

Abstract
Mutations in cardiac myosin binding protein C (cMyBP-C) are prevalent causes of hypertrophic cardiomyopathy (HCM). Although HCM-causing truncation mutations in cMyBP-C are well studied, the growing number of disease-related cMyBP-C missense mutations remain poorly understood. Our objective was to define the primary contractile effect and molecular disease mechanisms of the prevalent cMyBP-C E258K HCM-causing mutation in nonremodeled murine engineered cardiac tissue (mECT). Wild-type and human E258K cMyBP-C were expressed in mECT lacking endogenous mouse cMyBP-C through adenoviral-mediated gene transfer. Expression of E258K cMyBP-C did not affect cardiac cell survival and was appropriately incorporated into the cardiac sarcomere. Functionally, expression of E258K cMyBP-C caused accelerated contractile kinetics and severely compromised twitch force amplitude in mECT. Yeast two-hybrid analysis revealed that E258K cMyBP-C abolished interaction between the N terminal of cMyBP-C and myosin heavy chain sub-fragment 2 (S2). Furthermore, this mutation increased the affinity between the N terminal of cMyBP-C and actin. Assessment of phosphorylation of three serine residues in cMyBP-C showed that aberrant phosphorylation of cMyBP-C is unlikely to be responsible for altering these interactions. We show that the E258K mutation in cMyBP-C abolishes interaction between N-terminal cMyBP-C and myosin S2 by directly disrupting the cMyBP-C–S2 interface, independent of cMyBP-C phosphorylation. Similar to cMyBP-C ablation or phosphorylation, abolition of this inhibitory interaction accelerates contractile kinetics. Additionally, the E258K mutation impaired force production of mECT, which suggests that in addition to the loss of physiological function, this mutation disrupts contractility possibly by tethering the thick and thin filament or acting as an internal load.
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is a primary cardiac disease inherited in an autosomal dominant fashion (Spirito et al., 1997). HCM is the most prevalent cause of sudden cardiac death in apparently healthy young individuals (Fananapazir and Epstein, 1991), and is estimated to affect about one in five hundred people (Maron et al., 1995; Seidman and Seidman, 2001). Globally, mutations in cardiac myosin binding protein C (cMyBP-C), encoded by the human cardiac myosin binding protein C gene (MYBPC3) gene, are among the most prevalent causes of HCM, accounting for ∼34% of all mutations identified (Richard et al., 2003; Olivotto et al., 2008). To date, most recognized HCM-causing mutations in cMyBP-C are either frame-shift or splice-site mutations, resulting in the translation of a truncated protein. Evidence suggests that mutations of this type adversely affect sarcomeric function through a mechanism of haploinsufficiency (Niimura et al., 1998; Marston et al., 2009; Marston et al., 2012). Though slightly less common than truncation mutations, a growing number of HCM-causing single amino acid substitutions (missense mutations) in cMyBP-C have been identified (Olivotto et al., 2008; Page et al., 2012). Interestingly, missense mutations are frequently associated with early onset HCM (Morita et al., 2008), which indicates that missense mutations in MYBPC3 may have a profound functional consequence. However, the mechanisms through which mutations of this type alter sarcomeric function and lead to disease remain poorly understood.
The E258K mutation results in a substitution of the amino acid lysine for glutamic acid at position 258 in cMyBP-C. It is one of the most prevalent disease-causing mutations in MYBPC3, with 39 probands identified in 11 independent studies (Niimura et al., 1998; Alders et al., 2003; Nanni et al., 2003; Richard et al., 2003; Van Driest et al., 2004; Song et al., 2005; Ehlermann et al., 2008; Olivotto et al., 2008; Page et al., 2012). The E258K mutation is highly penetrant, with six out of seven mutation carriers in one well-studied family showing clinical symptoms of HCM (Niimura et al., 1998). The phenotype is often severe with a high incidence of HCM-related death occurring in carriers of the mutation (Niimura et al., 1998; Page et al., 2012). Although HCM is often a disease that manifests during adulthood, this mutation has also resulted in childhood onset of HCM (Morita et al., 2008). The E258K mutation affects the last residue of the C1 IgC2 domain, at the junction between C1 and the MyBPC motif (Fig. 1; Carrier et al., 1997; Niimura et al., 1998; Ababou et al., 2008; Govada et al., 2008).
Figure 1.
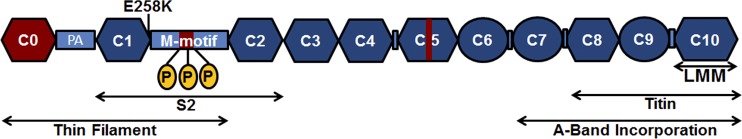
Diagrammatic representation of the domain structure of cMyBP-C. Ig domains are shown as hexagons, fibronectin-domains are shown as circles, and regions with a less organized domain structure are shown as light blue filled rectangles. Cardiac-specific regions are shown in red. Ig and fibronectin domains are numbered as C0–C10 from the N terminus to the C terminus. The positions of the three physiologically relevant phosphorylation sites (Ser275, Ser284, and Ser304) in the MyBPC motif (M-motif) are indicated by the yellow ovals. The position of the E258K mutation in the C1 domain, adjacent to the MyBPC motif is shown above the diagram. The binding sites for light meromyosin (LMM), myosin-S2 (S2), and titin, as well as the region essential for correct A-band incorporation and the region of N-terminal cMyBP-C implicated in interactions with components of the thin filament, are indicated by the double-headed arrows below the diagram.
Phosphorylation of three serine residues in the MyBPC motif (S275, S284, and S304) by PKA and other kinases (Gautel et al., 1995; Barefield and Sadayappan, 2010) causes the N-terminal domains (C1-C2) to dissociate from myosin heavy chain sub-fragment 2 (S2) and/or components of the thin filament (Gruen and Gautel, 1999; Gruen et al., 1999), thereby increasing the probability of cross-bridge formation and accelerating contractile kinetics (Gruen and Gautel, 1999; Gruen et al., 1999; Razumova et al., 2006; Shaffer et al., 2009; Barefield and Sadayappan, 2010; Kensler et al., 2011; Sadayappan et al., 2011). The highly conserved glutamic acid residue at position 258 of cMyBP-C forms part of a strongly acidic molecular structure at the junction of the C1 domain thought to constitute the MyBPC-C1–S2 interface (Ababou et al., 2008; Govada et al., 2008). Substitution of this acidic residue with a basic lysine residue may weaken or abrogate the interaction between C1 of cMyBPC and S2 (Ababou et al., 2008; Govada et al., 2008). Alternatively, because of the proximity of this mutation to phosphorylation sites in the MyBPC motif, the E258K mutation may alter cMyBP-C phosphorylation, indirectly disrupting the interaction between the N-terminal cMyBP-C and S2 (Niimura et al., 1998). Whether direct or indirect, disruption of the cMyBP-C–S2 interaction has been postulated to lead to accelerated contractile kinetics and an increase in twitch force, resulting from an increased probability of cross-bridge formation, similar to that observed after cMyBP-C phosphorylation or ablation (de Lange et al., 2011, 2013).
To explore the effects of the E258K mutation on contractility, we generated murine engineered cardiac tissue constructs (mECT) lacking endogenous mouse cMyBP-C in which we expressed human WT E258K cMyBP-C (knockout [KO] adWT and KO ad258 mECT). Consistent with the hypothesis that the E258K mutation disrupts cMyBP-C interaction with S2, contractile kinetics were significantly faster in KO ad258 than in KO adWT mECT. Yeast two-hybrid (Y2H) experiments confirmed that the E258K mutation decreased the affinity of N-terminal cMyBP-C for myosin S2. The observed acceleration in contractile kinetics did not correlate with relevant changes in cMyBP-C phosphorylation, which suggests that the E258K mutation has a direct effect on the cMyBP-C–S2 interaction. Surprisingly, KO ad258 mECT produced significantly less force than KO adWT mECT, which indicates that E258K may not cause disease through a simple loss-of-function mechanism.
MATERIALS AND METHODS
Animals
Ventricular tissue was harvested from 1-d-old homozygous cMyBP-C−/− mouse pups that were previously generated on the E129X1/SvJ background (Harris et al., 2002). Pups were anesthetized using inhaled isoflurane before harvesting of ventricular tissue. This study was approved by the Animal Care and Use Committee of the School of Medicine and Public Health at the University of Wisconsin-Madison in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23).
Isolation of neonatal mouse ventricular cardiac cells
Ventricular cardiac cells were isolated using enzymatic dissociation as described previously (de Lange et al., 2011). In brief, excised ventricular tissue was minced, suspended in 3 mg/ml collagenase type II (Gibco) in KG buffer, pH 7.4, and incubated at 37°C for 20 min with gentle agitation. KG buffer consisted of: 127 mmol/liter l-glutamic acid potassium salt monohydrate (Sigma-Aldrich), 0.1335% (wt/vol) NaHCO3 (Gibco), 16.5 mmol/liter d-glucose (Sigma-Aldrich), 0.42 mmol/liter Na2HPO4 (Sigma-Aldrich), and 25 mmol/liter HEPES (Sigma-Aldrich). Ventricular tissue was subsequently resuspended and agitated in 0.025% trypsin (Gibco) in KG buffer for 10-min intervals until dispersed. Cells were pelleted at 200 g and resuspended in mouse culture media consisting of: 60.3% high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco), 20% F12 nutrient mix (Gibco) supplemented with 1 mg/ml gentamicin (Sigma-Aldrich), 8.75% fetal bovine serum (HyClone), 6.25% horse serum (HyClone), 1% Hepes (Sigma-Aldrich), 1× nonessential amino acid cocktail (Gibco), 3 mmol/liter sodium pyruvate (Gibco), 0.00384% (wt/vol) NaHCO3 (Gibco), and 1 µg/ml insulin (Sigma-Aldrich). Cell suspensions were preplated into P100 cell culture dishes and incubated at 37°C for 45 min to allow preferential attachment of nonmyocyte cell populations and enrichment of cardiomyocyte population. Cardiac cells remaining in suspension were collected, checked for viability by dye-exclusion, counted, and prepared for subsequent ECT construction.
