recovered cell pellets were portioned into aliquots, washed with PBS by centrifugation, and frozen in liquid nitrogen until protein or DNA was extracted. The remaining half of the liver pieces were kept in 10% formalin in PBS overnight at 4°C, transferred to 70% ethanol, paraffin embedded, and sectioned for histology.
Immunoblotting
A cell pellet representing ∼10–15% of the entire mouse liver was resuspended in 750 μl of hypertonic buffer [20 mM Tris-HCl (Promega), pH 7.4; 10 mM NaCl (Fisher, Fair Lawn, NJ, USA), and 3 mM MgCl2 (Sigma, St. Louis, MO, USA)] and rotated at 4°C for 30 min, followed by addition of 37.5 μl of 10% IGEPAL (Sigma) detergent. The cells were vortexed for 10 s and centrifuged for 10 min at 3000 rpm at 4°C. The supernatant containing the cytoplasmic fraction was saved, and the nuclear pellet was resuspended in 75 μl of complete cell extraction buffer [100 mM Tris-HCl, pH 7.4; 2 mM Na3VO4 (Sigma); 100 mM NaCl; 1% Triton X-100 (Sigma); 1 mM EDTA (EMD Biosciences, Gibbstown, NJ, USA); 10% glycerol (Fisher); 1 mM EGTA (Sigma); 0.1% SDS (Pierce-Thermo, Rockford, IL, USA); 1 mM NaF (Sigma); 0.5% C24H39NaO4 (Sigma); 20 mM Na4P2O7 (Sigma); 1 mM PMSF (Sigma); and 1× protease inhibitor cocktail (Roche)] and kept on ice for 30 min with vortexing every 10 min. After centrifugation at 14,000 g for 30 min at 4°C, the nuclear fraction was transferred to another tube, and all protein was frozen in liquid nitrogen and stored at −80°C. The BCA test (Pierce-Thermo) was performed in triplicate according to manufacturer's directions to determine protein concentration. Each sample was diluted to 2 mg/ml in complete cell extraction buffer. Laemmli Sample Buffer (Bio-Rad, Richmond, CA, USA) and β-mercaptoethanol (EMD Millipore, Darmstadt, Germany) were added to the samples and were heated at 99°C for 2 min; 30 μg was loaded onto 10% Bis-Tris 1.5 mm Gels (Invitrogen). Protein was transferred from the gel onto nitrocellulose with an iBlot device (Invitrogen), cut at the 50-kDa marker into 2 pieces, and blocked overnight at 4°C in blocking buffer (LI-COR, Lincoln, NE, USA) diluted 1:1 with PBS. Mouse α-HA (diluted 1:1000; Covance, Alice, TX, USA) or mouse α-β-actin (diluted 1:7000; Novus Biologicals, Littleton, CO, USA) was added to the blocking buffer for each part and incubated with the blots for 90 min with shaking. After four 5-min washes with PBS + 0.1% Tween 20 (PBST; Sigma), the blot was incubated with goat anti-mouse IR 800 CW secondary antibody (diluted 1:15,000; LI-COR) for 2 h with shaking and protected from light. The blot was washed 4 times for 5 min with PBST, followed by one PBS wash for 5 min, and images were acquired with an Odyssey imaging device (LI-COR).
Immunofluorescence
Slides were treated with Trilogy reagent (CellMarque, Rocklin, CA, USA) for 15 min on low in a pressure cooker and rinsed in hot Trilogy followed by PBS for 5 min each. Tissues were circled with the Elite Mini PAP pen (Diagnostic Biosystems, Pleasanton, CA, USA) and PBST was added to the tissues for 30 min. Tissues were blocked using Background Sniper (Biocare Medical, Concord, CA, USA) for 20 min followed by three washes of PBST. Rat α-HA antibody (clone 3F10; Roche) or rat IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was diluted 1:500 in serum buffer [10% donkey serum (Jackson Immunoresearch, West Grove, PA, USA) and 10% 10× PBST] and left at 4°C overnight. The primary antibody was removed with three 5-min washes of PBST. Donkey α-rat AlexaFluor 594 (Invitrogen) was diluted 1:500 in serum buffer and incubated on the tissues for 1 h. The secondary antibody was removed with 3 PBST washes, and slides were immersed in deionized water to remove residual salt, quickly dried, and mounted in ProLong Gold Antifade Reagent + DAPI (Invitrogen). Slides were sealed with nail polish the following day. Images were acquired on a Nikon microscope (Nikon, Tokyo, Japan).
Excision copy number assay by qPCR
DNA was purified from strained mouse liver cells with the DNeasy kit (Qiagen) according to manufacturer's directions. For excision product quantification, the standard curve was based on the plasmid pPCR2.1-ZeoExc. The primers to the excision product were pBZeoExcF3, 5′-GCCATGAGGGTTTAGTTC-3′, and pBZeoExcR4, 5′-CTTAGCCTTTGTCGACTC-3′. For Itk, the standard curve was based on plasmid pItk, and the primers to determine genomic copy number were Itk-F, 5′-GCCGTAAATGAACAGGTGGTGA-3′, and Itk-R, 5′-TGCTCCAGACTGTGAGAGTCG-3′ (34). The PCR program was 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 60°C for 15 s, 72°C for 20 s, and plate read, with a final extension of 72°C for 3 min and melting curve read (65–95°C: increment 0.5°C for 5 s). Each well contained 12 ng of liver DNA template. All other experimental conditions, instruments, and data analysis have been previously described (35). The triplicate reactions were based on iQ SYBR Green Master Mix (Bio-Rad), and melting curve analysis confirmed single PCR products only in the expected wells. The experiment was repeated 4 times with the same statistical result.
