

Abstract
Much of the morbidity following traumatic brain injury (TBI) is associated with traumatic axonal injury (TAI). Although most TAI studies focus on corpus callosum white matter, the visual system has received increased interest. To assess visual system TAI, we developed a mouse model of optic nerve TAI. It is unknown, however, whether this TAI causes retinal ganglion cell (RGC) death. To address this issue, YFP-16 transgenic mice were subjected to mild TBI and followed from 2 to 28 days. Neither TUNEL-positive or cleaved caspase-3 immunoreactive RGCs were observed from 2 to 28 days post-TBI. Quantification of immunoreactivity of Brn3a, an RGC marker, demonstrated no RGC loss; parallel electron microscopic analysis confirmed RGC viability. Persistent RGC survival was also consistent with the finding of reorganization in the proximal axonal segments following TAI wherein microglia/macrophages remained inactive. In contrast, activated microglia/macrophages closely enveloped the distal disconnected, degenerating axonal segments at 7 to 28 days post-injury, thereby confirming that this model consistently evoked TAI followed by disconnection. Collectively, these data provide novel insight into the evolving pathobiology associated with TAI that will form a foundation for future studies exploring TAI therapy and its downstream consequences.
Keywords: Microglia/macrophage, Retinal ganglion cell survival, Traumatic axonal injury, Traumatic brain injury, Visual system, YFP-16 transgenic mice
INTRODUCTION
Traumatic brain injury (TBI) is a major national and global health problem that can lead to death and serious neurological dysfunction. Much of the mortality and morbidity of TBI is associated with diffuse traumatic axonal injury (TAI) (1–4). Although previous studies of TAI have focused on the corpus callosum and rostral subcortical white matter, there is increased recognition that visual system white matter is equally sensitive to injury (5–10). This is reflected in both the military and civilian populations; for example, a study on the incidence of combat-related eye injuries by the Defense Veterans Brain Injury Center reporting that 80% of over 3,900 troops who had sustained TBI had some form of visual problem (11).
To date, few studies have addressed this issue in either animal models or humans to improve insight into the pathobiology of this visual damage and its overall functional implications. To address this issue, we have recently characterized a model of diffuse TAI in the mouse visual system in which we found diffuse/scattered axonal swellings, together with their disconnection, proximal and distal dieback and reorganization over time (12).
In view of the TAI observed in this system, an obvious question relevant to any future investigation centers on the fate of the cell bodies of origin of diffusely damaged optic nerve axons. Historically, it has been reported that most forms of damage to optic nerve result in retinal ganglion cell (RGC) loss. Complete intraorbital optic nerve transection has been shown to result in the loss of the majority of RGCs within 2 weeks (13–15). Dibas et al reported that after optic nerve crush injury in rats, the number of RGCs decreased by 47% at 7 days post-injury, reaching a 76% loss at 2 weeks post-injury (16). In a guinea pig optic nerve stretch model, a model assumed to replicate the important features of TAI, Mohammed Sulaiman et al observed a continued loss of RGCs beginning at 1 week and increasing over the next 12 weeks, with only 60% of RGCs remaining at 12 weeks post-TAI (17). In these studies they recognized that this cell loss was associated with RGC apoptosis, with the peak RGC apoptosis occurring at 3 weeks post-injury (17). In a mouse optic nerve stretch model, Saatman et al similarly reported that the number of RGCs decreased dramatically by over 50% by 1 day after stretch injury, a finding that was again confirmed at 2 weeks post-injury (18).
Based upon these studies, it would seem that retinal ganglion cell death would likely follow from diffuse TAI in the optic nerve. However, our previous work following diffuse traumatic injury in the optic nerve was not consistent with this premise (12). Specifically, we demonstrated that once damaged axons disconnected, the proximal segments underwent a rapid transition from an enlarged and swollen axonal profile to a thinned and truncated appendage (which we operationally defined as a club-shaped process) that differed from spheroid swellings on the distal detached segments. This transition was not related to evolving degeneration. Rather, it displayed features consistent with axonal reorganization and repair (12). Collectively, these observations in the proximal axotomized segment appeared consistent with an intact axon that maintained continuity with a viable RGC capable of continued protein synthesis and cellular function.
Moreover, in our previous studies evaluating neuronal cell bodies in the neocortex linked to diffuse axonal injury, we demonstrated that TAI did not result in either neocortical or thalamic neuronal death; rather, it triggered atrophic change (19–22). In these studies, it was assumed that the subtle axonal changes precipitated by TAI did not trigger the massive ionic influx and dysregulation associated with axonal transection or crushing, thereby protecting the neuronal cell body; this premise may also be applicable to diffusely injured optic nerves.
Because of the importance of these issues to understanding of the pathobiology and treatment of TAI in the visual system and because of the predominance of the literature suggesting that RGC loss follows from many forms of optic nerve damage, we evaluated the response of RGCs to diffuse TAI. Specifically, using multiple markers of neuronal cell death and qualitative and quantitative assessments of RGC integrity, we demonstrate that TAI in this model is not associated with ganglionic cell death. Further, to confirm that this model system always generated parallel optic nerve damage, we probed the optic nerve of the same injured animals with antibodies to microglia/macrophages. The premise was that optic nerve damage and fiber disconnection would be confirmed by the presence of microglia/macrophage activation and fiber engulfment in the distal disconnected optic nerve segments. Similarly, we posited that if the RGCs remained intact, the proximal disconnected segments would remain viable and not evoke comparable activation with fiber engulfment. Through this approach we recognized that microglia/macrophage activation and alteration were consistent findings with the microglia/macrophages differentially found in distal degenerating vs. proximal preserved axonal segments, consistent with the maintenance of RGC integrity.
MATERIALS AND METHODS
Mice
Thy1-YFP-16 transgenic mice express yellow fluorescent protein (YFP) under the promoter Thy1 within the retina and the optic nerve fibers (23). The mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and were bred, genotyped and maintained as heterozygotes, as previously described (12). All protocols used in this study were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Animal Surgery and Perfusion
Adult 2-month-old Thy1-YFP-16 mice (20–25 g) were subjected to mild central fluid percussion injury (1.40 ± 0.05 atmospheres), as previously described (12, 24). The fluid pressure pulse in this model resulted in a brief deformation of the brain and induced diffuse TAI in the optic nerve approximately 1 mm proximal to the chiasm (12). Sham-injured animals received the same surgeries, with the absence of the injury. Because virtually all published work evaluating the RGC response to optic nerve injury have been performed within the first 30 days post-injury and major RGC loss has been detected within the first 3 weeks post-injury (16–18), the mice were allowed to survive for 2 days, 7 days, 14 days and 28 days post-injury. At these survival times, animals were killed via an intraperitoneal overdose of sodium pentobarbital and then transcardially perfused with 100 ml heparinized normal saline, followed by 200 ml 4% paraformaldehyde in Millonig’s buffer. For electron microscopy (EM) analysis, animals were perfused with 200 ml 2% paraformaldehyde and 2.5 % glutaraldehyde in Millonig’s buffer. At each time point, 9 animals were injured; 2 animals were prepared for retinal whole mount observation to allow for a qualitative assessment of any potential loss of YFP-expressing cells in the RGC layer; 5 mice were prepared for immunocytochemical processing for markers of cell death while also allowing for the quantitative assessment of any potential RGC loss; 2 mice were prepared for EM analysis to identify any potential neuronal damage or loss in the RGC layer. The injured animals were prepared together with 6 sham-injured mice per time point (1 for retinal whole mount observation, 3 for qualitative immunocytochemistry and quantitative assessment, and 2 for EM).