Generation of human WT MYBPC3-expressing adenovirus (adWT)
Generation of adWT was previously described in detail (de Lange et al., 2011). In brief, full-length human MYBPC3 cDNA in pCMV-SPORT6 (pCMV-MYBPC3) was obtained from Thermo Fisher Scientific. The full-length cMyBP-C open reading frame was subsequently PCR amplified using PfuUltra (Agilent Technologies) according to the manufacturer’s instructions using the following primer sequences: hMYBPC3 C0 Myc F, 5′-CACCATGGAACAAAAACTTATTTCTGAAGAAGATCTGATGCCTGAGCCGGGGAAG-3′; and hMYBPC3 C10R, 5′-TCACTGAGGCACTCGCACCTCCAGG-3′. During amplification, a Myc epitope tag was added in-frame with the MYBPC3 coding region, allowing for subsequent production of N-terminally Myc-tagged WT cMyBP-C. The 3,855-bp PCR product thus produced was subsequently cloned into the pENTR/D-TOPO entry vector (Invitrogen) according to the manufacturer’s instructions. After characterization by DNA sequencing, the Myc-tagged human cMyBP-C encoding inserts were subcloned into the pAd/CMV/V5-DEST adenoviral shuttle vector (a component of the ViraPower Adenoviral Expression System; Invitrogen), using the Gateway LR Clonase recombination system (Invitrogen) to form the pAdWT plasmid. adWT adenoviral particles were produced using the ViraPower Adenoviral Expression System (Invitrogen), according to the manufacturer’s instructions. In brief, the pAdWT plasmid was digested with PacI (New England BioLabs, Inc.) to expose the left and right viral inverted terminal repeat sequences before transfection into the 293A cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. Viral particles were harvested from 293A cells by repeated rapid freeze-thaw, 18 d after transfection. The crude, low-titer adWT-containing lysates were subsequently used to reinfect fresh 293A cells, from which adWT was harvested, purified, and titrated according to the manufacturer’s instructions (Invitrogen).
Generation of human E258K MYBPC3-expressing adenovirus (ad258)
In vitro mutagenesis was performed on pCMV-MYBPC3WT (Open Biosystems) using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer’s instructions, using the following E258K mutagenesis primers E258K F, 5′-CCAACTTCAATCTCACTGTCCACAAGGCCATGGGCACCG-3′; and E258K R, 5′-CGGTGCCCATGGCCTTGTGGACAGTGAGATTGAAGTTGG-3′; to generate pCMV-MYBPC3E258K. After in vitro mutagenesis, the full-length E258K cMyBP-C open reading frame was PCR amplified using PfuUltra (Agilent Technologies) according to the manufacturer’s instructions using the following primer sequences: hMYBPC3 C0 hemagglutinin (HA) F, 5′-CACCATGTACCCATACGACGTCCCAGACTACGCTATGCCTGAGCCGGGGAAG-3′; and hMYBPC3 C10R, 5′-TCACTGAGGCACTCGCACCTCCAGG-3′. During amplification, an HA epitope tag was added in-frame with the MYBPC3 coding region, allowing for subsequent production of N-terminally HA-tagged E258K cMyBP-C. The 3,855-bp PCR product thus produced was subsequently cloned into the pENTR/D-TOPO entry vector (Invitrogen) according to the manufacturer’s instructions. After characterization by DNA sequencing, the HA-tagged human E258K cMyBP-C encoding inserts were subcloned into the pAd/CMV/V5-DEST adenoviral shuttle vector (component of the ViraPower Adenoviral Expression System; Invitrogen), using the Gateway LR Clonase recombination system (Invitrogen) to form the pAd258 plasmid. ad258 adenoviral particles were produced using the ViraPower Adenoviral Expression System (Invitrogen), according to the manufacturer’s instructions. In brief, the pAd258 plasmid was digested with PacI (New England BioLabs, Inc.) to expose the left and right viral inverted terminal repeat sequences before transfection into the 293A cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Viral particles were harvested from 293A cells by repeated rapid freeze-thaw, 18 d after transfection. The crude, low-titer ad258-containing lysates were subsequently used to reinfect fresh 293A cells, from which ad258 was harvested, purified, and titrated according to the manufacturer’s instructions (Invitrogen).
mECT construction
cMyBP-C−/− neonatal mouse ventricular cells suspended in mouse media were rotated at 50 rpm on a gyratory shaker (195 mm diameter) for 6–8 h at 37°C and 5% CO2 to allow aggregation of small, uniform clusters of viable cells. For production of KO adWT and KO ad258 mECT, cMyBP-C−/− cardiac cells were transduced with adWT or ad258 at a multiplicity of infection (MOI) of 20 during the rotational culture period. After rotational culture, ∼8.0 × 105 CMs were suspended in 83.3 µl mouse media and added to 116.7 µl of an mECT matrix mixture consisting of 66.7 µl 2 mg/ml acid soluble rat-tail collagen type I, pH 3.0, (Sigma-Aldrich), 8.3 µl 10× MEM (Gibco), 8.3 µl reconstitution buffer (200 mmol/liter NaHCO3 [Gibco], 200 mmol/liter Hepes [Sigma-Aldrich], and 100 mmol/liter NaOH [Sigma-Aldrich]), and 33.3 µl Matrigel (BD). 200 µl of the cell/matrix suspension were cast into 20 mm × 3 mm cylinder constructs using a Flexcell Tissue Train silicone membrane culture plate (Flexcell International) and incubated under preprogrammed vacuum conditions for 120 min (37°C, 5% CO2) to form cylindrical-shaped mECT constructs that were attached at each end to fibrinous tabs. Upon matrix polymerization, mouse media was added to the mECT within the 6-well culture dish. mECT constructs were maintained in culture for 7 d with media changes every other day.
Adenoviral transduction of cardiac cells in 2D primary culture
To assess adenoviral transduction efficiency, cMyBP-C−/− neonatal mouse ventricular cardiac cells suspended in mouse media were plated in 12-well flat bottom tissue culture plates (Falcon; BD) at a density of 2 × 105 CMs per well. CMs were allowed to adhere to the surface of the dish for 48 h before transduction with adWT or ad258 at MOIs of 0, 5, 10, 20, 50, and 100. The adenovirus-containing media was removed and replaced with mouse culture media 24 h after transduction, and cells were cultured for an additional 48 h before harvesting of RNA or protein.
Mechanical testing of mECT
Isometric force generated by mECT was measured using protocols similar to those described previously (Tobita et al., 2006; de Lange et al., 2011). In brief, each mECT construct was transferred from the culture dish to a model 801B small intact fiber test apparatus (Aurora Scientific) in Krebs-Henseleit buffer (119 mmol/liter NaCl, 12 mmol/liter glucose, 4.6 mmol/liter KCl, 25 mmol/liter NaHCO3, 1.2 mmol/liter KH2PO4, 1.2 mmol/liter MgCl2, and 1.8 mmol/liter CaCl2, gassed with 95% O2/5% CO2, pH 7.4). mECT constructs were attached with sutures between a model 403A force transducer (Aurora Scientific) and a model 322C high-speed length controller (Aurora Scientific). mECT were perfused with 24°C Krebs-Henseleit buffer at a rate of 1 ml/min, and field stimulation was initiated at 2 Hz (2.5 ms, 12.5 V). The longitudinal length of each construct was increased stepwise until maximal twitch force (Fmax) was achieved. mECT were then equilibrated for 10–20 min or until a stable level of passive tension was achieved while being paced at 2 Hz. The pacing frequency was increased to 4 Hz and the temperature of the perfusate in the chamber was adjusted to 37°C before force measurements were made at stimulation frequencies of 6 Hz and 9 Hz. Data from force measurements were analyzed and averaged using IonWizard 6.0 software (IonOptix). For each stimulation frequency, the forces of 80 to 160 successive contractions were collected and averaged. Contraction data were exported to Excel (Microsoft) and the magnitude and kinetics of force generation were calculated.
MTT assay
To establish whether expression of E258K cMyBP-C in mouse cardiac cells adversely affected cell survival, cMyBP-C−/− cardiac cells were transduced with adWT and ad258 in 2D culture at MOIs of 0, 5, 10, 20, 50, and 100. The virus was removed and the media was changed after 24 h. After an additional 48 h in culture, the media was changed and an equal volume of 2 µg/ml 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma-Aldrich) diluted in 1× PBS was added to cells. Cells were subsequently incubated for 30 min at 37°C, after which an equal volume of cell lysis buffer (20% [wt/vol] SDS [Sigma-Aldrich] and 50% [vol/vol] dimethyl formamide [Thermo Fischer Scientific], pH 4.7) was added. Cell lysates were incubated at room temperature for 3–4 h, whereafter optical densities at 550 nm were measured.
RNA extraction
Total RNA was extracted from cardiac cells transduced with adWT or ad258 at MOIs of 0, 5, 10, 20, 50, and 100 in 2D culture and KO adWT or KO ad258 mECT using standard protocols. In brief, tissue/cells were homogenized in TRIZOL reagent (Invitrogen) according to the manufacturer’s instructions. After addition of an appropriate amount of chloroform (Sigma-Aldrich), mixing, incubation, and centrifugation according to the TRIZOL reagent protocol, the RNA containing aqueous phase was collected and treated with DNaseI (RNase-free DNase set; QIAGEN) for 1 h. RNA was subsequently purified using the RNeasy mini kit (QIAGEN) according to the manufacturer’s instructions.