Interferon γ (IFNγ) enzme-linked immunosorbent spot (ELISPOT)
The indicated plasmids were delivered by hydrodynamic injection as before and luciferase expression was monitored by in vivo imaging. T cells were isolated from the spleens of treated mice using a mouse Pan-T Cell Isolation Kit II and MACS cell separation columns (Miltenyi Biotec, Cologne, Germany).
Dendritic cells were used as antigen-presenting cells. Dendritic cells were generated from the bone marrow of uninjected mice 7 d prior to use by culturing in complete RPMI in the presence of 20 ng/ml of IL4 and GM-CSF. Dendritic cells were transduced with Ad5-CMV-luciferase at an MOI of 100 and stimulated with 30ng/ml LPS 48 h before use. Luciferase expression was confirmed by luciferase assay (Promega).
Immunoprecipitation-type ELISPOT plates were precoated overnight with anti-IFNγ capture antibody (Mabtech, Nacka Strand, Sweden). T cells (1×106/well) were plated with or without 1 × 104 luciferase-expressing dendritic cells, and the plates were incubated overnight at 37°C. The plates were thoroughly washed and coated with biotinylated anti-IFNγ detection antibody (Mabtech). The plates were then developed and dried overnight at room temperature in the dark, then sent for quantification by personnel blinded to experimental conditions (Zellnet Consulting, New York, NY, USA).
RESULTS
Regulatable transposon-mediated transgene expression in human cells
Hydrodynamic tail vein injection of plasmid DNA leads to an initial spike of transgene expression, which then decreases by several orders of magnitude to a steady-state level (15, 36). We hypothesized that eliminating the initial peak in transgene expression would ameliorate the formation of a transgene-specific immune response. We chose to accomplish this through the use of the Tet-KRAB Tet-regulated inducible expression system (28), specifically the optimized Tet-off version of this system (rtTR-KRAB-2SM2). By using the Tet-KRAB system, other transgenes can easily be tested by exchanging the transgene cDNA in the regulatable transposon vector.
The Tet-KRAB system uses fusions of either a forward Tet repressor (TetR) or reverse TetR (rTetR) Tet-responsive DNA-binding domain fused to the KRAB domain of Kox1, a human transcriptional repressor (29). The KRAB domain exhibits promiscuous repression of transcription (37) and, when tethered to TetR and guided to the gene of interest by the addition of TREs, strongly represses expression from any DNA polymerase II promoter including tissue-specific promoters. Since the KRAB domain is derived from a mammalian protein, it is also theoretically less likely to provoke a host immune response than the Tet-VP16 fusion, which uses an activation domain from human herpes simplex virus (26).
We examined the ability of the rtTR-KRAB Tet-off repressor construct to regulate gene expression in our system. The Tet-KRAB system was initially described as a 2-part system and characterized by transient and stable expression in human cells (29), but the rtTR-KRAB version of the system has thus far been exclusively used in lentiviral vectors (28, 38). The rtTR-KRAB repressor construct from pLCVT-rtTR-KRAB-2SM2 was cloned into plasmid expression vectors. Since the mechanism of the Tet-KRAB system allows for strong gene expression in the absence of the Tet-regulated fusion protein (in contrast to the more commonly used TetR-VP16 system; refs. 26, 27), we cloned the repressor into both an integrating (i.e., a piggyBac transposon) and nonintegrating plasmid vector (Fig. 1A). This provides an option of selecting whether it would be preferable for the repressor to be stably integrated or only transiently expressed.
Figure 1.

In vitro gene repression by rtTR-KRAB as a 2-part nonviral system. A) Plasmid constructs used in this study. Liver-specific luciferase reporter constructs without (pT-PEPCK-luc) and with (pT-TRE-PEPCK-TRE-luc) Tet-regulatable elements, constitutive luciferase piggyBac transposon plasmid (pTCAGluc); Tet-off repressor expression constructs in a piggyBac transposon (pT-CMV-rtTR-KRAB) and nonintegrating vector (pCMV-rtTRKRAB); hyperactive piggyBac transposase expression plasmid (pCMV-m7pB). CMV, cytomegalovirus immediate early promoter; PEPCK, phosphoenolpyruvate carboxykinase promoter; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element; SV40 pA, simian virus 40 polyadenylation signal; hGH pA, human growth hormone polyadenyaltion signal. B) Comparison of luciferase expression from vectors with no binding site (pT-PEPCK-luc) or 2 binding sites (pT-TRE-PEPCK-TRE-luc for the rtTR-KRAB repressor construct. HeLa cells were transfected with 200 ng of luciferase plasmid and the indicated ratios of rtTR-KRAB transposon plasmid (pT-CMV-rtTR-KRAB). Luciferase activity was assayed 48 h after transfection. While both luciferase constructs showed ∼75% reduction in signal with rtTR-KRAB in the absence of doxycycline, only the construct with binding sites for the repressor (pT-TRE-PEPCK-TRE-luc) had specific repression of expression in the presence of doxycycline (n=3±sd). C) Characterization of nonspecific repression by rtTR-KRAB with different promoters. Cells were transfected with 200 ng of PEPCK promoter-driven luciferase plasmid with or without repressor binding sites or CAG promoter-driven luciferase plasmid (pTCAGluc) in the absence and presence of a 4-fold excess of rtTR-KRAB transposon plasmid. Luciferase activity was assayed 48 h post-transfection (n=3±sd).