Retinal Whole Mount Preparation
Following transcardial perfusion, the extraocular muscles were cut from the globe, the optic nerve was transected immediately behind the globe, and the eye was dissected from the orbit. The cornea was punctured with an 18-gauge needle, cut into 4 quadrants, and opened to the limbus using 2 sets of forceps. The lens was removed and the retina was then separated from the pigmented layer, and immersed in 4% paraformaldehyde in Millonig’s buffer for 4 hours. Next, the retina was washed thoroughly with phosphate buffered saline (PBS) 5 × 10 minutes. The retina was then cut into 4 quadrants and mounted flat on a glass slide with Vectashield Hardset Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA), and cover-slipped for image capture with a Leica TCS-SP2 AOBS confocal microscope (Wetzlar, Germany).
Retinal Cryostat Section Preparation
After the retina was separated from the pigmented layer as described above, fixation was continued in 4% paraformaldehyde in Millonig’s buffer overnight. After washing in PBS 5 × 10 min, retinas were immersed in 30% sucrose in PBS overnight. The pair of retinal cups from the same animal was then embedded side-to-side and frozen to −20°C in optimal cutting temperature compound (Electron Microscopy Sciences, Cat No.62550–01, Hatfield, PA). Twelve-μm-thick sections were then cut with a cryotome (Shandon Scientific Ltd., Cheshire, UK). Serial sections were collected on Superfrost/Plus microscopic glass slides (Fisher Scientific, catalog no.12-550-15, USA), with 4 serial sections of each eye on each slide for a total of 8 sections on each slide. Approximately 60 slides were collected for each animal and stored at −20°C until further use.
Optic Nerve Preparation
After the animal perfusion and overnight incubation in the perfusion fixative solution, the optic nerve, chiasm and optic tract, together with the brain were removed from the skull. The optic nerves were blocked to include the entire optic nerve, the chiasm, the initial part of the optic tract, and related brain tissue, and post-fixed in the same fixative solution overnight. The optic nerve blocks were then cryoprotected and cut longitudinally into 10-μm sections. Serial sections revealing the entire length of the optic nerve were then collected in a serial order in Millonig’s buffer in 24-well culture plates (Falcon, Newark, DE), and stored at 4°C.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling
To detect nuclear fragmentation within RGCs, 24 frozen retinal sections on 3 slides (8 sections each slide) were collected from each injured and sham injured animal. Of the 3 slides, 1 was harvested from the center of the retina with other 2 slides flanking this central slide by 500 μm. Thus, the slides contained sections through both the central and peripheral retina. These sections were processed using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit (ApoTag In Situ Apoptosis Detection Kit; S7101, S7160 and S7165, Millipore, Billerica, MA), using modification of the manufacturer’s protocol. Frozen retinal sections were obtained and warmed to room temperature (RT) for 30 minutes. They were then rinsed 2 × 5 minutes in PBS, and post-fixed in precooled ethanol: acetic acid (2:1) solution for 5 minutes at −20°C in a humidified chamber. After washing 2 × 5 minutes in PBS, the sections were incubated in 50 μg/ml proteinase K (Invitrogen, Cat No. 25530-015, Billerica, MA) in PBS for 30 minutes at RT. The sections were then washed 2 × 5 minutes in PBS, and incubated in the proprietary equilibration buffer for 30 minutes at RT. After clearing the equilibration buffer, the sections were incubated with working strength terminal deoxynucleotidyl transferase (TdT) enzyme in a humidified chamber for 1.5 hour at 37°C. The slides were then rinsed by the proprietary stop/wash buffer 2 × 10 minutes at RT, and washed in PBS 3 × 5 minutes. The sections were then incubated in the mouse anti-Digoxigenin (1:250, Roche, Cat. No. 11333062910) at 4°C in a humidified chamber overnight. After rinse in PBS 4 × 5 minutes, the sections were incubated with secondary antibody Alexa Fluor 594-conjugated goat anti-mouse IgG (1:250, Invitrogen) for 1 hour at RT. Then the slides were washed 4 × 2 minutes in PBS, cover-slipped with Vectashield Hardset Mounting Medium with DAPI (Vector Laboratories) for confocal microscopic analysis. Sham-injured retinal sections pretreated with deoxyribonuclease (DNAase, 1,000 U/ml) at RT for 10 minutes were used as a positive control; TdT enzyme treatment was omitted as a negative control.
Cleaved-Caspase-3 Immunohistochemistry
Frozen retinal slides adjacent to those prepared for TUNEL staining (24 sections on 3 slides per animal) were selected for cleaved-caspase-3 immunofluorescent staining (25–27). Sections were warmed to RT for 30 minutes and then rinsed 3 × 10 minutes in PBS. Following pretreatment with 10% normal goat serum (NGS) with 2% bovine serum albumin (BSA) and 0.5% Triton X in PBS at RT for 2 hours they were incubated overnight at 4°C rabbit anti-cleaved-caspase-3(Asp175) (1:500, Cell Signaling Technology, Cat. no. 9661) in 10% NGS with 2% BSA and 0.5% Triton X in PBS. The sections were washed 6 × 10 minutes with 1% NGS with 1% BSA and 0.2% Triton X in PBS at RT, followed by incubation with the secondary antibody Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:250) in 1% NGS with 1% BSA and 0.2% Triton X in PBS for 2 hours at RT. Then the sections were washed 4 × 5 minutes in PBS and 2 × 5 minutes in 0.1 M sodium phosphate buffer, and cover-slipped with Vectashield Hardset Mounting Medium with DAPI (Vector Laboratories) for confocal microscopic analysis. To generate a positive control, retinal sections from neonatal P2 mouse pups were utilized because apoptosis predominates during this developmental time frame.
Brn3a Immunohistochemistry
Semiquantitative evaluation of the number of RGC was performed using an anti-Brn3a antibody, which specifically detects rodent RGCs (28, 29). This approach provided advantages over routine YFP analysis in that, unlike the YFP (which can be found in RGCs as well as amacrine cells [23]), Brn3a is confined to RGCs (30). For Brn3a immunostaining, 3 slides containing 24 sections through both the central as well as the peripheral retina adjacent to those utilized for TUNEL and Caspase-3 immunostaining were employed. The slides were warmed to RT, rinsed as in PBS and pretreated as described above. Endogenous mouse IgG was blocked by anti-mouse Ig reagent (Mouse on Mouse Kit, Vector Laboratories). The sections were then washed 3 × 10 minutes in 10% NGS with 2% BSA and 0.5% Triton X in PBS, and incubated overnight at 4°C with the mouse anti-Brn3a monoclonal antibody (1:125, Millipore, MAB1585) in 10% NGS with 2% BSA and 0.5% Triton X in PBS. The sections were then washed 6 × 10 minutes with 1% NGS with 1% BSA and 0.2% Triton X in PBS at RT, followed by incubation with the secondary antibody Alexa Fluor 568-conjugated goat anti-mouse IgG (1:500) in 1% NGS with 1% BSA and 0.2% Triton X in PBS for 2 hours at RT. The sections were then washed 4 × 5 minutes in PBS and 2 × 5 minutes in 0.1 M sodium phosphate buffer, and cover-slipped with Vectashield Hardset Mounting Medium with DAPI (Vector Laboratories) for epifluorescent microscopic analysis. A Nikon Eclipse 800 microscope (Tokyo, Japan) fitted with an Olympus DP71 digital camera (Olympus, Center Valley, PA) was utilized with the appropriate excitation/emission filters.