DNA extraction
Genomic DNA was extracted from KO adWT or KO ad258 mECT using standard protocols. In brief, tissues were homogenized in TRIZOL reagent (Invitrogen). After phase separation and RNA extraction from the aqueous phase, DNA was extracted from the interphase according to the manufacturer’s instructions.
Protein analysis
Total protein lysates were prepared from adWT and ad258 2D cell cultures and mECT. In brief, cMyBP-C−/− cardiac cells in 2D culture transduced with adWT or ad258 were incubated in 37°C mouse media with and without 5 µM dobutamine (Hospira Inc.) for 5 min, then briefly washed in cold 1× PBS and lysed in RIPA isolation buffer. KO adWT and KO ad258 mECT were cut in half; one half was treated with dobutamine as described, the other half was placed immediately in RIPA buffer for protein isolation. Lysates were also prepared from ex vivo dobutamine-treated hearts excised from neonatal and 5-wk-old WT mice. Protein lysates were electrophoresed using precast 4–20% Criterion Gels (Bio-Rad Laboratories; 30 µg for lysates from cells in 2D culture and mECT, 1 µg for hearts). After electrophoresis, proteins were transferred to Immobilon-FL membranes (EMD Millipore) using standard techniques. Membranes were subsequently blocked for 1 h with Odyssey blocking buffer (LI-COR Biosciences) before overnight hybridization with primary specific antibodies. Membranes were incubated with a goat anti–cMyBP-C polyclonal antibody diluted at 1:200 (sc-50115; Santa Cruz Biotechnology, Inc.) that was able to detect both human and mouse cMyBP-C. Additionally, the three phosphorylation sites of cMyBP-C were detected with rabbit anti–phospho-Ser275, -Ser284 (ALX-215-057-R050; Enzo Life Sciences), and -Ser304 polyclonal antibodies diluted at 1:1,000 (anti–phospho-Ser275 and -Ser304 were provided by S. Sadayappan, Loyola University, Chicago, IL). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), used as a loading control, was detected with a rabbit anti-GAPDH polyclonal antibody diluted at 1:2,000 (G9545; Sigma-Aldrich). Primary antibodies were subsequently visualized by incubating the membranes for 1 h in donkey anti–goat IgG (Alexa Fluor 546; Life Technologies) or goat anti–rabbit IgG (Alexa Fluor 647; Life Technologies) immunofluorescent secondary antibodies diluted at 1:10,000 using a ChemiDoc MP imaging system (Bio-Rad Laboratories) according to the manufacturer’s instructions.
Immunohistochemistry
For immunohistochemistry, mECT constructs were rinsed 1 × 1 min in 0.1 mol/liter KCl to relax the sarcomeres and then 1 × 1 min in 1× PBS, pH 7.6, before being fixed in Dent’s fixative (80% methanol:20% DMSO) for 2 h at 4°C. Constructs were then dehydrated in methanol series and stored at −20°C for at least 24 h and up to 1 wk. For sectioning, fixed constructs were warmed to room temperature, transferred directly to xylenes, and allowed to equilibrate for 5 min before being placed in paraffin and maintained at 60°C for 90 min. They were subsequently embedded in fresh, hot (68°C) paraffin in disposable plastic molds and allowed to cool before being sectioned at 8 µm on a microtome (RM2165; Leica), mounted on Superfrost Plus glass sides (Thermo Fisher Scientific), and dried overnight on a flattening plate at 37°C. After drying, slides were heated to 68°C for 1 h, placed 2 × 5 min in xylenes, rehydrated through ethanol series to water, and rinsed twice in 1× PBS. Blocking to minimize nonspecific binding of antibodies was performed in 1× PBS, pH 7.6, containing 5% (vol/vol) sheep serum, 2 mg/ml bovine serum albumin, and 0.1% (vol/vol) Tween 20. Sections were incubated with 1:200 anti-cMyBPC rabbit polyclonal antibody (Harris et al., 2002) and anti-tropomyosin (CH-1) monoclonal IgG1 antibody (Developmental Studies Hybridoma Bank), 1:200 anti-cMyBPC rabbit polyclonal antibody (Harris et al., 2002) and 1:200 anti-Myc Tag mouse monoclonal IgG1 antibody (MMS-150R; Covance), or 1:200 anti-cMyBPC rabbit polyclonal antibody (Harris et al., 2002) and 1:200 anti-HA Tag rat IgG1 monoclonal antibody (11867423001; Roche) in PBS containing 0.1% (vol/vol) Tween 20 (PBST). After rinsing 2 × 2 min in PBST, sections were incubated in the dark with appropriate Alexa Fluor 488 and 568 antibodies (Molecular Probes) at a concentration of 1:200 in PBST. All antibody incubations were performed for 1 h at room temperature. After rinsing 2 × 2 min in PBST, sections were coverslipped using warmed ProLong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (DAPI). Imaging was performed using a fluorescent microscope (ECLIPSE 90i; Nikon) using a 40× oil-immersion objective lens and 405, 488, and 568 nm to excite DAPI and Alexa Fluor dyes, respectively. Images were acquired using NIS-Elements software suite (Nikon).
Y2H analysis
We performed Y2H analysis to study the effect of the E258K mutation in the C1 domain of cMyBP-C on its ability to interact with either the distal portion of myosin S2 or cardiac actin. Y2H was preferred over other biochemical analysis because it allows expression of distinct protein domains (a necessity in the current study given the size of cMyBP-C and MyHC, and the existence of multiple interactions between these proteins) in a eukaryotic system that is more likely to support appropriate domain folding than bacterial or in vitro expression systems used to express domains for techniques such as coimmunoprecipitation. Furthermore, it should be noted that interactions between N-terminal cMyBP-C and S2 and/or actin have been well-established using both Y2H and multiple other biochemical techniques. The C1C2 region of human cMyBP-C was PCR amplified using PfuUltra (Agilent Technologies) according to the manufacturer’s instructions from both pCMV-MYBPC3WT and pCMV-MYBPC3E258K using the following primers: cMyBPC pGBK C1FIn-Fusion, 5′-CATGGAGGCCGAATTCATGCCCATTGGCCTCTTCGTGA-3′; and cMyBPC pGBK C2RIn-Fusion, 5′-GCAGGTCGACGGATCCCTAGGGCTCTTTAAAGAGCT-3′. The 126 distal amino acids of S2 of human β myosin heavy chain were PCR amplified using PfuUltra (Agilent Technologies) according to the manufacturer’s instructions, amplified from pDONR-MYH7 using the following primers: MYH7 GAD S2FIn-Fusion, 5′-GGAGGCCATGGAATTCATGCCGCTGCTGAAGAGTGCAG-3′; and MYH7 GAD S2RIn-Fusion, 5′-CGAGCTCGATGGATCCCTATTTGGCCAGTGTCAGCTCCAG-3′. Full-length human α cardiac actin (ACTC1) was amplified from human cardiac cDNA using the following primers: ACTC1 GAD FIn-Fusion, 5′-GGAGGCCAGTGAATTCATGTGTGACGACGAGGAGAC-3′; and ACTC1 GAD RIn-Fusion, 5′-CGAGCTCGATGGATCCTTAGAAGCATTTGCGGTGGAC-3′. The WT and E258K C1C2 PCR products were cloned into pGBKT7 using the In-Fusion HD cloning kit (Takara Bio Inc.) according to the manufacturer’s instructions. The S2 and ACTC1 PCR products were similarly cloned into pGADT7. The plasmids thus generated were sequenced before transformation into the Y2HGold (pGBKT7-C1C2WT/E258K, as well as control bait constructs) or Y187 (pGADT7-S2, pGADT7-ACTC1 as well as control prey construct) yeast strains (Takara Bio Inc.) according to the manufacturer’s instructions. After yeast mating between bait-containing Y2HGold and prey-containing Y187 yeast, diploid yeast colonies were selected on dropout selection media lacking leucine and tryptophan plates. We subsequently assayed for activation of the HIS3 and ADE2 nutritional reporter genes by sequentially plating diploid yeast colonies on dropout selection media lacking leucine, tryptophan, and histidine (TDO) and on dropout selection media lacking leucine, tryptophan, histidine, and adenine (QDO) plates. Growth on selection media was assessed and recorded 7 d after plating on respective media. Additionally, diploid yeast were subjected to liquid culture quantitative β-galactosidase assays using ONPG as a substrate according to the manufacturer’s instructions (Takara Bio Inc.). All Y2H assays were performed at least three times from individual diploid yeast colonies.
Statistical analysis
IBM SPSS software (version 21) was used to perform statistical analysis. Student’s t tests were used for two way comparisons, while one-way ANOVA with Tukey’s test correction for post-hoc analysis was used for multiple comparisons. All error bars indicate SEM. Statistical significance was set at P < 0.05.
Online supplemental material
Fig. S1 show DNA, RNA, and protein content in cMyBP-C−/− mECT and monolayer cardiomyocytes transduced with adWT and ad258. Figs. S2–S4 show immunofluorescent histology of KO adWT and KO ad258 ECT confirming appropriate incorporation of exogenous human cMyBP-C into the mouse sarcomere. Fig. S5 shows the phosphorylation status of Ser275, Ser284, and Ser304 in monolayer cMyBP-C−/− cardiomyocytes transduced with adWT and ad258. Fig. S6 show the phosphorylation status of Ser284 in KO adWT and KO ad258 mECT, as well as in WT neonatal and adult mouse hearts. Detailed data regarding mECT twitch force production and cMyBP-C phosphorylation depicted in Figs. 4, 5, and S5 are provided in Tables S1 and S2. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201311018/DC1.
Figure 4.