Initial characterization of the system was performed in HeLa cells using the Tet-off transposon construct pT-CMV-rtTR-KRAB and luciferase reporters pT-PEPCK-luc (no repressor binding sites) and pT-TRE-PEPCK-TRE-luc (TRE binding sites for the repressor flanking the transposon; Fig. 1B). We cotransfected 200 ng of the luciferase reporters with the indicated ratios of repressor plasmid, and the cells were lysed and luciferase expression was assayed 48 h later (Fig. 1B).
In the presence of doxycycline, rtTR-KRAB was found to efficiently repress gene expression. At both ratios used, luciferase activity was <1% of that found in the no-repressor control. However, significant repression was also noted in the absence of doxycycline and of the TRE binding site. Expression from pT-PEPCK-luc decreased by ∼80% from control both in the presence and absence of doxycycline, as did pT-TRE-PEPCK-TRE-luc expression in the absence of doxycycline.
Nonspecific repression has not been previously reported with the Tet-KRAB system (29), however, our observation could be due to gene dosage effects or different cell types or assays employed. To further examine nonspecific repression and exclude the possibility of specific repression of the PEPCK promoter by rtTR-KRAB, we examined the effects of the repressor on the constitutive CAG promoter (a chicken β-actin promoter with CMV IE enhancer element; ref. 39) using the plasmid pTCAGluc. The three luciferase plasmids were transfected with and without a 4-fold excess of rtTR-KRAB transposon (Fig. 1C). All three had significantly decreased luciferase expression when cotransfected with rtTR-KRAB; however, only the construct with the repressor binding sites, pT-TRE-PEPCK-TRE-luc, had expression further decreased in the presence of doxycycline.
Because of the nonspecific repression observed in the absence of doxycycline, we decided to use a transiently expressed rtTR-KRAB construct rather than integrate the repressor transposon. By removing the inverted repeat elements flanking the repressor, which serve as recognition sites for the transposase and are required to stably integrate the transposon into the genome, the plasmid remains episomal and is lost as the cell divides. For all future experiments, rtTR-KRAB in a nontransposon plasmid was used (pCMV-rtTR-KRAB; Fig. 1A).
To better define the optimal ratio of repressor to regulated transgene, we examined gene expression at a range of rtTR-KRAB plasmid doses. We cotransfected 200 ng of luciferase plasmid with a wide range of rtTR-KRAB plasmid doses (3–800 ng), and expression was measured by luciferase assay (Fig. 2). Both with and without doxycycline, luciferase expression increased with decreasing repressor dose. In the presence of doxycycline, repression was notable at all doses, with luciferase expression only 0.27% of the no-repressor control at the highest rtTR-KRAB dose and 10% at the lowest dose. All transfections with a repressor to luciferase plasmid ratio ≥ 1:1 decreased expression to <1% of control in the presence of doxycycline. Observable repression was noted in the absence of doxycycline, with luciferase expression at the lowest repressor dose (1:64 ratio of repressor to luciferase) only ∼60% of that in the no repressor control. At the highest dose, expression without doxycycline was <5% of control.
Figure 2.

Optimization of rtTR-KRAB to luciferase plasmid ratio in vitro. HeLa cells were transfected with 200 ng of luciferase plasmid (pT-TRE-PEPCK-TRE-luc) and varying amounts of nonintegrating repressor plasmid (pCMV-rtTR-KRAB) in a constant total plasmid DNA amount of 1 μg by using pUC19 as inert filler plasmid DNA. Luciferase expression was assayed after 48 h (n=3±sem).
Improved in vivo transgene expression with a regulatable transposon
We next examined the ability of rtTR-KRAB to regulate transgene expression after gene transfer in vivo. All mice were pretreated with doxycycline for 7 d prior to injection. For initial experimentation, 3 mice/group were injected with 25 μg of Tet-responsive luciferase plasmid (pT-TRE-PEPCK-TRE-luc) and 5 μg hyperactive piggyBac transposase plasmid (pCMV-m7pB), with varying doses of rtTR-KRAB plasmid to determine the optimal ratio of plasmids (Supplemental Fig. S1). We found that the highest dose of repressor, 100 μg, was best for repressing early gene expression and led to significantly greater luciferase expression long-term when followed to 6 mo by in vivo imaging.