Phosphorylated c-Jun Immunohistochemistry
Based on our recent experience with neocortical axonal injury (19), we employed antibodies to phospho-c-Jun. Three slides adjacent to those slides used for TUNEL, cleaved-caspase-3 staining and Brn3a staining, were chosen from each animal for immunostaining with polyclonal rabbit anti-phospho-c-Jun (Ser63) (1:100; Cat. no. 9261, Cell Signaling, Danvers, MA), followed by the secondary antibody Alexa Fluor 633 or Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:200). The same protocol for Brn3a immunostaining was utilized, except for the replacement of PBS by Tris-buffered saline. Adjacent slides were chosen for Brn3a and phospho-c-Jun double labeling and observation under confocal microscopy.
Microglia/Macrophage Immunohistochemistry
The optic nerves from the same animals utilized to evaluate the fate of RGCs, were also used to study the relationship between YFP-linked axonal dieback and activation of microglia/macrophages at the sites of injury. Labeling of the optic nerves was performed utilizing an antibody targeting the microglia/macrophage population (Iba1). Although not a specific marker for activated microglia, when coupled with their morphological appearance it was possible to determine if these immunoreactive microglia were in either an activated or inactive/resting state (31–33). YFP fluorescent optic nerve sections from 2 days to 28 days post-injury were selected and rinsed 3 × 10 minutes in PBS and pretreated with 10% NGS with 2% BSA and 0.5% Triton X in PBS at RT for 2 hours. Endogenous mouse IgG was blocked using the Mouse on Mouse Kit reagent. The sections were then washed 3 × 10 minutes in 10% NGS with 2% BSA and 0.5% Triton X in PBS, and then incubated overnight at 4°C with rabbit anti-Iba1 (1:1000; Zymed, South San Francisco, CA). Next, the sections were washed 6 × 10 minutes with 1% NGS with 1% BSA and 0.2% Triton X in PBS at RT, followed by incubation with the secondary antibody Alexa 568-conjugated goat anti-rabbit IgG (1:500). The sections were then washed 4 × 5 minutes in PBS and 2 × 5 minutes in 0.1 M sodium phosphate buffer, and cover-slipped as described above. Three-dimensional reconstructions were obtained with the Volocity Software package (Perkin Elmer; Waltham, MA).
EM
Retinas from paraformaldehyde/glutaraldehyde-perfused animals were immersed in 2% paraformaldehyde and 2.5% glutaraldehyde in Millonig’s buffer overnight, together with the brain and the optic nerve. The retinas were osmicated in 2 % OsO4 in 0.1 M sodium phosphate buffer, dehydrated in alcohol with 1% uranyl acetate, embedded in epoxy resin (Embed-812; Electron Microscopy Sciences), mounted on plastic slides (Thomas Scientific Co., Swedesboro, NJ) and cover-slipped. After resin curing, the plastic slides were examined via routine light microscopy to identify RGCs, using a Nikon Eclipse E800 microscope fitted with an Olympus DP71 digital camera. Once identified, these sites were excised, mounted on plastic studs and thick-sectioned to the RGC layer using an ultramicrotome (Leica Ultracut R; Leica, Vienna, Austria). Serial 40-nm sections were cut and mounted onto Formvar-coated single-slotted grids. The grids were then stained with 5% uranyl acetate in 50% methanol for 2 minutes and 0.5% lead citrate for 1 minute and visualized using a JEM 1230 electron microscope (JEOL Ltd., Tokyo, Japan).
Quantification of RGCs and Statistical Analysis
To quantify the number of RGCs, the Brn3a-labeled retinal sections were captured using routine epifluorescent microscopy. Quantifications of RGCs were accomplished utilizing cross sections (34, 35). Specifically, retinas from each mouse were serially sectioned as described above (8 sections/slide). Approximately 60 slides were collected as described above and we analyzed 24 widely dispersed sections per mouse. Each Brn3a-immunoreactive retinal section was digitally acquired at 20 x magnification moving from side to side of the retina covering its entire extent, parallel to its curvature. Each retinal section was reconstructed using 7 to 9 images taken along the extent of the retina. To quantify RGCs, the central images were selected together with the other next adjacent image from both sides. For each selected image, the number of Brn3a positive RGCs was counted in a 0.05-mm2 (0.435 mm × 0.115 mm) grid expanding along the image parallel to the alignment of the RGCs. This process of acquisition and counting was repeated for each of the retinal sections on the slide for a total of 72 retinal images per animal (3 images/section x 8 sections/slide x 3 slides/animal = 72 images/animal). For each animal, the number of Brn3a-positive RGCs was calculated from the number of Brn3a-positive RGCs from these 72 images, expressing it as mean ± SD. Five injured animals and 3 sham injured animals were included for each time point. To test the difference of the number of Brn3a-positive RGCs over time post-injury, one-way ANOVA was conducted.
RESULTS
Whole Mount Retinal YFP Assessment following Optic Nerve TAI
The YFP expressing retinal whole mounts were well preserved; RGC axons converged towards the optic disc and showed no evidence of axonal swelling. In the injured animals, the YFP-positive cells in the RGC layer from both the central retina and its more peripheral regions did not reveal any evidence of overt loss or change in fluorescent intensity from 2 days, 7 days to 14 days and 28 days post-TBI as compared to retinas harvested from 2 day sham animals (Fig. 1).
Figure 1.
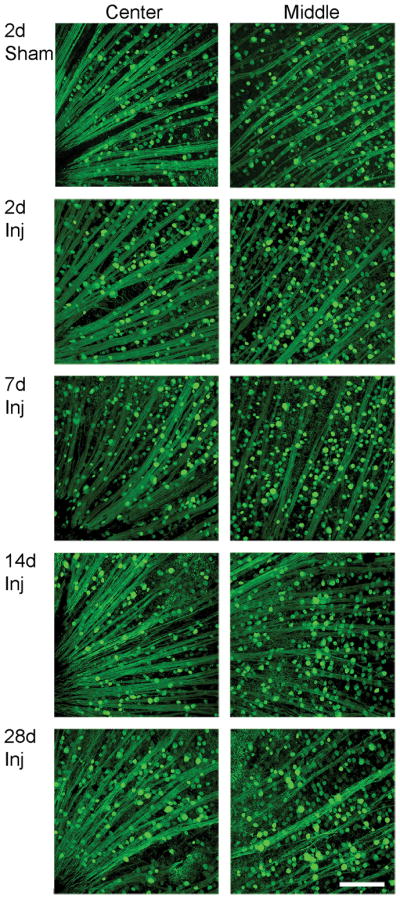
No overt loss of yellow fluorescent protein (YFP)-positive cells in the retinal ganglion cell (RGC) layer over time post-injury. These retinal whole mounts demonstrate similar YFP distribution in the sham and injured (Inj) animals at 2, 7, 14 and 28 days post-traumatic brain injury. The axons in the retina remain intact without any axonal swellings. The center and middle regions of the retina reveal no overt loss of YFP-positive cells in the RGC layer (the same layer as RGC axons) over time after injury. Scale bar: 100 μm.