Effect of the E258K cMyBP-C on twitch force production. (A and B) Absolute (A) and normalized (B) averaged force traces of KO adWT (blue line) and KO ad258 (red line). (C) FMax produced. (D) Contraction time from electrical stimulus to FMax (CT100). (E) Relaxation RT50. (F) Relaxation RT50–90. Blue bars, KO adWT mECT; red bars, KO ad258 mECT. *, P < 0.05 vs. WT (Student’s t test; KO adWT n = 11, KO ad258 n = 13). Error bars indicate SEM.
Figure 5.
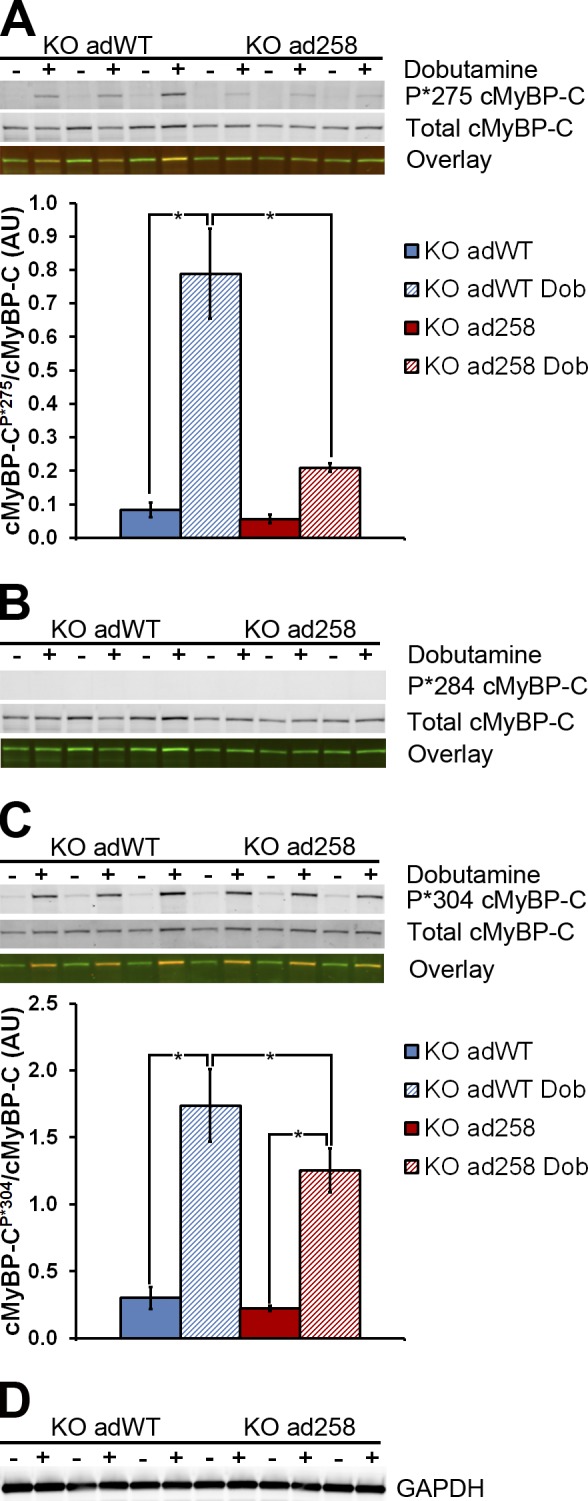
Effect of the E258K mutation on phosphorylation of cMyBP-C in mECT. (A–D) Phosphorylation of Ser275 (A), Ser284 (B), and Ser304 (C) in cMyBP-C−/− KO adWT and KO ad258 mECT. In A–C, the top Western blot rows show immunofluorescence after hybridization with the specific phospho-serine antibody and a goat anti–rabbit fluorescent secondary antibody, the middle rows show immunofluorescence after hybridization with a total cMyBP-C and a donkey anti–goat fluorescent secondary antibody, and the bottom rows show the dual fluorescence overlay, with the phospho-serine fluorescence in red and the total cMyBP-C fluorescence in green. In all cases, phospho-serine and total cMyBP-C migrated at ∼150 kD. (D) GAPDH (migrating at ∼37 kD) was used as an additional loading control for the same samples. +, mECT treated with 5 µM dobutamine 5 min before cell harvest; −, mECT not treated with dobutamine. The bar graphs show densitometric quantification of the Ser275 and Ser304 Western blots. *, P < 0.05 (one-way ANOVA with a Tukey’s post-hoc test; n = 6). Error bars indicate SEM.
RESULTS
Macroscopic properties of KO adWT and KO ad258 mECT
KO adWT mECT were produced by transducing neonatal cMyBP-C−/− cardiac cells (lacking endogenous mouse cMyBP-C) with adenovirus encoding WT human cMyBP-C at an MOI of 20 before generation of mECT. Using this approach, we previously showed that mECT expressed amounts of cMyBP-C comparable to WT mECT with appropriate incorporation into the mouse sarcomere; and furthermore that expression of human cMyBP-C effectively rescued contractile abnormalities caused by ablation of mouse cMyBP-C in mECT (de Lange et al., 2011). KO ad258 mECT were similarly generated using adenovirus encoding human cMyBP-C into which the E258K missense mutation was introduced by in vitro mutagenesis. For the generation of both KO adWT and KO ad258 mECT, neonatal cMyBPC−/− cardiomyocytes were transduced at an MOI of 20 before generation of mECT. Spontaneous contraction in both KO adWT and KO ad258 mECT began in an uncoordinated manner on the second day after mECT construction and transitioned to coordinated contraction by day 4. Spontaneous contraction rates were similar in KO adWT and KO ad258 mECT (4.6 ± 0.3 Hz and 5.1 ± 0.2 Hz for KO adWT and KO ad258 mECT, respectively; P = 0.16). During 7 d in culture, mECT diameter decreased from the mold width of 3 mm to ∼1 mm as the construct formed and compacted. Although KO adWT and KO ad258 mECT remodeled similarly and appeared indistinguishable by gross inspection, KO ad258 mECT were marginally but significantly narrower than KO adWT mECT (974 ± 24 nm vs. 1,097 ± 41 nm; P = 0.018). The absence of hypertrophy in our model may reflect that we use neonatal cardiomyocytes isolated from cMyBP-C–null newborn mice that have yet to develop the hypertrophy that develops later (de Lange et al., 2013). The acute expression of the E258K mutation on this background provides the opportunity to evaluate the contractile phenotype, free of remodeling. Although we cannot predict how our mECT would behave over a long period, the lack of overt hypertrophy is consistent with data suggesting that 40% of patients with mutations in cMyBP-C present without hypertrophy (Niimura et al., 1998).
Effect of E258K cMyBP-C expression in cardiac cells and mECT
We previously demonstrated that transduction of cardiac cells with adWT or a scrambled control adenovirus at an MOI up to fivefold higher than used in these experiments did not adversely affect cell survival (de Lange et al., 2011). To confirm this finding and to assess the effect of E258K cMyBP-C on cell survival, 2D monolayer cultures of mouse neonatal cMyBP-C−/− cardiac cells were transduced with adWT and ad258 at MOIs of 0, 5, 10, 20, 50, and 100. Cardiac cell number and viability were assessed by both total RNA yield/105 cells and the MTT viability assay, and showed no significant differences between cardiac cells transduced with either virus at an MOI of up to 100 (Fig. 2 A and Fig. S1 A), which suggests that neither the virus nor expression of E258K cMyBP-C adversely affect cardiac cell survival. Because these data agreed with observations in similar experiments with the adWT virus (de Lange et al., 2011), KO adWT and KO ad258 mECT were produced from KO cardiac cells transduced at an MOI of 20 during the gyration culture. We subsequently assessed expression levels of human cMyBP-C in KO adWT and KO ad258 mECT and compared it to the levels of endogenous mouse cMyBP-C in untransduced WT mECT. Our data show equivalent expression levels of cMyBP-C between these three groups, confirming appropriate cMyBP-C levels in KO adWT and KO ad258 mECT (Fig. 2, B and C).
Figure 2.
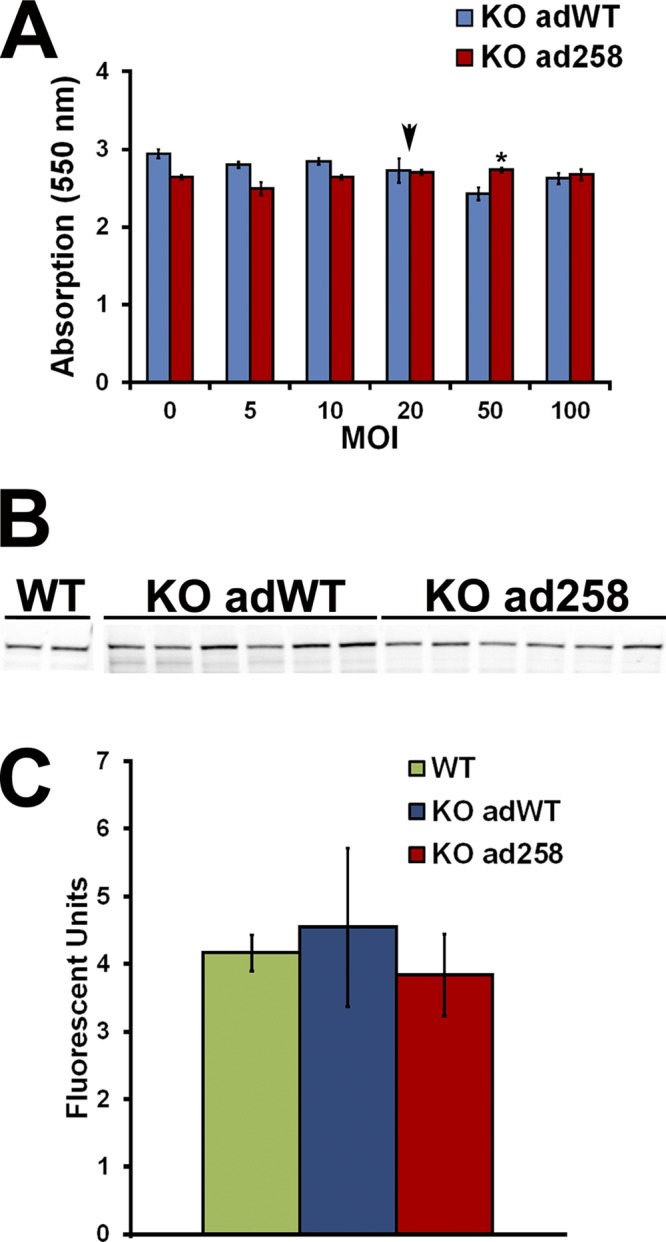
Expression of WT and E258K cMyBP-C in cardiomyocytes and mECT. (A) Effect of adenoviral transduction on cardiac cell viability in neonatal mouse cMyBP-C−/− cardiac cells in monolayer culture, transduced with adWT (blue bars) and ad258 (red bars) at MOIs of 0, 5, 10, 20, 50, and 100 as assessed by an MTT assay. (B) Western blot showing total cMyBP-C protein levels in WT, KO adWT, and KO ad258 mECT. (C) Densitometric quantification of cMyBP-C protein levels in WT, KO adWT, and KO ad258 mECT. The arrow in A indicates an MOI = 20 used to transduce mECT. *, P < 0.05 (one-way ANOVA with a Tukey’s post-hoc test; n = 3). Error bars indicate SEM.
