

Abstract
In all domains of life, initiator tRNA functions exclusively at the first step of protein synthesis while elongator tRNAs extend the polypeptide chain. Unique features of initiator tRNA enable it to preferentially bind the ribosomal P site and initiate translation. Recently, we showed that the abundance of initiator tRNA also contributes to its specialized role. This motivates the question, can a cell also use elongator tRNA to initiate translation under certain conditions? To address this, we introduced non-AUG initiation codons CCC (Pro), GAG (Glu), GGU (Gly), UCU (Ser), UGU (Cys), ACG (Thr), AAU (Asn), and AGA (Arg) into the uracil DNA glycosylase gene (ung) used as a reporter gene. Enzyme assays from log-phase cells revealed initiation from non-AUG codons when intracellular initiator tRNA levels were reduced. The activity increased significantly in stationary phase. Further increases in initiation from non-AUG codons occurred in both growth phases upon introduction of plasmid-borne genes of cognate elongator tRNAs. Since purine-rich Shine-Dalgarno sequences occur frequently on mRNAs (in places other than the canonical AUG codon initiation contexts), initiation with elongator tRNAs from the alternate contexts may generate proteome diversity under stress without compromising genomic integrity. Thus, by changing the relative amounts of initiator and elongator tRNAs within the cell, we have blurred the distinction between the two classes of tRNAs thought to be frozen through years of evolution.
INTRODUCTION
Most organisms possess two kinds of tRNAs, the initiator tRNA, which decodes the initiation codon, and the elongator tRNAs, which decode the subsequent codons within the open reading frame (ORF). In Escherichia coli, there are four copies of the initiator tRNA genes; three of these, metZ, metW, and metV, are present at 63.5 min, whereas the fourth one, metY, is found at 71.5 min in the genome (1). It is also known that either (but not both) of these loci may be deleted from E. coli. The strain deleted for metZWV becomes cold sensitive. However, at temperatures of 30°C or higher the strain grows normally (2, 3).
Initiator and elongator tRNAs are believed to perform their respective decoding functions without functional interference from one another. Although they share the same cloverleaf-like backbone structure, years of evolution have led to their separate and distinct functions, now held in place by an intricate support system comprising translation factors, quality control mechanisms, and the ribosome itself. Elongator tRNAs are excluded from participation at the start of protein synthesis by sequence features specific to the initiator tRNA that are recognized exclusively at the P site of the ribosome (4–6). Various other details, such as the fact that the initiation and elongation factors bind specifically to their cognate species of tRNAs, further aid the specificity of their functions.
However, earlier studies have enabled initiation by elongator tRNAs, by transplanting the critical features of initiator tRNA onto them and creating a hybrid initiator-elongator molecule (7). More recently it was reported that while expression of an initiator tRNA mutant with the CUA anticodon results in efficient initiation of protein synthesis from the cognate UAG initiation codon of a reporter mRNA, expression of the supE gene encoding an elongator tRNAGln (also with CUA anticodon) does not. However, as expected of an elongator tRNA, supE expression led to efficient suppression of an in-frame amber (UAG) codon when present internally in the open reading frame of the same reporter in a wild-type E. coli background (3). Intriguingly, when the metZWV locus that harbors three of the four initiator tRNA genes was deleted, overexpression of supE resulted in a significant increase in initiation activity, showing that in spite of being an elongator tRNA, the supE tRNAGln could initiate protein synthesis when the cell's initiator tRNA content was reduced (3). It should, however, be emphasized that suppressor species of tRNA are not naturally present in organisms and occur only as occasional mutants. We do not know how common a property it is for the elongator tRNAs to initiate under stressful conditions, such as during initiator tRNA deficiency. Likewise, how common it is for nonmethionine codons to be utilized in initiation (as would happen if elongator tRNAs initiated) is also not known.
To examine the significance of the phenomenon of initiation with elongator tRNAs in a cellular context, we have investigated the impact of reduction of initiator tRNA gene numbers on the versatility of native elongator tRNAs in initiation from their cognate codons. By knocking out three of the four genes that encode initiator tRNA, we were able to uncover initiation by elongator tRNAs as a widespread phenomenon. This suggests a novel mechanism to generate proteomic diversity, with non-AUG initiation potentially occurring at many places on one mRNA. Such a system, somewhat reminiscent of the SOS response at the DNA level, could help the cell generate variety (by proteome remodeling) without compromising genomic integrity.
MATERIALS AND METHODS
Media and growth conditions.