TUNEL and Caspase 3 Assessments in the RGC Layer
To assess the potential for axonal damage-induced death of RGCs, the TUNEL method was utilized to identify nuclear fragmentation within the RGC layer. Positive control retinal sections from DNase-treated adult mouse YFP retinal sections demonstrated nuclear staining in the different layers of the retina (Fig. 2). In retina from sham-injured animals, no TUNEL-positive cells were observed in the RGC layer (data not shown) or at any time point from 2 days to 28 days post-injury (Fig. 2).
Figure 2.

Negative TUNEL staining of retinal ganglion cells from 2 days to 28 days post-traumatic brain injury (Inj); the control section treated with DNAase shows positive TUNEL staining in multiple retinal layers. Scale bar: 50 μm.
Caspase-3 immunostaining was also used to identify apoptosis in the RGC layer after axonal damage. Positive control retinal sections from P2 mice pups demonstrated positive immunostaining for cleaved caspase-3 (Fig. 3). In retinas from sham-injured animals, no immunostaining for cleaved caspase-3 was observed in the RGC layer (data not shown) or within the RGC layer from 2 days to 28 days post-TBI (Fig. 3).
Figure 3.

Absence of cleaved caspase-3 expression in the retinal ganglion cell layer from 2 days to 28 days post-injury. The control retinal section from a P2 pup shows positive staining for cleaved caspase-3 in several retinal layers. Scale bar: 100 μm.
Quantitative Analysis of RGCs
Quantitative analyses utilizing Brn3a immunostaining revealed a subtle time-dependent decrease of RGCs in both sham-injured and TBI-injured animals from 7 days to 28 days post-injury (Fig. 4C). Notably, there were no significant decreases in numbers of RGCs at each time point post-injury. At 7 days post-injury, 2620.6 ± 102.3 (mean ± SD) RGCs survived vs. 2610.7 ± 103.5 in the sham group; at 14 days post-injury, 2358.8 ± 148.1 RGCs survived vs. 2509.5 ± 65.8 in the sham group; at 28 days post-injury, 2378.8 ± 93.2 RGCs survived vs. 2465.5 ± 65.8 in the sham group. Despite the subtle loss of RGCs over time, the majority of RGCs survived at each time point post-injury, consistent with the TUNEL and caspase-3 immunostaining, which did not support dramatic RGC death following diffuse TAI of the optic nerve.
Figure 4.
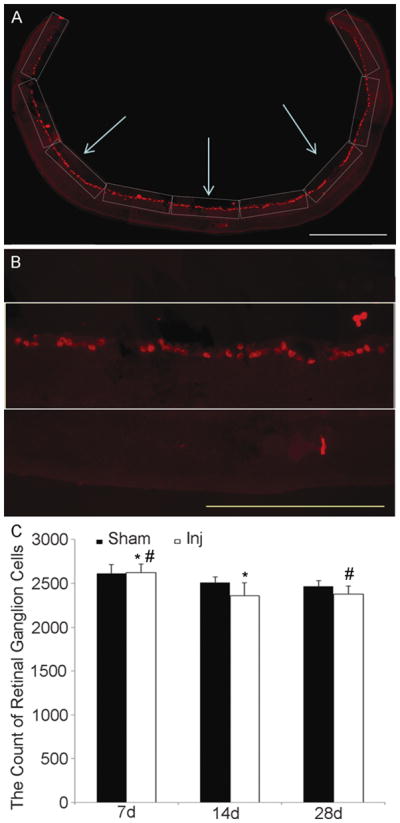
Analysis of retinal ganglion cell (RGC) number following traumatic axonal injury (TAI). (A) Images from side to side of the retinal section immunostained with anti-Brn3a antibody. (B) Placement of a grid (0.435 mm x 0.115 mm = 0.05 mm2) expanding along the image parallel to the alignment of the RGCs and covering all RGCs that were counted. (C) Quantitative assessment reveals a subtle time-dependent decrease of RGCs in both sham and injured animals from 7 days to 14 days and 28 days post-injury (* p < 0.05, # p < 0.05). At each time point there is no difference in numbers of RGC between the sham and the injured groups. Scale bars: A, 500 μm; B, 200 μm.
phospho-c-Jun Expression
We also employed an antibody to phospho-c-Jun, which has been linked to neocortical neuronal survival and axonal regeneration following diffuse TAI (19). We observed that phospho-c-Jun immunostaining was found specifically within the RGCs, based upon its colocalization with Brn3a (Fig. 5). phospho-c-Jun-positive RGCs were interspersed with other RGCs lacking phospho-c-Jun expression. Phospho-c-Jun expression in RGCs persisted from 2 days to 28 days post-TBI (Fig. 6), consistent with RGC survival.
Figure 5.

Expression of phospho-c-Jun, a regulator of neuronal repair and regeneration, in the retinal ganglion cell (RGC) layer colocalizes with Brn3a-positive RGCs (arrows). There are phospho-c-Jun-positive RGCs (purple) scattered among adjacent phospho-c-Jun-negative RGCs. Scale bar: 50 μm.
Figure 6.
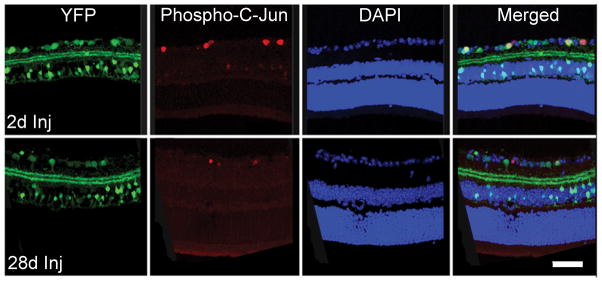
Yellow fluorescent protein (YFP)-positive retinal ganglion cell (RGC) layer with concomitant labeling with immunostaining for phospho-c-Jun from 2 days to 28 days post-injury (Inj). Phospho-c-Jun-positive RGCs are scattered among other phospho-c-Jun-negative RGCs. Scale bar: 50 μm.
EM
Ultrastructural analysis of cells within the RGC layer undertaken from 7 days to 28 days post-injury confirmed the absence of overt cell death; throughout this period, no apoptotic or necrotic RGC profiles were seen (Fig. 7). Additionally, no evidence of chromatin clumping, nuclear eccentricity or increased neuronal electron density was found, again consistent with neuronal preservation. Although occasional mitochondrial dilation was observed, this dilation did not progress over time, suggesting that this was a fixation artifact (Fig. 7).
Figure 7.
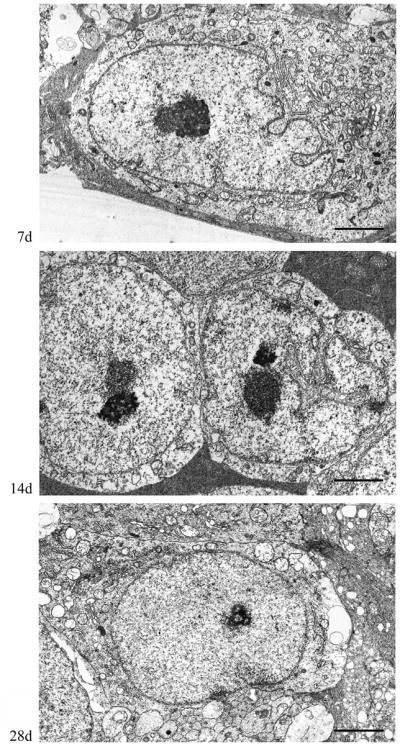
Retinal ganglion cell (RGC) ultrastructure at 7 days, 14 days and 28 days post-injury. At all time points, the RGCs show normal ultrastructural features characteristic of healthy neurons. Scale bar: 2 μm.