We then injected 5 mice/group with 25 μg pT-TRE-PEPCK-TRE-luc with and without 100 μg pCMV-rtTR-KRAB. Luciferase signal in the mice that received the repressor construct was decreased by two orders of magnitude at 24 h postinjection as compared to those that received luciferase plasmid alone (4.3×107 photons/s/cm2/sr vs. 4.2×109 photons/s/cm2/sr; Fig. 3A). Expression in the luciferase-only group peaked around 1 × 1010 photons/s/cm2/sr at d 4 and decreased to a steady state around 1 × 107 photons/s/cm2/sr, while the group with rtTR-KRAB reached maximal expression at d 18 at 3 × 108 photons/s/cm2/sr and remained near that level for the duration of the experiments (10 wk; Fig. 3B).
Figure 3.

Luciferase expression in vivo with rtTR-KRAB. Luciferase transposon plasmid (25 μg; pT-TRE-PEPCK-TRE-luc) with and without pCMV-rtTR-KRAB (100 μg) and pCMV-m7pB transposase plasmid (5 μg) was delivered to wild-type mice by hydrodynamic tail vein injection. All mice were pretreated for 7 d with doxycycline, which continued until d 14 postinjection. A) Luciferase expression 24 h postinjection (n=5). B) Luciferase expression was followed in vivo (n=5±sem). At later time points, luciferase expression in the group that received rtTR-KRAB plasmid (shaded triangles) was more than an order of magnitude greater than the no repressor group (solid circles) (P=0.002 at wk 10 by ANOVA).
Temporal transgene repression diminishes immune response without affecting transposition
Increased long-term luciferase expression after early repression of gene expression conforms well to the hypothesis that eliminating the peak of expression that occurs after hydrodynamic tail vein injection can help avoid an immune response to the transgene. However, the repression of transposase expression could in theory improve long-term expression via a mechanism related to overproduction inhibition, as has been seen with some transposon systems (40). To test this, we evaluated for piggyBac transposase repression in vivo. Immunofluorescent analysis of liver sections revealed repressed transposase expression in animals injected with repressor activated with doxycycline (Fig. 4A). We also observed repression of transposase expression by immunoblot (Fig. 4B), which was consistent with non-Tet-responsive transgene repression we observed in vitro (Fig. 1C). Nonetheless, we observed no difference in transposase activity as measured by transposon excision analysis in vivo using qPCR (Fig. 4C and Supplemental Fig. S2). Although we observed transposase repression, this had no effect on the ability of transposase to integrate transposon DNA. Therefore, it is unlikely that we observed improved long-term transgene expression due to effects on transposase activity.
Figure 4.

Effect of rtTR-KRAB on hyperactive piggyBac transposase expression and transposon integration in vivo. Mice were treated pretreated with doxycycline and euthanized 1 d after hydrodynamic tail vein injection of 25 μg pT-TRE-PEPCK-TRE-Luc and 5 μg pCMV-HA-m7pB with or without pCMV-rtTR-KRAB (n=4). Livers were harvested for protein, immunofluorescence, or DNA. A) Immunofluorescent detection of HA-m7pB (red) and nuclei (stained with DAPI, blue) in mouse liver sections. Each image is representative of the liver of one mouse. B) Western blot detection of HA-m7pB and β-actin in the cytoplasmic and nuclear liver protein fractions. The mice are in the same order as shown in panel A. C) qPCR analysis of transposon excision with or without rtTR-KRAB expression. Number of copies of the postexcision transposon plasmid product was normalized to the number of haploid genomes (by mouse genomic Itk copy number) for both groups. Comparison of the groups with and without rtTA-KRAB with respect to the number of postexcision plasmids per haploid genome for total DNA recovered did not reach significance (t test). Error bars = sem.
We next examined the T-cell response to luciferase with and without transgene repression in vivo by performing an IFN-γ ELISPOT assay. The immune response to luciferase gene transfer has been examined after adenovirus-mediated (8) and lentivirus-mediated (10) gene transfer, as well as after intradermal delivery of a luciferase plasmid (7). However, the T-cell response to luciferase delivered via hydrodynamic injection has not yet been studied. Mice were injected as before (Fig. 3) and euthanized on d 15, and T cells (both CD4+ and CD8+) were isolated from spleens. Cells were stimulated with syngeneic bone marrow-derived dendritic cells that were transduced with a luciferase adenovirus. Activation was monitored by detection of IFNγ secretion. T cells isolated from mice injected with pT-TRE-PEPCK-TRE-luc and rtTR-KRAB exhibited diminished responsiveness to luciferase compared to mice injected with luciferase plasmid alone; however, mice given the repressor plasmid were no different from control mice (P=0.026; ANOVA and Bonferroni's multiple comparison posttest; Fig. 5). Cells from mice injected with rtTR-KRAB and TRE-PEPCK-TRE-luc had on average 25.1 spot-forming cells (SFCs)/well, while those from mice injected with PEPCK-luc alone had 46.2 SFCs/well, and control mice had 32 SFCs/well. Therefore, although plasmid delivery of luciferase to mouse liver does not elicit a strong immune response, the T-cell response was diminished via temporal transgene repression without the use of immunosuppressive medications.
Figure 5.