We subsequently assessed the effect of expression of human WT and E258K cMyBP-C on cardiac cell survival in mECT using several complimentary approaches. Total DNA, RNA, and protein content were similar in KO adWT and KO ad258 mECT (Fig. S1, B–D). These findings suggest that expression of human WT or E258K cMyBP-C does not adversely affect cardiac cell survival in mECT and are consistent with those of a previous study in which we demonstrated that transduction of cardiac cells with either adWT or a control virus did not affect cardiac cell viability (de Lange et al., 2011).
Immunohistochemical staining of KO adWT and KO ad258 mECT with cMyBP-C antibody show that all mouse cardiomyocytes present in the sections express human cMyBP-C, indicating transduction efficiency at or near 100%. Furthermore, similar to the WT human cMyBP-C (de Lange et al., 2011), E258K human cMyBP-C incorporates appropriately into the C-zone of the cardiac sarcomere as indicated by the characteristic doublet immunofluorescence pattern surrounding the sarcomeric M-line with no visual evidence of protein accumulation in other cellular compartments (Fig. 3, A and B; and Figs. S2–S4).
Figure 3.

Immunohistochemical analysis of a KO ad258 mECT. Red, α-Tropomyosin; green, cMyBP-C; blue, nuclei stained with DAPI. B shows a higher magnification of the boxed area in A and shows the characteristic doublet staining pattern caused by appropriate incorporation of mutant human E258K cMyBP-C into the C-zone of the mouse sarcomere.
The effect of the E258K cMyBP-C on twitch force production
To determine the impact of E258K cMyBP-C on cardiac contractility, twitch force production was measured in KO adWT and KO ad258 mECT according to methods described previously (de Lange et al., 2011, 2013). FMax produced by KO ad258 mECT was reduced by >80% compared with KO adWT mECT (Fig. 4, A and C; and Table S1). This was in marked contrast to previous data in which cMyBP-C−/− mECT demonstrated an ∼80% increase in twitch force production compared with WT and KO adWT mECT (de Lange et al., 2011). Furthermore, time from electrical stimulation to FMax (CT100) was significantly shortened in the KO ad258 compared with KO adWT mECT, which indicates faster twitch kinetics in KO ad258 mECT (Fig. 4, B and D; and Table S1). Time from FMax to 50% twitch force decay (RT50) was reduced by ∼25% in KO ad258 mECT (Fig. 4, B and E; and Table S1), which indicates accelerated kinetics during the initial phase of relaxation compared with KO adWT mECT. These kinetic changes brought about by expression of E258K cMyBP-C were strikingly similar to those previously observed in cMyBP-C−/− mECT, in which cMyBP-C ablation caused 18% and 24% decreases in CT100 and RT50, respectively, compared with WT and KO adWT mECT (de Lange et al., 2011). Time from 50–90% twitch force decay (RT50–90) was unaffected or marginally prolonged by the presence of the E258K mutation (Fig. 4, B and F; and Table S1).
Effect of the E258K mutation on phosphorylation of cMyBP-C
Because phosphorylation of cMyBP-C has been recognized to accelerate contractile kinetics, we assessed the effect of the E258K mutation on phosphorylation of human cMyBP-C. Phosphorylation status was determined in KO adWT and KO ad258 mECT using three phospho-serine–specific antibodies (Sadayappan et al., 2011; Fraysse et al., 2012) in conjunction with an antibody detecting total cMyBP-C (Fig. 5 and Table S2). Baseline phosphorylation levels of Ser275 were similar in KO adWT and KO ad258 mECT. Dobutamine stimulation significantly increased phosphorylation at Ser275 in KO adWT as expected; however, in the KO ad258 mECT, the magnitude of the increase was significantly decreased (Fig. 5 A). Although we were able to show baseline phosphorylation of Ser284 in neonatal and adult WT mouse hearts (Fig. S4), we were unable to detect baseline or dobutamine-induced phosphorylation of this residue in KO adWT or KO ad258 mECT (Fig. 5 B). Phosphorylation of Ser304 was not significantly affected by the presence of the E258K mutation under nonstimulated conditions, whereas dobutamine-mediated phosphorylation of this site was marginally decreased in KO ad258 mECT (Fig. 5 C). Similar results were obtained when baseline and dobutamine-induced phosphorylation were assessed in neonatal cardiac cells grown and transduced in monolayers, which indicates that the observed phosphorylation of each of these residues was not influenced by the ECT configuration (Fig. S5 and Table S2). Together, these data suggest that the phosphorylation status of the KO ad258 mECT does not account for the observed acceleration in contractile kinetics because a decrease in PKA-mediated phosphorylation of these residues should slow, rather than accelerate, contraction kinetics.
Effect of the E258K mutation on the C1C2–S2 interaction
The effect of the E258K missense mutation on the interaction between N-terminal cMyBP-C and myosin S2 was tested using a Y2H interaction assay. In these tests, either WT human C1C2 (C1C2WT) or human C1C2 into which the E258K missense mutation was engineered (C1C2E258K) were fused to the GAL4-DNA binding domain (C1C2WT/C1C2E258K-BD). The 126 distal amino acids of myosin S2 were also expressed as a fusion peptide with the GAL4 activation domain (S2-AD). Additionally, as myosin S2 is known to form coiled-coil dimers (Blankenfeldt et al., 2006), we also expressed an S2-BD fusion peptide. Coexpression of BD and AD fusion peptides was achieved through yeast mating, and selection for interaction between fusion peptides was assessed by the ability of yeast to grow on minimal selection media lacking leucine, tryptophan, histidine, and adenine (QDO). Robust growth, resulting from activation of the HIS3 and ADE2 nutritional reporter genes, was noted when positive control 53-BD and T-AD fusion peptides were expressed (Fig. 6 A). Additionally, consistent with the known dimerization of myosin S2 (Blankenfeldt et al., 2006), coexpression of S2-BD and S2-AD also resulted in growth (Fig. 6 B). Coexpression of unfused BD and AD peptides did not result in reporter gene activation, as indicated by lack of growth on QDO selection media (Fig. 6 C). Coexpression of unfused BD and S2-AD, or C1C2WT/C1C2E258K-BD and AD, did not result in growth on QDO selection media (Fig. 6, D–F), which suggests that S2-AD, C1C2WT-BD, and C1C2E258K-BD do not auto-activate expression of nutritional reporter genes. Coexpression of C1C2WT-BD and S2-AD fusion peptides resulted in robust growth on QDO selection media, which indicates a strong interaction between C1C2WT and S2, resulting in robust activation of the HIS3 and ADE2 nutritional reporter genes (Fig. 6 G). In contrast, no growth was observed when C1C2E258K-BD and S2-AD were coexpressed (Fig. 6 H), which suggests that the C1C2E258K is unable to interact with myosin-S2.
Figure 6.

Effect of the E258K mutation on the interactions of N-terminal cMyBP-C with myosin S2 and cardiac actin. (A–H) Y2H interaction tests between the WT and E258K C1-C2 region of human cMyBP-C and the 126 distal amino acids of human myosin S2. (A) pGBKT7-53 × pGADT7-T (positive control of a robust interaction). (B) pGBKT7-S2 × pGADT7-S2 (positive control of a relatively weak interaction). (C) pGBKT7 × pGADT7 (negative control). (D) pGBKT7 × pGADT7-S2 (negative control). (E) pGBKT7-C1C2WT × pGADT7 (negative control). (F) pGBKT7-C1C2E258K × pGADT7 (negative control). (G) pGBKT7-C1C2WT × pGADT7-S2 (interaction of interest). (H) pGBKT7-C1C2E258K × pGADT7-S2 (interaction of interest). (I–N) Y2H interaction tests between the WT and E258K C1-C2 region of human cMyBP-C and α cardiac actin (ACTC1), as well as their appropriate control interaction tests. (I) pGBKT7-53 × pGADT7-T (positive control of a robust interaction). (J) pGBKT7-S2 × pGADT7-S2 (positive control of a relatively weak interaction). (K) pGBKT7 × pGADT7-ACTC1 (negative control). (L) pGBKT7-53 × pGADT7-ACTC1 (negative control). (M) pGBKT7-C1C2WT × pGADT7-ACTC1 (interaction of interest). (N) pGBKT7-C1C2E258K × pGADT7-ACTC1 (interaction of interest). Yeast were grown on minimal selection media lacking leucine, tryptophan, histidine, and adenine. (O) Quantitative β-galactosidase assay performed in diploid yeast containing pGBKT7-C1C2WT × pGADT7-S2 (blue bar) and pGBKT7-C1C2E258K × pGADT7-S2 (red bar). n = 3. Error bars indicate SEM.