Microglia/Macrophages in Sham Injury
In the optic nerves of sham-injured animals, YFP-positive axons maintained continuity without any evidence of axonal swelling. Along the length of the optic nerve, no activation of microglia/macrophages was observed at any time post-injury (Fig. 8A). The microglia/macrophages demonstrated a resting morphology, indicated by in their small cell bodies and tightly ramified processes that ran parallel to rather than intact axons (Fig. 8B, C). These morphological features are consistent with a resting microglia phenotype, as observed in ex vivo brain slices (36).
Figure 8.

Microglia/macrophages show morphologic features of a resting state at 2 days after sham injury. (A) Microglia/macrophages are distributed throughout the length of the optic nerve. (B, C) Enlarged images of (A) show scattered microglia/macrophages with slender processes parallel to the normal yellow fluorescent protein (YFP)-expressing axons. (Blue: DAPI; YFP: axon; Red: microglia/macrophage). Scale bar: 300 μm in A.
Microglia/Macrophages at 2 Days Post-injury
Consistent with our previous report, by day 2 post-injury there was dramatic axonal damage that was readily observed in all optic nerves evaluated. Numerous YFP-positive swellings were identified with most seen in the early stages of disconnection and degeneration. In these preparations, activated microglia/macrophages were found scattered among diffusely injured axons within the optic nerve. They were largely confined to sites of initial axonal separation and degeneration and were easily identified in relation to YFP-positive axonal swellings (Fig. 9A). At this time post-injury, there was only limited direct microglia association with the disconnected, distal axonal segments (Fig. 9B). As described previously (12), the proximal axonal segments appeared truncated, apparently undergoing reorganization. These proximal swellings were not associated with or attached to the activated microglia/macrophage population (Fig. 9C).
Figure 9.

Activated microglia/macrophages in the optic nerve at 2 days post-traumatic brain injury (Inj). (A, B) Microglia/macrophages with an active morphology are prominent between injured axons at the site of initial axonal injury (arrow in A). Note that scattered microglia/macrophages with increased cytoplasm are adjacent to distal swollen axonal segments dying back towards the chiasm (B, arrowhead). (C) In proximal axonal segments, there is no association of microglia/macrophages and reorganizing axonal segments. (Blue: DAPI; yellow fluorescent protein (YFP): axon; Red: microglia/macrophage). Scale bar: A, 300 μm; B, C, 20 μm.
Microglia/Macrophages at 7 to 28 Days Post-injury
By 7 days post-injury, microglia/macrophage activation reflected in their amoeboid forms was dramatic and easily identified in the injured optic nerves (Fig. 10). The YFP-positive proximal axonal segments, which had truncated configurations (Fig. 11), retained an axonal profile distinct from the distal disconnected and degenerating axonal segments. In relation to the proximal axonal segments, the microglia/macrophages remained morphologically non-reactive, with their processes paralleling but not engulfing the axonal profiles (Fig. 11). This phenotype was maintained up to 28 days post-injury.
Figure 10.
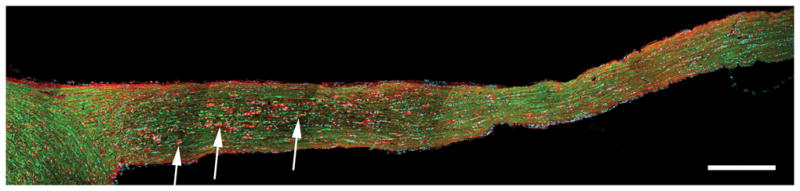
Activated microglia/macrophages predominate in the optic nerve at 7 days post-injury. Activated microglia/macrophages appear to attach and engulf distal, disconnected degenerating axonal segments (arrow). (Blue: DAPI; YFP: axon; Red: microglia/macrophage). Scale bar: 300 μm.
Figure 11.
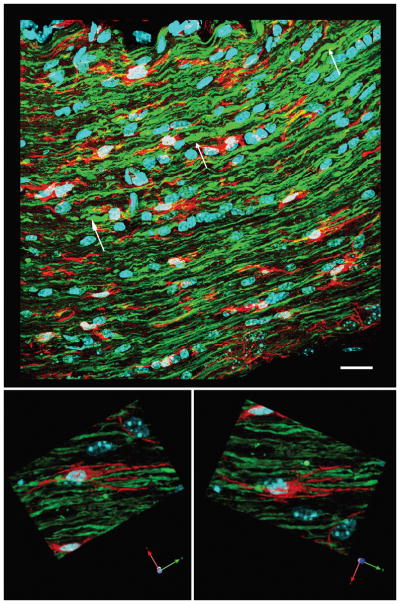
Microglia/macrophages appear less activated in the proximal axonal segments of the optic nerve at 7 days post-injury. The top image shows truncated proximal axonal segments (white arrows), consistent with their reorganization. Microglia/macrophages are present between these proximal axonal segments. At a higher magnification in the lower images, the microglia/macrophages (red) adjacent to the abnormal axons maintain a resting state with processes paralleling the alignment of the axonal fibers. Scale bar in upper panel: 20 μm.
In contrast to the reorganization of the proximal axonal segments, the distal disconnected axonal segments retained their rounded/spheroidal shape, and in relation to these swellings the microglia/macrophages revealed dramatic activation, i.e. their cell bodies were rounded (Fig. 12). These activated microglia/macrophages were in direct contact with the degenerating, distal axonal segments and their processes attached to and engulfed the swollen, distal axonal segments (Fig. 12). This microglia/macrophage activation persisted up to 28 days post-injury in relation to these distal disconnected, degenerating axonal segments, consistent with their role in clearing the degenerating axonal debris generated by Wallerian degeneration.
Figure 12.
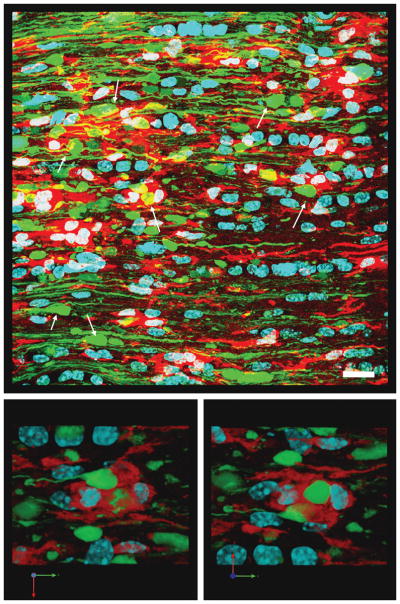
Activated microglia/macrophages appear to approach and engulf the distal, disconnected swollen axonal segments in the optic nerve at 7 days post-injury. The top image shows numerous persistent, disconnected distal swollen axonal segments (white arrows). Activated microglia/macrophages (red) are seen in this area and their processes contact and appear to engulf swollen axonal segments. The lower 2 images show at a higher magnification that the distal swollen axonal segments (yellow fluorescent protein-positive) appear to be enveloped by the surrounding activated microglia/macrophages. Scale bar in upper panel: 20 μm.