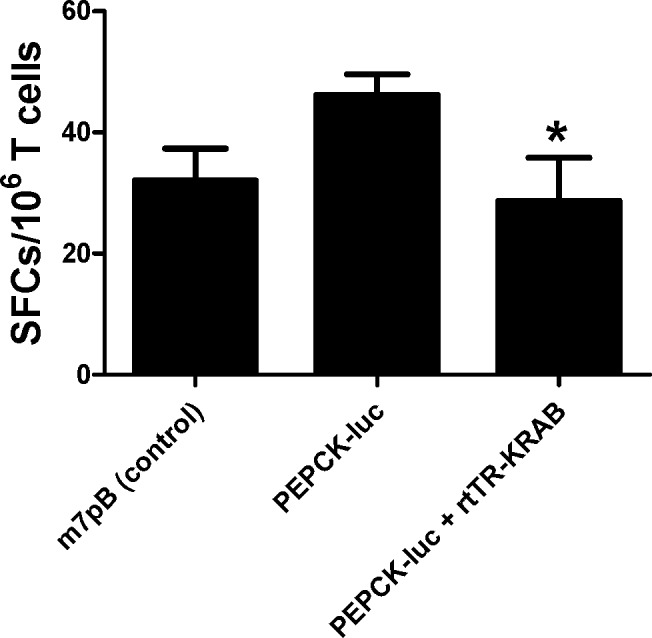
T-cell response to luciferase after hydrodynamic tail vein injection. Luciferase (25 μg) transposon plasmid and transposase plasmid (5 μg) without repressor or with nonintegrating rtTR-KRAB repressor plasmid (100 μg) were delivered by hydrodynamic tail vein injection to wild-type mice pretreated with doxycycline. Control mice received transposase plasmid alone. Spleens were collected from the mice 15 d postinjection, and T cells were isolated for use in ELISPOT. Immune response to luciferase was examined by assaying IFNγ secretion in response to coculture with dendritic cells expressing luciferase. Mice that received rtTR-KRAB plasmid in addition to PEPCK-luc had significantly fewer luciferase-specific T cells than mice that were injected with PEPCK-luc alone, and they did not differ from mice that received sham injections (P=0.026 by ANOVA and Bonferroni's multiple comparison posttest). Five mice per group were injected; 1 × 106 T cells from each mouse were assayed in triplicate. Data are presented as means ± sem. SFCs, spot-forming cells per well, corrected for background of dendritic cells alone.
DISCUSSION
We used a standardized gene delivery method in combination with an integrating but regulatable nonviral transposon system to deliver a widely used reporter gene in live animals. In this context, temporal transgene repression immediately after gene delivery improved long-term transgene expression by >200-fold. Temporal transgene repression mitigated the T-cell mediated immune response to the transgene without affecting the activity of the integration mechanism (i.e., transposition).
Delaying gene expression to avoid “danger signals” resulting from gene delivery has been proposed by others (1), but few applications have been reported in vivo. Hydrodynamic injection relies on physical damage for DNA delivery (19), which may cause inflammation that can predispose to an immune response to the transgene. Indeed, it has even been demonstrated that the degree of rise in liver enzymes correlates with the efficiency of gene delivery (41). Minimization of transgene expression during this inflammation could mitigate transgene-specific immune response. Previously, investigators have used α-galactosidase A-specific small interfering RNA (siRNA) to repress α-galactosidase A expression from delivered plasmid in mouse liver thereby reducing immune responses (42). However, these investigators used nonintegrating; i.e., nonpermanent, plasmid vector, and not all transgenes have known siRNA analogs capable of repression.
Hydrodynamic injection has been used to deliver a variety of different types of molecules to the liver (43), including RNA, proteins, and dyes, in addition to plasmid DNA. Therefore, it is unlikely that a specific cellular uptake mechanism is responsible for the uptake of injected DNA and the subsequent gene expression. The damage caused by hydrodynamic tail vein injection has been shown to precipitate a nonspecific inflammatory response, including induction of the JAK/STAT signaling pathway (which leads to interferon production) and elevation of plasma IL-12 within 6 h postinjection (30). The resultant inflammation may lead to a “bystander effect” (44, 45), which predisposes the animals to development of a specific immune response to the transgene, in our case to luciferase.
A number of other immunomodulatory strategies could be attempted to avoid an immune response to transgenes in the setting of gene delivery. The liver is thought to be a naturally tolerogenic organ (46), and hydrodynamic tail vein injection specifically targets gene expression to hepatocytes (47). Others have had success at achieving tolerance to transgenes by careful vector design to direct tissue-specific expression (2, 48) and avoid expression in antigen-presenting cells (49). Our expression vector uses the PEPCK promoter, which mediates gene expression specifically in hepatocytes (50). However, no promoter or delivery is absolutely cell-type specific. After hydrodynamic injection, low levels of gene expression can be detected in a number of organs besides the liver (36), including the spleen, which may promote an immune response. A wide variety of immunosuppressive strategies, both general and targeted to particular arms of the host response, have also been studied for use in achieving tolerance to the transgene (1). In this study, we chose to use modulation of gene expression via a regulatable expression system rather than attempting to interfere with the host response. This, in combination with nonviral gene therapy technologies, allowed us to use a completely plasmid-based approach, as well as avoiding the use of immunosuppressive drugs.