To quantitate the interaction, we performed quantitative β-galactosidase assays on diploid yeast coexpressing C1C2WT-BD and S2-AD fusion peptides or C1C2E258K-BD and S2-AD fusion peptides to assess activation of the LacZ reporter gene. Although coexpression of C1C2WT-BD and S2-AD fusion peptides resulted in robust color development, coexpression of C1C2E258K-BD and S2-AD fusion peptides did not (Fig. 6 I). Collectively, these results suggest that in our ECT system, the E258K missense mutation dramatically weakens or abolishes the interaction between the C1 domain of cMyBP-C and myosin S2. This finding provides experimental data supporting the previously proposed hypothesis that was based on the location of this mutation within cMyBP-C and the expected residue charge change (Ababou et al., 2008; Govada et al., 2008).
Effect of the E258K mutation on C1C2–actin binding affinity
Although disruption of the cMyBP-C–S2 interaction adequately explains the accelerated kinetics observed in KO ad258 mECT, it does not explain the reduction in twitch force. We hypothesized that the E258K mutation may increase the affinity for N-terminal cMyBP-C for actin, because in addition to interacting with S2, the same surface of cMyBP-C at the C1:MyBPC motif interface housing the E258K mutation is also implicated in actin binding (Bhuiyan et al., 2012). To test this hypothesis, we performed Y2H analysis by coexpressing either C1C2WT-BD or C1C2E258K-BD with human α cardiac actin fused to the GAL4 activation domain (ACTC1-AD). Robust growth, resulting from activation of the HIS3 and ADE2 nutritional reporter genes, was noted when positive control 53-BD and T-AD fusion peptides were expressed (Fig. 6 I), while coexpression of S2-BD and S2-AD also resulted in growth (Fig. 6 J). Coexpression of ACTC1-AD and either unfused BD or 53-BD did not result in growth on QDO selection media (Fig. 6, K and L), which suggests that the ACTC1-AD does not auto-activate expression of nutritional reporter genes. Interestingly, although coexpression of C1-C1WT-BD and ACTC1-AD resulted in relatively modest growth (Fig. 6 M), coexpression of C1-C1E258K-BD and ACTC1-AD resulted in more robust growth. These results suggest that the charge reversal brought about by the E258K mutation increases the affinity of N-terminal cMyBP-C for actin, independent of its effect on the cMyBP-C–S2 interaction.
DISCUSSION
The objective of this study was to establish the effect of the HCM-causing E258K mutation on cardiac contractility and to define the underlying mechanisms. Similar to a previous study in which we showed that expression of WT human cMyBP-C in nonremodeled mECT lacking endogenous mouse cMyBP-C restored contractile function (de Lange et al., 2011), in the current experiments we exclusively expressed the E258K mutant allele to establish the impact of this mutation on mechanical function. Expression of cMyBP-CE258K resulted in significantly accelerated twitch kinetics and reduction in force production compared with our control mECT expressing human WT cMyBP-C. Our data indicate that accelerated twitch kinetics result from the abrogation of an inhibitory interaction between cMyBP-C C1C2 and myosin S2, independent of cMyBP-C phosphorylation. Although previous studies have shown that HCM-causing mutations at the C1C2–S2 interface in either cMyBP-C or β-myosin heavy chain can cause a weakening or abolition of this interaction, this is the first study that directly correlates this mechanism with changes in contractility.
The accelerated twitch kinetics, as indicated by abbreviation of the time to peak twitch force development and the initial phase of relaxation in KO ad258 mECT, were strikingly similar to those previously reported in both mECT, in which cMyBP-C was absent, and WT mECT treated with dobutamine (de Lange et al., 2011, 2013). The proposed mechanism responsible for accelerated kinetics in either the absence of cMyBP-C or in the presence of maximally phosphorylated cMyBP-C is the loss of the inhibitory interaction between the C1C2 region of cMyBP-C and myosin S2 (Gautel et al., 1995; Gruen et al., 1999). Loss of this inhibitory interaction results in an increased probability of cross-bridge formation between the thick and thin filaments (Hofmann et al., 1991; Weisberg and Winegrad, 1996; Colson et al., 2008, 2010, 2012). We therefore postulated that the E258K mutation may constitutively reduce the strength of the interaction between the C1C2 region of cMyBP-C and myosin S2. Our Y2H analysis supports this hypothesis, revealing that although C1C2WT readily interacts with myosin S2, C1C2E258K is unable to do so. The eukaryotic yeast-based assay was chosen for these experiments over in vitro or bacterial-based expression systems and coimmunoprecipitation because of the increased likelihood of appropriate posttranslational modifications, protein folding, and S2 dimerization, which might impact the C1C2–S2 binding affinity.
Based on the localization of the E258K mutation in close proximity to physiologically relevant phosphorylation sites in the MyBPC motif (Ser275, Ser284, and Ser304; Gautel et al., 1995), Niimura et al. (1998) postulated that this mutation may adversely affect MyBP-C phosphorylation. Because phosphorylation of these sites causes dissociation of C1C2 from S2 (Gautel et al., 1995; Gruen et al., 1999), hyperphosphorylation of cMyBP-CE258K could explain the observed acceleration of contractile kinetics in the intact cardiomyocyte. Our finding that baseline phosphorylation, using phospho-specific antibodies (Copeland et al., 2010; Sadayappan et al., 2011; Fraysse et al., 2012), of the three physiologically relevant serine residues in the MyBPC motif was not elevated by the presence of the E258K mutation argues against the hypothesis that C1C2–S2 interaction is abolished as a result of baseline hyperphosphorylation of cMyBP-CE258K. Dobutamine stimulation increased phosphorylation levels of Ser273 and Ser304 in both KO adWT and KOad258 mECT. The magnitude of increased phosphorylation of Ser275 and Ser304 were, however, significantly blunted by the E258K mutation. Because phosphorylation of these sites in the MyBPC motif is primarily associated with a decreased affinity for S2 (Gruen and Gautel, 1999; Gruen et al., 1999), an interaction abolished by the presence of the E258K mutation, decreased PKA-mediated phosphorylation of these residues is most likely inconsequential to the C1C2–S2 interaction. Surprisingly, we failed to detect phosphorylation of Ser284 in cultured neonatal cardiac cells or mECT at either baseline or after dobutamine stimulation. This intriguing finding requires further investigation, as phosphorylation of Ser284 is thought to be a prerequisite for phosphorylation of Ser275 (Gautel et al., 1995) and Ser304 (Gautel et al., 1995; Sadayappan et al., 2011).
Collectively our phosphorylation data and Y2H data suggest a model in which the E258K mutation abolishes the C1C2–S2 interaction by altering the intrinsic binding properties of C1C2 (Ababou et al., 2008). The importance of the glutamic acid residue at position 258 cMyBP-C is supported by its 100% conservation among species and among the MyBP-C isoforms (Govada et al., 2008). The glutamic acid residue at position 258 of WT human cMyBP-C forms part of an acidic patch on the surface of the C-terminal end of the C1 domain that constitutes the MyBPC-C1–S2 interface (Fig. 7; Ababou et al., 2008; Govada et al., 2008). E258 is thought to form a hydrogen bond with T857 of myosin (Fig. 7 A), and the importance of this bond is indicated by the fact that the chemical shift perturbation at this residue as a result of S2 binding is among the highest of any residues in the C1 domain (Ababou et al., 2008). It is therefore plausible that the charge reversal brought about by the E258K may, in addition to abolishing this one hydrogen bond, also cause a repulsion between the C1 binding surface and myosin S2. Furthermore, as the binding site for the C2 domain has been mapped to the region directly adjacent to E258 (E258 in C1 interacting with T857 of S2 and D431 in C2 interacting with R858; Ababou et al., 2007, 2008), a charge reversal in this region may destabilize the whole C1C2–S2 interaction. Although the present study is the first to show that the E258K mutation abolishes the C1C2–S2 interaction, abolition or weakening of this interaction as a result of HCM-causing mutations in cMyBP-C and myosin S2 is not unprecedented (Gruen and Gautel, 1999; Ababou et al., 2007, 2008). Another example can be found with the charge reversal caused by the E924K mutation in S2 abolishing its interaction with N-terminal cMyBP-C (Gruen and Gautel, 1999). Additionally, the myosin S2 E846K, R870H, and cMyBP-C (C1) D228N mutations severely weakened the interaction (Gruen and Gautel, 1999; Ababou et al., 2007, 2008). Finally, introduction of negative charges through phosphorylation of serine residues 275, 284, and 304 in the MyBPC motif cause a repulsion of the C1C2–S2 interaction (Gruen et al., 1999).
Figure 7.

Model of the interaction between cMyBP-C C1-C2 and the distal 126 aa of S2. (A) Detailed view of the interactions of C1 and S2. C1 is shown in blue and S2 is shown in red. Important side chains in the interaction are colored by atom type (carbon, green; oxygen, red; nitrogen, blue), and amino acid labels are colored by protein. Amino acids at the C1–S2 interface that, when mutated, are implicated in the pathogenesis of HCM are underlined, and the position of the cMyBP-C E258K mutation is highlighted by the arrow. (B) Diagrammatic representation of the proposed orientation of the C1C2 region of cMyBP-C on S2. Domains C1 and C2 of MyBP-C are shown in orange and the MyBPC-motif in gray. The three phosphorylation sites in the linker are shown in purple, residues in the MyBPC-motif mutated in HCM are shown in yellow with labels, and charged residues are colored according to their charge. S2 is shown with solvent-accessible surface colored by a simple electrostatic potential with the N terminus on the left (hidden by C1) and the C terminus on the right. Reproduced from Ababou et al. (2008) with permission from Elsevier.