DISCUSSION
In this study, both qualitative (TUNEL and caspase-3 immunostaining) and quantitative (Brn3a immunostaining) analyses demonstrate no evidence of RGC death following diffuse TAI in the optic nerve. The parallel finding from 2 days to 28 days post-injury of the expression of phospho-c-Jun further argues for RGC survival rather than death; similarly, EM did not reveal RGC death. By contrast, the TAI consistently evoked optic nerve injury, indicated by dramatic microglia/macrophage activation in relation to diffuse axonal injury within portions of the optic nerve proximal to the chiasm. Importantly, while the distal axonal segments showed Wallerian change with microglia/macrophage engulfment, the proximal axonal segments remained intact with no evidence of progressive change or microglia/macrophage activation.
These findings stand in contrast to studies reporting that damage to optic nerve can result in dramatic RGC loss 1 to 3 weeks post-injury (13–18). The reasons for this different response to TAI in comparison to other models of optic nerve injury are not entirely clear but most likely reside in the diffuse nature of the injury, together with the major differences associated with the pathogenesis of axonal injury following diffuse traumatic vs. crush/transection injury. Optic nerve transection and crush constitute a primary axonal injury whereas TAI involves delayed axonal damage and disconnection over several hours (37). Primary axonal injury (transection) directly disrupts the axolemma, immediately exposing the injured axonal cylinder to the extracellular environment, resulting in sodium and calcium influx and the activation of proteases that then could cause further axonal damage progressing to neuronal death (38–42). RGC death observed following optic nerve transection or crush has been linked to the activation of the superoxide anion-initiated apoptotic signaling (43), caspase-3 induced apoptotic signaling (44), and tumor necrosis factor (45) pathways. Unlike optic nerve transection/crush injury, the central fluid percussion brain injury used in the current study induces acute transit acceleration-deceleration and compression of the optic nerve, resulting in diffuse TAI. This results in subtle, progressive changes in the axonal cylinder in the optic nerve and in other fiber systems (12, 37). These more subtle and progressive axonal changes do not lead to the massive ionic disruption associated with transection thereby permitting RGC survival.
In addition to the differences between optic nerve TAI and transection/crush injury, the survival of RGCs following diffuse TAI of the optic nerve, also differs from the RGC response to optic nerve stretch injury, which also typically evokes RGC death, involving necrotic and/or apoptotic pathways. This RGC response is most likely due to the mechanical stretch injury itself, during which the globe and optic nerve are stretched by a sling placed behind the globe (17, 18, 46–49). In this model, the force of stretch is applied directly to the globe, and then transferred from the globe to the optic nerve. This approach most likely injures both RGCs and the axons within the optic nerve via the mechanical forces acting on the retina, its intrinsic vasculature and downstream fibers. In contrast, with central fluid percussion injury, the fluid injury pulse impacts the dorsal cortex, causing its elastic deformation along a rostral caudal path to reach the brainstem (24). This deformation compresses the optic nerve transiently, stretching it, inducing the diffuse TAI found approximately 1 mm proximal to the chiasm (12). Importantly, in this model, the globe itself is shielded from the injury pulse, most likely sparing the retina while generating TAI in a more remote segment of the optic nerve than previously assessed by others.
In relation to this issue, the majority of optic nerve transection and crush lesions have been performed in the intraorbital segment of the optic nerve, which directly approximates the RGCs (16, 50, 51). Similarly with optic nerve stretch, the utilized sling triggered axonal damage in the proximal optic nerve segment, with the caveat that the proximity of the axonal injury to the RGC of origin may also be a confounding factor in the fate of the RGC cell body. Previous studies evaluating the survival of rubrospinal neurons following axonal transection at the brainstem vs. the C2 level (52, 53), and the survival of the corticospinal motor neuron after the intracortical axonal transection at different depth below layer V cortex (54) have shown that more neuronal sparing occurs as the distance between the neuronal somata and the downstream lesion increases, with proportionally more neuronal death occurring when the primary axonal lesion occurs adjacent to the neuronal cell body of origin (52–54). These results are consistent with our observations in more RGCs surviving TAI in the remote intracranial segment of the optic nerve, compared to less RGCs surviving transection/crush or stretch of the more proximal segment of the optic nerve.
While the above-described differences in animal modeling as well as our failure to demonstrate evidence of cell loss/death support our conclusions, confirmation of RGC survival is further supported by our immunocytochemical studies utilizing anti-phospho-c-Jun. Previous studies have demonstrated that the activation of the c-Jun pathway is associated with scattered RGC survival, axonal sprouting and regeneration in various models of optic nerve injury in fish and rodents (55–62). Furthermore, RGC expression of phospho-c-Jun is consistent with its expression in the neocortical neurons sustaining TAI, which also do not die (19). Importantly, ATF-3, another transcription factor associated with axonal regeneration, is activated in neocortical neurons following TAI (19).
One unexpected finding in the current study was is a subtle decrease of RGC number in both the sham and injured animals over time, as detected through the use of anti-Brn3a. Because no difference was found between the numbers of RGC in sham vs. injured animals at each time point, the subtle RGC loss in the injured group was not likely due to TAI. Recently, Comley et al demonstrated that the YFP fluorescent protein expression in thy1-YFP mice was not biologically inactive and, indeed, it upregulated multiple genes and proteins associated with various cell stress responses, such as DNA damage and repair, inflammation as well as oxidative or metabolic stress (63). The high neuronal expression of YFP in YFP-16 transgenic mouse also subtly increased the incidence of the neuronal morphological abnormality and altered the time-course of dying-back pathology (63). Thus, these effects of YFP fluorescent protein within the neurons might explain the subtle decrease of RGC in both the sham and injured animals.
Lastly, our finding of persistent RGC survival following optic nerve TAI is also consistent with our observation of the continued reorganization of the proximal axonal segments, wherein their related microglia/macrophages remained inactive. This was in stark contrast to the disconnected distal axonal segments, which exhibited degenerative changes first at 2 days post-injury, becoming dominant by 7 days, and persisting up to 28 days post-TBI. These varied responses in the distal vs. the proximal axonal segments most likely reflect the different signaling pathways involved in these 2 regions that translated to different microglia/macrophage responses.
The mechanisms underlining these different responses are most likely linked to the rapid resealing of the proximal axonal membrane and its retention of continuity with a viable RGC. Proximal axonal reorganization/repair has been routinely associated with a rapid resealing of the axolemmal membrane (64–67). Thus, such axonal reorganization and repair most likely does not trigger a reactive microglia/macrophage response. In contrast, the persistent activation of microglia/macrophages observed in the distal disconnect axonal segments is most likely linked to the persistent membrane disruption and the production of myelin debris during Wallerian degeneration (68, 69). Such microglia/macrophage activation is also consistent with the clearance of the degenerating axonal segments following brain and spinal cord injury (70–73). Irrespective of the factors involved in their activation, the robust activation of microglia/macrophages found in the current study support the premise of extensive axonal damage and disconnection.
While the current communication in the visual system significantly extends our understanding of TAI and its implication for concomitant retinal change, it also offers other potential applications and benefits. The preservation of the RGCs and their proximal axonal appendages in a relatively intact optic nerve offer an unprecedented opportunity for evaluating possible regeneration and repair. Further, the fact that the optic nerve itself is accessible to both blood-borne and CSF- applied therapies, this model may be valuable in multiple drug screening approaches targeting TAI. Accordingly, in light of the current findings on the preservation of the RGCs in response to TAI, our model system takes on increased utility and importance.
Acknowledgments
This study was supported by NIH grants HD055813 and NS047463.
The authors thank Dr. Robert T. Hamm, Dr. Scott Henderson, Susan A. Walker, Carol L. Davis and Jesse A. Sims for their invaluable assistance.