A concern about use of inducible expression systems for gene therapy is the possibility of an immune response to the regulatory protein itself. While this possibility has not been investigated for the Tet-KRAB fusion used in the present work, an immune response to the Tet-on transactivator rtTA has been observed in a number of nonhuman primate studies (51). Theoretically, the Tet-KRAB system should be less immunogenic than the Tet-VP16 system in mammals because it uses a human regulatory domain rather than a viral domain; an immune response to the bacterial TetR domain that is shared by the two systems has been observed in mice (52). The risk of an immune response to the regulatory system could be lessened by use of a transcriptional regulatory system completely derived from mammalian components (23) or by avoiding use of regulatory proteins altogether, such as using a regulated expression system that relies on modulation of mRNA stability (53).
It is important to better understand the immune response to foreign proteins in order to improve methods for gene therapy. A more complete understanding of the immunology behind rejection of gene products and vectors could guide rational design of these components to avoid complications and failure of therapy. Notably, our approach described herein achieves improved long-term gene expression and mitigates the immune response to the transgene without the use of immunosuppressive medications.
As most in vivo gene transfer methodologies elicit inflammation and immune response, our results should have notable effects throughout the realm of gene delivery. Our transposon vectors can easily be adapted to other transgenes by swapping out the reporter gene for any transgene of interest. Therefore, our approach could be used in a variety of ways to increase transgene, even therapeutic transgene, expression after in vivo gene delivery.
Supplementary Material
Acknowledgments
J.E.D. was supported by U.S. National Institutes of Health (NIH) Medical Scientist Training Program grant T32GM007330, and subsequently by NIH Cell and Gene Therapy Training grant T32DK064717 and the generous support of Bill and Mary Jo Robbins. L.E.W. was supported by NIH grant T32DK062706 and subsequently by NIH grant T32DK060445. M.H.W. was supported in part by a career development award from the U.S. Department of Veterans Affairs and NIH grant R01 DK093660. This work was also made possible by the generous support of Dr. and Mrs. Harold M. Selzman.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ELISPOT
- enzme-linked immunosorbent spot
- IFNγ
- interferon γ
- KRAB
- Krüppel-associated box
- ORF
- open reading frame
- PBS
- phosphate-buffered saline
- PEPCK
- phosphoenolpyruvate carboxykinase
- siRNA
- small interfering RNA
- Tet
- tetracycline
- TetR
- tetracycline repressor
- TRE
- tetracycline-responsive element
REFERENCES
- 1. Arruda V. R., Favaro P., Finn J. D. (2009) Strategies to modulate immune responses: a new frontier for gene therapy. Mol. Ther. 17, 1492–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zaldumbide A., Hoeben R. C. (2008) How not to be seen: immune-evasion strategies in gene therapy. Gene Ther. 15, 239–246 [DOI] [PubMed] [Google Scholar]
- 3. Isenmann S., Engel S., Kugler S., Gravel C., Weller M., Bahr M. (2001) Intravitreal adenoviral gene transfer evokes an immune response in the retina that is directed against the heterologous lacZ transgene product but does not limit transgene expression. Brain Res. 892, 229–240 [DOI] [PubMed] [Google Scholar]
- 4. Abina M. A., Lee M. G., Descamps V., Cordier L., Lopez M., Perricaudet M., Haddada H. (1996) LacZ gene transfer into tumor cells abrogates tumorigenicity and protects mice against the development of further tumors. Gene Ther. 3, 212–216 [PubMed] [Google Scholar]
- 5. Stripecke R., Carmen V. M., Skelton D., Satake N., Halene S., Kohn D. (1999) Immune response to green fluorescent protein: implications for gene therapy. Gene Ther. 6, 1305–1312 [DOI] [PubMed] [Google Scholar]
- 6. Morris J. C., Conerly M., Thomasson B., Storek J., Riddell S. R., Kiem H. P. (2004) Induction of cytotoxic T-lymphocyte responses to enhanced green and yellow fluorescent proteins after myeloablative conditioning. Blood 103, 492–499 [DOI] [PubMed] [Google Scholar]
- 7. Jeon Y. H., Choi Y., Kang J. H., Kim C. W., Jeong J. M., Lee D. S., Chung J. K. (2007) Immune response to firefly luciferase as a naked DNA. Cancer Biol. Ther. 6, 781–786 [DOI] [PubMed] [Google Scholar]
- 8. Limberis M. P., Bell C. L., Wilson J. M. (2009) Identification of the murine firefly luciferase-specific CD8 T-cell epitopes. Gene Ther. 16, 441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Contag P. R., Olomu I. N., Stevenson D. K., Contag C. H. (1998) Bioluminescent indicators in living mammals. Nat. Med. 4, 245–247 [DOI] [PubMed] [Google Scholar]
- 10. Limberis M. P., Bell C. L., Heath J., Wilson J. M. (2010) Activation of transgene-specific T cells following lentivirus-mediated gene delivery to mouse lung. Mol. Ther. 18, 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raper S. E., Chirmule N., Lee F. S., Wivel N. A., Bagg A., Gao G. P., Wilson J. M., Batshaw M. L. (2003) Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 80, 148–158 [DOI] [PubMed] [Google Scholar]