Another important finding of this study is the negative impact that the E258K mutation has on twitch force amplitude. Reduced force production could potentially stem from the negative force–velocity relationship in cardiac muscle that has been previously reported (Gao et al., 1996; McDonald et al., 1998; Hiranandani et al., 2006). However, data showing that cMyBP-C ablation results in increased twitch force production in mECT (de Lange et al., 2011) and increased power output in skinned fibers (Korte et al., 2003) argues against this mechanism as the sole determinant of the decreased twitch force produced by KO ad285 mECT. Additionally, we show that the reduction in force production of KO ad258 mECT is not caused by cell death, protein accumulation, or misincorporation. An alternative mechanism for the decreased twitch force in KO ad258 mECT is that the E258K mutation facilitates or strengthens interaction with other proteins in addition to abrogating the C1C2–S2 interaction. Although controversial, the same N-terminal cMyBP-C domains that interact with S2 also have the ability to interact with F-actin (Razumova et al., 2006; Shaffer et al., 2009; Kensler et al., 2011; Lu et al., 2011; Rybakova et al., 2011; Bhuiyan et al., 2012). Recent studies showed significant overlap between the actin and S2 binding sites in N-terminal cMyBP-C and, more importantly, found that the interactions between the N terminus and either S2 or actin are mutually exclusive (Lu et al., 2011; Bhuiyan et al., 2012). Furthermore, lysine residues in N-terminal cMyBP-C are essential to facilitating its interaction with actin (Bhuiyan et al., 2012). As the E258K mutation introduces an additional lysine residue at this interface, it is possible that this mutation may strengthen the cMyBP-C–actin interaction. A high-affinity interaction between N-terminal cMyBP-C and the thin filament could potentially explain the dramatic reduction in twitch force produced by KO ad258 mECT, as cMyBP-CE258K would effectively act as a leash tethering thick and thin filaments or impose an internal load on contraction. This interaction is suggested by our Y2H data, which shows greater interaction between the C1C2 domain and actin in the E258K mutant versus WT. Though Y2H analysis was previously used to study the C1C2–S2 interaction (Bhuiyan et al., 2012), caution should be used when interpreting this data, as the polymerization status of actin Y2H fusion peptides are unclear.
It is important to note that the experimental approach used in this study involved adenoviral mediated gene transfer to express equivalent amounts of full-length WT or E258K MYBPC3 cDNA in cardiac cells and mECT. Adenoviral-mediated expression of the E258K mutation achieved incorporation of a full-length mutant cMyBP-C without adversely affecting cell survival. The existence in vivo of the full-length mutant protein has been debated within the literature. The mutation is a change of a G to an A at nucleotide position 5,256 of MYBPC3, affecting the exon 6 splice donor site, which prompts speculation that exon splicing may be altered (Niimura et al., 1998; Andersen et al., 2004; Marston et al., 2009; Fraysse et al., 2012). One study in which MYBPC3 was ectopically expressed in lymphocytes did identify an aberrantly spliced transcript lacking exon 6 (Andersen et al., 2004). Additionally, a murine model showed that a mutation in the exon 6 splice donor site of mouse Mybpc3 resulted in a mixture of transcripts including full-length and truncated forms with an overall reduction in total cMyBP-C protein level (Vignier et al., 2009; Fraysse et al., 2012). Human cardiomyocyte data, however, remains incomplete, with one study of human myocardial samples from E258K mutation carriers showing no evidence of transcripts that lacked exon 6 or other truncated forms (Marston et al., 2009). Additionally, although known MYBPC3 truncation mutations result in a significant reduction in cMyBP-C protein levels in myocardium (Marston et al., 2009, 2012; van Dijk et al., 2009), total cMyBP-C levels were only marginally reduced in the myocardium of E258K mutation carriers (Marston et al., 2009).
The finding that the phenotypic effect of exclusive E258K cMyBP-C expression was more severe than that of homozygous cMyBP-C ablation (de Lange et al., 2011) was intriguing. Interestingly, previous studies have shown that cMyBP-C missense mutations, and particularly those in the C1C2 region, may cause a more malignant form of HCM than cMyBP-C truncation mutations that cause HCM through haploinsufficiency (Niimura et al., 1998; Morita et al., 2008). Together, these findings suggest that some HCM-causing missense mutations in cMyBP-C, including E258K, can act as true poison peptides resulting in greater disruption of sarcomeric function than cMyBP-C haploinsufficiency.
In conclusion, the major findings of this study were that the E258K mutation in cMYBP-C: (1) reduced the strength of interaction between the C1C2 region of cMyBP-C and the S2 domain of myosin, (2) accelerated the kinetics of the twitch in a manner similar to both the phosphorylated state of cMyBP-C and its complete absence, and (3) reduced the magnitude of the twitch force, presumably by enhancing interaction between cMyBP-C and the thin filament. Future studies will evaluate the impact of heterozygous expression of E258K on contractility to more precisely model the heterozygous expression typical of human familial HCM. Thorough characterization of myectomy samples from humans with E258K HCM will be helpful in defining the status of the mutant protein within the human cardiomyocyte. Finally, the present study joins a growing body of work that supports the application of various configurations of engineered cardiac preparations as efficient and cost-effective alternatives to genetically modified animal models to study the functional effects of relatively rare HCM-causing missense mutations.
Supplementary Material
Acknowledgments
Phospho-serine specific antibodies detecting phosphorylation of Ser275 and Ser304 were kind gifts from Dr. Sakthivel Sadayappan. The authors are grateful to Drs. Richard Moss and Michael Wilhelm for critical review of the manuscript.
This work was supported by NIH grant R01HL107367-01 (J.C. Ralphe) and Children’s Cardiomyopathy Foundation grant 133-PRJ32ZU (J.C. Ralphe).
Angus C. Nairn served as editor.
Footnotes
Abbreviations used in this paper:
- AD
- GAL4 activation domain
- BD
- GAL4-DNA binding domain
- cMyBP-C
- cardiac myosin binding protein C
- CT100
- time to maximal twitch force
- DAPI
- 4′,6-diamidino-2-phenylindole
- DMEM
- Dulbecco’s modified Eagle medium
- FMax
- maximal twitch force
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- HA
- hemagglutinin
- HCM
- hypertrophic cardiomyopathy
- KO
- knockout
- mECT
- murine engineered cardiac tissue
- MOI
- multiplicity of infection
- MTT
- 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- MYBPC3
- human cardiac myosin binding protein C gene
- RT50
- time from FMax to 50% twitch force decay
- RT50–90
- time from 50–90% twitch force decay
- S2
- myosin heavy chain sub-fragment 2
- Y2H
- yeast two-hybrid
References
- Ababou A., Gautel M., Pfuhl M. 2007. Dissecting the N-terminal myosin binding site of human cardiac myosin-binding protein C. Structure and myosin binding of domain C2. J. Biol. Chem. 282:9204–9215 10.1074/jbc.M610899200 [DOI] [PubMed] [Google Scholar]
- Ababou A., Rostkova E., Mistry S., Le Masurier C., Gautel M., Pfuhl M. 2008. Myosin binding protein C positioned to play a key role in regulation of muscle contraction: structure and interactions of domain C1. J. Mol. Biol. 384:615–630 10.1016/j.jmb.2008.09.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alders M., Jongbloed R., Deelen W., van den Wijngaard A., Doevendans P., Ten Cate F., Regitz-Zagrosek V., Vosberg H.P., van Langen I., Wilde A., et al. 2003. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur. Heart J. 24:1848–1853 10.1016/S0195-668X(03)00466-4 [DOI] [PubMed] [Google Scholar]
- Andersen P.S., Havndrup O., Bundgaard H., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M. 2004. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur. J. Hum. Genet. 12:673–677 10.1038/sj.ejhg.5201190 [DOI] [PubMed] [Google Scholar]
- Barefield D., Sadayappan S. 2010. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 48:866–875 10.1016/j.yjmcc.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan M.S., Gulick J., Osinska H., Gupta M., Robbins J. 2012. Determination of the critical residues responsible for cardiac myosin binding protein C’s interactions. J. Mol. Cell. Cardiol. 53:838–847 10.1016/j.yjmcc.2012.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenfeldt W., Thomä N.H., Wray J.S., Gautel M., Schlichting I. 2006. Crystal structures of human cardiac β-myosin II S2-Δ provide insight into the functional role of the S2 subfragment. Proc. Natl. Acad. Sci. USA. 103:17713–17717 10.1073/pnas.0606741103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier L., Bonne G., Bährend E., Yu B., Richard P., Niel F., Hainque B., Cruaud C., Gary F., Labeit S., et al. 1997. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ. Res. 80:427–434 [PubMed] [Google Scholar]