References
- 1.Christman CW, Grady MS, Walker SA, et al. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–86. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- 2.Li XY, Feng DF. Diffuse axonal injury: novel insights into detection and treatment. J Clin Neurosci. 2009;16:614–9. doi: 10.1016/j.jocn.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Li XY, Feng DF, et al. Biomarkers associated with diffuse traumatic axonal injury: exploring pathogenesis, early diagnosis, and prognosis. J Trauma. 2010;69:1610–18. doi: 10.1097/TA.0b013e3181f5a9ed. [DOI] [PubMed] [Google Scholar]
- 4.Zemlan FP, Rosenberg WS, Luebbe PA, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]
- 5.Ding Y, Yao B, Lai Q, et al. Impaired motor learning and diffuse axonal damage in motor and visual systems of the rat following traumatic brain injury. Neurol Res. 2001;23:193–202. doi: 10.1179/016164101101198334. [DOI] [PubMed] [Google Scholar]
- 6.Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg. 1994;80:301–13. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- 7.Gleckman AM, Evans RJ, Bell MD, et al. Optic nerve damage in shaken baby syndrome: detection by beta-amyloid precursor protein immunohistochemistry. Arch Pathol Lab Med. 2000;124:251–6. doi: 10.5858/2000-124-0251-ONDISB. [DOI] [PubMed] [Google Scholar]
- 8.Koliatsos VE, Cernak I, Xu L, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol. 2011;70:399–416. doi: 10.1097/NEN.0b013e3182189f06. [DOI] [PubMed] [Google Scholar]
- 9.Palmer HS, Garzon B, Xu J, et al. Reduced fractional anisotropy does not change the shape of the hemodynamic response in survivors of severe traumatic brain injury. J Neurotrauma. 2010;27:853–62. doi: 10.1089/neu.2009.1225. [DOI] [PubMed] [Google Scholar]
- 10.Smith DH, Dr, Hicks R, Povlishock J. Therapy Development for Diffuse Axonal Injury. J Neurotrauma. 2013;30:307–23. doi: 10.1089/neu.2012.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zampieri T. [Accessed July 28, 2012];Blinded veterans association testimony. 2007 Oct 4; Available at: http://www.eyeresearch.org/pdf/1004_zampieri_testimony.pdf.
- 12.Wang J, Hamm RJ, Povlishock JT. Traumatic axonal injury in the optic nerve: evidence for axonal swelling, disconnection, dieback, and reorganization. J Neurotrauma. 2011;28:1185–98. doi: 10.1089/neu.2011.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berkelaar M, Clarke DB, Wang YC, et al. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–74. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kielczewski JL, Pease ME, Quigley HA. The effect of experimental glaucoma and optic nerve transection on amacrine cells in the rat retina. Invest Ophthalmol Vis Sci. 2005;46:3188–96. doi: 10.1167/iovs.05-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peinado-Ramon P, Salvador M, Villegas-Perez MP, et al. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Invest Ophthalmol Vis Sci. 1996;37:489–500. [PubMed] [Google Scholar]
- 16.Dibas A, Oku H, Fukuhara M, et al. Changes in ocular aquaporin expression following optic nerve crush. Mol Vis. 2010;16:330–40. [PMC free article] [PubMed] [Google Scholar]
- 17.Mohammed Sulaiman A, Denman N, Buchanan S, et al. Stereology and ultrastructure of chronic phase axonal and cell soma pathology in stretch-injured central nerve fibers. J Neurotrauma. 2011;28:383–400. doi: 10.1089/neu.2010.1707. [DOI] [PubMed] [Google Scholar]
- 18.Saatman KE, Abai B, Grosvenor A, et al. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab. 2003;23:34–42. doi: 10.1097/01.WCB.0000035040.10031.B0. [DOI] [PubMed] [Google Scholar]
- 19.Greer JE, McGinn MJ, Povlishock JT. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci. 2011;31:5089–105. doi: 10.1523/JNEUROSCI.5103-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greer JE, Povlishock JT, Jacobs KM. Electrophysiological abnormalities in both axotomized and nonaxotomized pyramidal neurons following mild traumatic brain injury. J Neurosci. 2012;32:6682–7. doi: 10.1523/JNEUROSCI.0881-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lifshitz J, Kelley BJ, Povlishock JT. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol. 2007;66:218–29. doi: 10.1097/01.jnen.0000248558.75950.4d. [DOI] [PubMed] [Google Scholar]
- 22.Singleton RH, Zhu J, Stone JR, et al. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791–802. doi: 10.1523/JNEUROSCI.22-03-00791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng G, Mellor RH, Bernstein M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 24.Dixon CE, Lyeth BG, Povlishock JT, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–19. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–4. [PubMed] [Google Scholar]
- 26.Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem. 2002;50:449–54. doi: 10.1177/002215540205000401. [DOI] [PubMed] [Google Scholar]
- 27.Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 28.Galindo-Romero C, Avilés-Trigueros M, Jiménez-López M, et al. Axotomy-induced retinal ganglion cell death in adult mice: quantitative and topographic time course analyses. Exp Eye Res. 2011;92:377–87. doi: 10.1016/j.exer.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–8. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- 30.Xiang M, Zhou L, Macke JP, et al. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15:4762–85. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith PD, Bell MT, Puskas F, et al. Preservation of motor function after spinal cord ischemia and reperfusion injury through microglial inhibition. Ann Thorac Surg. 2013;95:1647–53. doi: 10.1016/j.athoracsur.2012.11.075. [DOI] [PubMed] [Google Scholar]
- 32.Viana LC, Lima CM, Oliveira MA, et al. Litter size, age-related memory impairments, and microglial changes in rat dentate gyrus: Stereological analysis and three dimensional morphometry. Neuroscience. 2013;238:280–96. doi: 10.1016/j.neuroscience.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 33.Bennett RE, Mac Donald CL, Brody DL. Diffusion tensor imaging detects axonal injury in a mouse model of repetitive closed-skull traumatic brain injury. Neurosci Lett. 2012;513:160–5. doi: 10.1016/j.neulet.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lefebvre JL, Zhang Y, Meister M, et al. Gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development. 2008;135:4141–51. doi: 10.1242/dev.027912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tezel G, Yang X, Yang J, et al. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996:202–12. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 36.Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–66. [PubMed] [Google Scholar]
- 37.Büki A, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–93. doi: 10.1007/s00701-005-0674-4. discussion 193–4. [DOI] [PubMed] [Google Scholar]
- 38.Emery DG, Lucas JH, Gross GW. Contributions of sodium and chloride to ultrastructural damage after dendrotomy. Exp Brain Res. 1991;86:60–72. doi: 10.1007/BF00231040. [DOI] [PubMed] [Google Scholar]
- 39.Leybaert L, de Hemptinne A. Changes of intracellular free calcium following mechanical injury in a spinal cord slice preparation. Exp Brain Res. 1996;112:392–40. doi: 10.1007/BF00227945. [DOI] [PubMed] [Google Scholar]