- 12. Marshall E. (1999) Gene therapy death prompts review of adenovirus vector. Science 286, 2244–2245 [DOI] [PubMed] [Google Scholar]
- 13. Mingozzi F., Maus M. V., Hui D. J., Sabatino D. E., Murphy S. L., Rasko J. E., Ragni M. V., Manno C. S., Sommer J., Jiang H., Pierce G. F., Ertl H. C., High K. A. (2007) CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422 [DOI] [PubMed] [Google Scholar]
- 14. Manno C. S., Pierce G. F., Arruda V. R., Glader B., Ragni M., Rasko J. J., Ozelo M. C., Hoots K., Blatt P., Konkle B., Dake M., Kaye R., Razavi M., Zajko A., Zehnder J., Rustagi P. K., Nakai H., Chew A., Leonard D., Wright J. F., Lessard R. R., Sommer J. M., Tigges M., Sabatino D., Luk A., Jiang H., Mingozzi F., Couto L., Ertl H. C., High K. A., Kay M. A. (2006) Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347 [DOI] [PubMed] [Google Scholar]
- 15. Saridey S. K., Liu L., Doherty J. E., Kaja A., Galvan D. L., Fletcher B. S., Wilson M. H. (2009) PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol. Ther. 17, 2115–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yusa K., Zhou L., Li M. A., Bradley A., Craig N. L. (2011) A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. U. S. A. 108, 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doherty J. E., Huye L. E., Yusa K., Zhou L., Craig N. L., Wilson M. H. (2012) Hyperactive piggyBac gene transfer in human cells and in vivo. Hum. Gene Ther. 23, 311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mates L., Chuah M. K., Belay E., Jerchow B., Manoj N., costa-Sanchez A., Grzela D. P., Schmitt A., Becker K., Matrai J., Ma L., Samara-Kuko E., Gysemans C., Pryputniewicz D., Miskey C., Fletcher B., Vandendriessche T., Ivics Z., Izsvak Z. (2009) Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 41, 753–761 [DOI] [PubMed] [Google Scholar]
- 19. Suda T., Liu D. (2007) Hydrodynamic gene delivery: its principles and applications. Mol. Ther. 15, 2063–2069 [DOI] [PubMed] [Google Scholar]
- 20. Nathwani A. C., Tuddenham E. G., Rangarajan S., Rosales C., McIntosh J., Linch D. C., Chowdary P., Riddell A., Pie A. J., Harrington C., O'Beirne J., Smith K., Pasi J., Glader B., Rustagi P., Ng C. Y., Kay M. A., Zhou J., Spence Y., Morton C. L., Allay J., Coleman J., Sleep S., Cunningham J. M., Srivastava D., Basner-Tschakarjan E., Mingozzi F., High K. A., Gray J. T., Reiss U. M., Nienhuis A. W., Davidoff A. M. (2011) Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365, 2357–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goverdhana S., Puntel M., Xiong W., Zirger J. M., Barcia C., Curtin J. F., Soffer E. B., Mondkar S., King G. D., Hu J., Sciascia S. A., Candolfi M., Greengold D. S., Lowenstein P. R., Castro M. G. (2005) Regulatable gene expression systems for gene therapy applications: progress and future challenges. Mol. Ther. 12, 189–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vilaboa N., Voellmy R. (2006) Regulatable gene expression systems for gene therapy. Curr. Gene Ther. 6, 421–438 [DOI] [PubMed] [Google Scholar]
- 23. Rivera V. M., Clackson T., Natesan S., Pollock R., Amara J. F., Keenan T., Magari S. R., Phillips T., Courage N. L., Cerasoli F., Jr., Holt D. A., Gilman M. (1996) A humanized system for pharmacologic control of gene expression. Nat. Med. 2, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y., O'Malley B. W., Jr., Tsai S. Y., O'Malley B. W. (1994) A regulatory system for use in gene transfer. Proc. Natl. Acad. Sci. U. S. A. 91, 8180–8184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. No D., Yao T. P., Evans R. M. (1996) Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 93, 3346–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gossen M., Freundlieb S., Bender G., Muller G., Hillen W., Bujard H. (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769 [DOI] [PubMed] [Google Scholar]
- 27. Gossen M., Bujard H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U. S. A. 89, 5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szulc J., Wiznerowicz M., Sauvain M. O., Trono D., Aebischer P. (2006) A versatile tool for conditional gene expression and knockdown. Nat. Methods 3, 109–116 [DOI] [PubMed] [Google Scholar]
- 29. Deuschle U., Meyer W. K., Thiesen H. J. (1995) Tetracycline-reversible silencing of eukaryotic promoters. Mol. Cell. Biol. 15, 1907–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Racz Z., Godo M., Revesz C., Hamar P. (2011) Immune activation and target organ damage are consequences of hydrodynamic treatment but not delivery of naked siRNAs in mice. Nucleic Acid Ther. 21, 215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerullo V., Seiler M. P., Mane V., Cela R., Clarke C., Kaufman R. J., Pipe S. W., Lee B. (2007) Correction of murine hemophilia A and immunological differences of factor VIII variants delivered by helper-dependent adenoviral vectors. Mol. Ther. 15, 2080–2087 [DOI] [PubMed] [Google Scholar]