- Colson B.A., Bekyarova T., Locher M.R., Fitzsimons D.P., Irving T.C., Moss R.L. 2008. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 103:244–251 10.1161/CIRCRESAHA.108.178996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson B.A., Locher M.R., Bekyarova T., Patel J.R., Fitzsimons D.P., Irving T.C., Moss R.L. 2010. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J. Physiol. 588:981–993 10.1113/jphysiol.2009.183897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson B.A., Patel J.R., Chen P.P., Bekyarova T., Abdalla M.I., Tong C.W., Fitzsimons D.P., Irving T.C., Moss R.L. 2012. Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J. Mol. Cell. Cardiol. 53:609–616 10.1016/j.yjmcc.2012.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland O., Sadayappan S., Messer A.E., Steinen G.J., van der Velden J., Marston S.B. 2010. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 49:1003–1011 10.1016/j.yjmcc.2010.09.007 [DOI] [PubMed] [Google Scholar]
- de Lange W.J., Hegge L.F., Grimes A.C., Tong C.W., Brost T.M., Moss R.L., Ralphe J.C. 2011. Neonatal mouse-derived engineered cardiac tissue: a novel model system for studying genetic heart disease. Circ. Res. 109:8–19 10.1161/CIRCRESAHA.111.242354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange W.J., Grimes A.C., Hegge L.F., Ralphe J.C. 2013. Ablation of cardiac myosin-binding protein-C accelerates contractile kinetics in engineered cardiac tissue. J. Gen. Physiol. 141:73–84 10.1085/jgp.201210837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlermann P., Weichenhan D., Zehelein J., Steen H., Pribe R., Zeller R., Lehrke S., Zugck C., Ivandic B.T., Katus H.A. 2008. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med. Genet. 9:95 10.1186/1471-2350-9-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fananapazir L., Epstein S.E. 1991. Value of electrophysiologic studies in hypertrophic cardiomyopathy treated with amiodarone. Am. J. Cardiol. 67:175–182 10.1016/0002-9149(91)90441-M [DOI] [PubMed] [Google Scholar]
- Fraysse B., Weinberger F., Bardswell S.C., Cuello F., Vignier N., Geertz B., Starbatty J., Krämer E., Coirault C., Eschenhagen T., et al. 2012. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J. Mol. Cell. Cardiol. 52:1299–1307 10.1016/j.yjmcc.2012.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.D., Liu Y., Marban E. 1996. Selective effects of oxygen free radicals on excitation-contraction coupling in ventricular muscle. Implications for the mechanism of stunned myocardium. Circulation. 94:2597–2604 10.1161/01.CIR.94.10.2597 [DOI] [PubMed] [Google Scholar]
- Gautel M., Zuffardi O., Freiburg A., Labeit S. 1995. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 14:1952–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govada L., Carpenter L., da Fonseca P.C., Helliwell J.R., Rizkallah P., Flashman E., Chayen N.E., Redwood C., Squire J.M. 2008. Crystal structure of the C1 domain of cardiac myosin binding protein-C: implications for hypertrophic cardiomyopathy. J. Mol. Biol. 378:387–397 10.1016/j.jmb.2008.02.044 [DOI] [PubMed] [Google Scholar]
- Gruen M., Gautel M. 1999. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 286:933–949 10.1006/jmbi.1998.2522 [DOI] [PubMed] [Google Scholar]
- Gruen M., Prinz H., Gautel M. 1999. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 453:254–259 10.1016/S0014-5793(99)00727-9 [DOI] [PubMed] [Google Scholar]
- Harris S.P., Bartley C.R., Hacker T.A., McDonald K.S., Douglas P.S., Greaser M.L., Powers P.A., Moss R.L. 2002. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 90:594–601 10.1161/01.RES.0000012222.70819.64 [DOI] [PubMed] [Google Scholar]
- Hiranandani N., Varian K.D., Monasky M.M., Janssen P.M.L. 2006. Frequency-dependent contractile response of isolated cardiac trabeculae under hypo-, normo-, and hyperthermic conditions. J. Appl. Physiol. 100:1727–1732 10.1152/japplphysiol.01244.2005 [DOI] [PubMed] [Google Scholar]
- Hofmann P.A., Greaser M.L., Moss R.L. 1991. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J. Physiol. 439:701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler R.W., Shaffer J.F., Harris S.P. 2011. Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 174:44–51 10.1016/j.jsb.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte F.S., McDonald K.S., Harris S.P., Moss R.L. 2003. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 93:752–758 10.1161/01.RES.0000096363.85588.9A [DOI] [PubMed] [Google Scholar]
- Lu Y., Kwan A.H., Trewhella J., Jeffries C.M. 2011. The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J. Mol. Biol. 413:908–913 10.1016/j.jmb.2011.09.026 [DOI] [PubMed] [Google Scholar]
- Maron B.J., Gardin J.M., Flack J.M., Gidding S.S., Kurosaki T.T., Bild D.E. 1995. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 92:785–789 10.1161/01.CIR.92.4.785 [DOI] [PubMed] [Google Scholar]
- Marston S., Copeland O., Jacques A., Livesey K., Tsang V., McKenna W.J., Jalilzadeh S., Carballo S., Redwood C., Watkins H. 2009. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 105:219–222 10.1161/CIRCRESAHA.109.202440 [DOI] [PubMed] [Google Scholar]
- Marston S., Copeland O., Gehmlich K., Schlossarek S., Carrier L. 2012. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 33:75–80 (published erratum appears in J. Muscle Res. Cell Motil. 2012. 33:81) 10.1007/s10974-011-9268-3 [DOI] [PubMed] [Google Scholar]
- McDonald K.S., Wolff M.R., Moss R.L. 1998. Force-velocity and power-load curves in rat skinned cardiac myocytes. J. Physiol. 511:519–531 10.1111/j.1469-7793.1998.519bh.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H., Rehm H.L., Menesses A., McDonough B., Roberts A.E., Kucherlapati R., Towbin J.A., Seidman J.G., Seidman C.E. 2008. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 358:1899–1908 10.1056/NEJMoa075463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni L., Pieroni M., Chimenti C., Simionati B., Zimbello R., Maseri A., Frustaci A., Lanfranchi G. 2003. Hypertrophic cardiomyopathy: two homozygous cases with “typical” hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 309:391–398 10.1016/j.bbrc.2003.08.014 [DOI] [PubMed] [Google Scholar]
- Niimura H., Bachinski L.L., Sangwatanaroj S., Watkins H., Chudley A.E., McKenna W., Kristinsson A., Roberts R., Sole M., Maron B.J., et al. 1998. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 338:1248–1257 10.1056/NEJM199804303381802 [DOI] [PubMed] [Google Scholar]
- Olivotto I., Girolami F., Ackerman M.J., Nistri S., Bos J.M., Zachara E., Ommen S.R., Theis J.L., Vaubel R.A., Re F., et al. 2008. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 83:630–638 [DOI] [PubMed] [Google Scholar]
- Page S.P., Kounas S., Syrris P., Christiansen M., Frank-Hansen R., Andersen P.S., Elliott P.M., McKenna W.J. 2012. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ. Cardiovasc. Genet. 5:156–166 10.1161/CIRCGENETICS.111.960831 [DOI] [PubMed] [Google Scholar]
- Razumova M.V., Shaffer J.F., Tu A.Y., Flint G.V., Regnier M., Harris S.P. 2006. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J. Biol. Chem. 281:35846–35854 10.1074/jbc.M606949200 [DOI] [PubMed] [Google Scholar]
- Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., et al. 2003. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 107:2227–2232 10.1161/01.CIR.0000066323.15244.54 [DOI] [PubMed] [Google Scholar]
- Rybakova I.N., Greaser M.L., Moss R.L. 2011. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J. Biol. Chem. 286:2008–2016 10.1074/jbc.M110.170605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S., Gulick J., Osinska H., Barefield D., Cuello F., Avkiran M., Lasko V.M., Lorenz J.N., Maillet M., Martin J.L., et al. 2011. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 109:141–150 10.1161/CIRCRESAHA.111.242560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman J.G., Seidman C. 2001. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 104:557–567 10.1016/S0092-8674(01)00242-2 [DOI] [PubMed] [Google Scholar]
- Shaffer J.F., Kensler R.W., Harris S.P. 2009. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 284:12318–12327 10.1074/jbc.M808850200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Zou Y., Wang J., Wang Z., Zhen Y., Lou K., Zhang Q., Wang X., Wang H., Li J., Hui R. 2005. Mutations profile in Chinese patients with hypertrophic cardiomyopathy. Clin. Chim. Acta. 351:209–216 10.1016/j.cccn.2004.09.016 [DOI] [PubMed] [Google Scholar]
- Spirito P., Seidman C.E., McKenna W.J., Maron B.J. 1997. The management of hypertrophic cardiomyopathy. N. Engl. J. Med. 336:775–785 10.1056/NEJM199703133361107 [DOI] [PubMed] [Google Scholar]
- Tobita K., Liu L.J., Janczewski A.M., Tinney J.P., Nonemaker J.M., Augustine S., Stolz D.B., Shroff S.G., Keller B.B. 2006. Engineered early embryonic cardiac tissue retains proliferative and contractile properties of developing embryonic myocardium. Am. J. Physiol. Heart Circ. Physiol. 291:H1829–H1837 10.1152/ajpheart.00205.2006 [DOI] [PubMed] [Google Scholar]
- van Dijk S.J., Dooijes D., dos Remedios C., Michels M., Lamers J.M., Winegrad S., Schlossarek S., Carrier L., ten Cate F.J., Stienen G.J., van der Velden J. 2009. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 119:1473–1483 10.1161/CIRCULATIONAHA.108.838672 [DOI] [PubMed] [Google Scholar]
- Van Driest S.L., Vasile V.C., Ommen S.R., Will M.L., Tajik A.J., Gersh B.J., Ackerman M.J. 2004. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 44:1903–1910 10.1016/j.jacc.2004.07.045 [DOI] [PubMed] [Google Scholar]
- Vignier N., Schlossarek S., Fraysse B., Mearini G., Krämer E., Pointu H., Mougenot N., Guiard J., Reimer R., Hohenberg H., et al. 2009. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ. Res. 105:239–248 10.1161/CIRCRESAHA.109.201251 [DOI] [PubMed] [Google Scholar]
- Weisberg A., Winegrad S. 1996. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc. Natl. Acad. Sci. USA. 93:8999–9003 10.1073/pnas.93.17.8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.