- 40.Lucas JH, Emery DG, Higgins ML, et al. Neuronal survival and dynamics of ultrastructural damage after dendrotomy in low calcium. J Neurotrauma. 1990;7:169–92. doi: 10.1089/neu.1990.7.169. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg LJ, Emery DG, Lucas JH. Effects of sodium and chloride on neuronal survival after neurite transection. J Neuropathol Exp Neurol. 2001;60:33–48. doi: 10.1093/jnen/60.1.33. [DOI] [PubMed] [Google Scholar]
- 42.Strautman AF, Cork RJ, Robinson KR. The distribution of free calcium in transected spinal axons and its modulation by applied electrical fields. J Neurosci. 1990;10:3564–75. doi: 10.1523/JNEUROSCI.10-11-03564.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Catrinescu MM, Chan W, Mahammed A, et al. Superoxide signaling and cell death in retinal ganglion cell axotomy: effects of metallocorroles. Exp Eye Res. 2012;97:31–35. doi: 10.1016/j.exer.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agudo M, Pérez-Marín MC, Lönngren U, et al. Time course profiling of the retinal transcriptome after optic nerve transection and optic nerve crush. Mol Vis. 2008;14:1050–63. [PMC free article] [PubMed] [Google Scholar]
- 45.Lukas TJ, Wang AL, Yuan M, et al. Early cellular signaling responses to axonal injury. Cell Commun Signal. 2009;7:5. doi: 10.1186/1478-811X-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gennarelli TA, Thibault LE, Tipperman R, et al. Axonal injury in the optic nerve: a model simulating diffuse axonal injury in the brain. J Neurosurg. 1989;71:244–53. doi: 10.3171/jns.1989.71.2.0244. [DOI] [PubMed] [Google Scholar]
- 47.Maxwell WL, Islam MN, Graham DI, et al. A qualitative and quantitative analysis of the response of the retinal ganglion cell soma after stretch injury to the adult guinea-pig optic nerve. J Neurocytol. 1994;23:379–92. doi: 10.1007/BF01666527. [DOI] [PubMed] [Google Scholar]
- 48.Maxwell WL, Graham DI. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma. 1997;14:603–14. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- 49.Serbest G, Burkhardt MF, Siman R, et al. Temporal profiles of cytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res. 2007;32:2006–14. doi: 10.1007/s11064-007-9318-9. [DOI] [PubMed] [Google Scholar]
- 50.Alarcón-Martínez L, Avilés-Trigueros M, Galindo-Romero C, et al. ERG changes in albino and pigmented mice after optic nerve transection. Vision Res. 2010;50:2176–87. doi: 10.1016/j.visres.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 51.Hu Y, Park KK, Yang L, et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73:445–52. doi: 10.1016/j.neuron.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu PH, Tsai HY, Chung YW, et al. The proximity of the lesion to cell bodies determines the free radical risk induced in rat rubrospinal neurons subjected to axonal injury. Anat Embryol (Berl) 2004;207:439–51. doi: 10.1007/s00429-003-0361-2. [DOI] [PubMed] [Google Scholar]
- 53.Liu PH, Wang YJ, Tseng GF. Close axonal injury of rubrospinal neurons induced transient perineuronal astrocytic and microglial reaction that coincided with their massive degeneration. Exp Neurol. 2003;179:111–26. doi: 10.1006/exnr.2002.8057. [DOI] [PubMed] [Google Scholar]
- 54.Dale SM, Kuang RZ, Wei X, et al. Corticospinal motor neurons in the adult rat: degeneration after intracortical axotomy and protection by ciliary neurotrophic factor (CNTF) Exp Neurol. 1995;135:67–73. doi: 10.1006/exnr.1995.1066. [DOI] [PubMed] [Google Scholar]
- 55.Fitzgerald M, Bartlett CA, Harvey AR, et al. Early events of secondary degeneration after partial optic nerve transection: an immunohistochemical study. J Neurotrauma. 2010;27:439–52. doi: 10.1089/neu.2009.1112. [DOI] [PubMed] [Google Scholar]
- 56.Fitzgerald M, Payne SC, Bartlett CA, et al. Secondary retinal ganglion cell death and the neuroprotective effects of the calcium channel blocker lomerizine. Invest Ophthalmol Vis Sci. 2009;50:5456–62. doi: 10.1167/iovs.09-3717. [DOI] [PubMed] [Google Scholar]
- 57.Herdegen T, Bastmeyer M, Bähr M, et al. Expression of JUN, KROX, and CREB transcription factors in goldfish and rat retinal ganglion cells following optic nerve lesion is related to axonal sprouting. J Neurobiol. 1993;24:528–43. doi: 10.1002/neu.480240410. [DOI] [PubMed] [Google Scholar]
- 58.Hüll M, Bähr M. Differential regulation of c-JUN expression in rat retinal ganglion cells after proximal and distal optic nerve transection. Neurosci Lett. 1994;178:39–42. doi: 10.1016/0304-3940(94)90284-4. [DOI] [PubMed] [Google Scholar]
- 59.Hüll M, Bähr M. Regulation of immediate-early gene expression in rat retinal ganglion cells after axotomy and during regeneration through a peripheral nerve graft. J Neurobiol. 1994;25:92–105. doi: 10.1002/neu.480250109. [DOI] [PubMed] [Google Scholar]
- 60.Lu Q, Cui Q, Yip HK, et al. c-Jun expression in surviving and regenerating retinal ganglion cells: effects of intravitreal neurotrophic supply. Invest Ophthalmol Vis Sci. 2003;44:5342–8. doi: 10.1167/iovs.03-0444. [DOI] [PubMed] [Google Scholar]
- 61.Robinson GA. Immediate early gene expression in axotomized and regenerating retinal ganglion cells of the adult rat. Brain Res Mol Brain Res. 1994;24:43–54. doi: 10.1016/0169-328x(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 62.Robinson GA. Axotomy-induced regulation of c-Jun expression in regenerating rat retinal ganglion cells. Brain Res Mol Brain Res. 1995;30:61–9. doi: 10.1016/0169-328x(94)00277-l. [DOI] [PubMed] [Google Scholar]
- 63.Comley LH, Wishart TM, Baxter B, et al. Induction of cell stress in neurons from transgenic mice expressing yellow fluorescent protein: implications for neurodegeneration research. PLoS One. 2011;6:e17639. doi: 10.1371/journal.pone.0017639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu K, Tedeschi A, Park KK, et al. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 2011;34:131–52. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- 65.Meiri H, Dormann A, Spira ME. Comparison of ultrastructural changes in proximal and distal segments of transected giant fibers of the cockroach Periplaneta americana. Brain Res. 1983;1263:1–14. doi: 10.1016/0006-8993(83)91195-2. [DOI] [PubMed] [Google Scholar]
- 66.Nehrt A, Rodgers R, Shapiro S, et al. The critical role of voltage-dependent calcium channel in axonal repair following mechanical trauma. Neuroscience. 2007;146:1504–12. doi: 10.1016/j.neuroscience.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca(2+)-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–67. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- 69.Brück W, Brück Y, Maruschak B, et al. Mechanisms of macrophage recruitment in Wallerian degeneration. Acta Neuropathol. 1995;89:363–67. doi: 10.1007/BF00309630. [DOI] [PubMed] [Google Scholar]
- 70.Busch SA, Horn KP, Silver DJ, et al. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009;29:9967–76. doi: 10.1523/JNEUROSCI.1151-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Busch SA, Horn KP, Cuascut FX, et al. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J Neurosci. 2010;30:255–65. doi: 10.1523/JNEUROSCI.3705-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horn KP, Busch SA, Hawthorne AL, et al. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–41. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Venkatesan C, Chrzaszcz M, Choi N, et al. Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation. 2010;7:32. doi: 10.1186/1742-2094-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]