- 32. Nakazawa Y., Saha S., Galvan D. L., Huye L., Rollins L., Rooney C. M., Wilson M. H. (2013) Evaluation of long-term transgene expression in piggyBac-modified human T lymphocytes. J. Immunother. 36, 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu F., Song Y., Liu D. (1999) Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266 [DOI] [PubMed] [Google Scholar]
- 34. Gomez-Rodriguez J., Washington V., Cheng J., Dutra A., Pak E., Liu P., McVicar D. W., Schwartzberg P. L. (2008) Advantages of q-PCR as a method of screening for gene targeting in mammalian cells using conventional and whole BAC-based constructs. Nucleic Acids Res. 36, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woodard L. E., Li X., Malani N., Kaja A., Hice R. H., Atkinson P. W., Bushman F. D., Craig N. L., Wilson M. H. (2012) Comparative analysis of the recently discovered hAT transposon TcBuster in human cells. PLoS One 7, e42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bell J. B., Podetz-Pedersen K. M., Aronovich E. L., Belur L. R., Mcivor R. S., Hackett P. B. (2007) Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat. Protoc. 2, 3153–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Margolin J. F., Friedman J. R., Meyer W. K., Vissing H., Thiesen H. J., Rauscher F. J., III (1994) Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. U. S. A. 91, 4509–4513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frka K., Facchinello N., Del V. C., Carpi A., Curtarello M., Venerando R., Angelin A., Parolin C., Bernardi P., Bonaldo P., Volpin D., Braghetta P., Bressan G. M. (2009) Lentiviral-mediated RNAi in vivo silencing of Col6a1, a gene with complex tissue specific expression pattern. J. Biotechnol. 141, 8–17 [DOI] [PubMed] [Google Scholar]
- 39. Niwa H., Yamamura K., Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 [DOI] [PubMed] [Google Scholar]
- 40. Geurts A. M., Yang Y., Clark K. J., Liu G. Y., Cui Z. B., Dupuy A. J., Bell J. B., Largaespada D. A., Hackett P. B. (2003) Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol. Ther. 8, 108–117 [DOI] [PubMed] [Google Scholar]
- 41. Sebestyen M. G., Budker V. G., Budker T., Subbotin V. M., Zhang G., Monahan S. D., Lewis D. L., Wong S. C., Hagstrom J. E., Wolff J. A. (2006) Mechanism of plasmid delivery by hydrodynamic tail vein injection. I. Hepatocyte uptake of various molecules. J. Gene Med. 8, 852–873 [DOI] [PubMed] [Google Scholar]
- 42. Chu Q., Joseph M., Przybylska M., Yew N. S., Scheule R. K. (2005) Transient siRNA-mediated attenuation of liver expression from an alpha-galactosidase A plasmid reduces subsequent humoral immune responses to the transgene product in mice. Mol. Ther. 12, 264–273 [DOI] [PubMed] [Google Scholar]
- 43. Herweijer H., Wolff J. A. (2007) Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther. 14, 99–107 [DOI] [PubMed] [Google Scholar]
- 44. Fujinami R. S., von Herrath M. G., Christen U., Whitton J. L. (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin. Microbiol. Rev. 19, 80–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khanna A., Morelli A. E., Zhong C., Takayama T., Lu L., Thomson A. W. (2000) Effects of liver-derived dendritic cell progenitors on Th1- and Th2-like cytokine responses in vitro and in vivo. J. Immunol. 164, 1346–1354 [DOI] [PubMed] [Google Scholar]
- 46. LoDuca P. A., Hoffman B. E., Herzog R. W. (2009) Hepatic gene transfer as a means of tolerance induction to transgene products. Curr. Gene Ther. 9, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suda T., Gao X., Stolz D. B., Liu D. (2007) Structural impact of hydrodynamic injection on mouse liver. Gene Ther. 14, 129–137 [DOI] [PubMed] [Google Scholar]
- 48. Pastore L., Morral N., Zhou H., Garcia R., Parks R. J., Kochanek S., Graham F. L., Lee B., Beaudet A. L. (1999) Use of a liver-specific promoter reduces immune response to the transgene in adenoviral vectors. Hum. Gene Ther. 10, 1773–1781 [DOI] [PubMed] [Google Scholar]
- 49. Seiler M. P., Cerullo V., Lee B. (2007) Immune response to helper dependent adenoviral mediated liver gene therapy: challenges and prospects. Curr. Gene Ther. 7, 297–305 [DOI] [PubMed] [Google Scholar]
- 50. Beale E. G., Clouthier D. E., Hammer R. E. (1992) Cell-specific expression of cytosolic phosphoenolpyruvate carboxykinase in transgenic mice. FASEB J. 6, 3330–3337 [DOI] [PubMed] [Google Scholar]
- 51. Le G. C., Stieger K., Snyder R. O., Rolling F., Moullier P. (2007) Immune responses to gene product of inducible promoters. Curr. Gene Ther. 7, 334–346 [DOI] [PubMed] [Google Scholar]
- 52. Ginhoux F., Turbant S., Gross D. A., Poupiot J., Marais T., Lone Y., Lemonnier F. A., Firat H., Perez N., Danos O., Davoust J. (2004) HLA-A*0201-restricted cytolytic responses to the rtTA transactivator dominant and cryptic epitopes compromise transgene expression induced by the tetracycline on system. Mol. Ther. 10, 279–289 [DOI] [PubMed] [Google Scholar]
- 53. Yen L., Svendsen J., Lee J. S., Gray J. T., Magnier M., Baba T., D'Amato R. J., Mulligan R. C. (2004) Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature 431, 471–476 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.