Bacterial strains (Table 1) were grown in Luria-Bertani (LB) broth or LB agar plates containing 1.8% Bacto-agar (Difco). Unless indicated otherwise, media were supplemented with ampicillin (Amp; 100 μg/ml), chloramphenicol (Cm; 30 μg/ml), kanamycin (Kan; 25 μg/ml), tetracycline (Tet; 7.5 μg/ml), or isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM) as required.
Table 1.
List of strains and plasmids
| Strain/plasmid | Genotype/details | Reference or source |
|---|---|---|
| KL16 | E. coli K-12 thi1 relA1 spoT1 | 41 |
| KL16 ΔmetZWV::kan | Derivative of KL16 carrying a deletion of the metZ, metW, and metV genes | This study |
| TG1 | E. coli K-12 supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM) | 42 |
| TG1 Δung::cm (TG1Δung) | Derivative of E. coli TG1 carrying a deletion of the ung gene | P. Kumar and U. Varshney, unpublished data |
| TG1 Δung::cm ΔmetZWV::kan (TG1 Δung ΔmetZWV) | Derivative of TG1Δung carrying a deletion of the metZWV genes | This study |
| LJ14 | E. coli MC1061 carrying the frr(Ts) allele (frr 14) | 43 |
| pTrc-ung | Derivative of pTrc99C vector carrying the ung gene, Ampr | 44 |
| pACDH | Plasmid carrying ACYC origin of replication, compatible with ColE1 origin of replication, Tetr | 45 |
| pACDH-metY | pACDH carrying the metY gene | This study |
| pACDH-Asn, -Glu, -Pro, -Thr, -Gly, -Ser, -Cys, or -Arg | pACDH carrying various elongator tRNA genes as indicated | This study |
P1 transduction.
P1 phage-mediated transductions were carried out as described previously (8).
Construction of ung gene reporters.
The plasmid pTrc-ung, carrying the ung gene encoding uracil DNA glycosylase, was used as a template for inverse PCR in order to change the start codon ATG of the reporter gene to a variety of different triplets at the first position. In all cases, a 50-μl PCR mixture containing the template (200 ng), 200 μM deoxyribonucleoside triphosphates (dNTPs), 20 pmol each of the cDNA oligomer set (ung mut fp and ung mut rp carrying NNN or any nucleotide at the ATG positions [Table 2]), 1× Pfu enzyme buffer as supplied by the vendor, and 2 U of Pfu DNA polymerase was set up. PCR conditions included initial heating at 94°C for 3 min followed by 29 cycles at 94°C for 1 min, 45°C for 30 s, and 70°C for 11 min. Upon completion of the PCR cycles, the reaction mixtures were heated at 72°C for 10 min and stored at 4°C. The PCR product was kept overnight for DpnI digestion and transformed into E. coli TG1 and plasmids prepared from single colonies. The mutants were confirmed by DNA sequencing (Macrogen) using appropriate primers.
Table 2.
List of the DNA oligomers
| DNA oligomer | Sequence |
|---|---|
| ung mut fp | 5′CACAGGAAACAGACCNNNGCTAACGAATTAACC3′ |
| ung mut rp | 5′GGTTAATTCGTTAGCNNNGGTCTGTTTCCTGTG3′ |
| Asn fp | 5′GCGTGAATTCTTAACAACTTTGCAG3′ |
| Asn rp | 5′GCTCCCATGGGGTCGAGGGCGCTTA3′ |
| Glu fp | 5′GCGTGAATTCGTTGGGAGTGAGGCT3′ |
| Glu rp | 5′GCGTCCATGGCAGCTTGATCCAGAT3′ |
| Gly fp | 5′GCGTGAATTCGGGCAGCAAGCCAAA3′ |
| Gly rp | 5′GCGTCCATGGTGAAAATAAGGTGTT3′ |
| Ser fp | 5′GCTTGAATTCAAGTTTGAATAAACG3′ |
| Ser rp | 5′GCTCCCATGGGTCGCCAGTTGTCGA3′ |
| Cys fp | 5′GCTTGAATTCCTTGCCAAGGTCGGG3′ |
| Cys rp | 5′GCTCCCATGGATGGTACCCGGAGCG3′ |
| Arg fp | 5′GCTTGAATTCGACGCGGTCGTTCAC3′ |
| Arg rp | 5′GCTCCCATGGTGCGAGGAAAATAG3′ |
| Pro fp | 5′CTCCCCATGGACGAGAAGCGTTTTATC3′ |
| Pro rp | 5′CGTTGAATTCACAGAAAATAAACAGGC3′ |
| Thr fp | 5′CTCCCCATGGTGCAAAAGTAGCCA3′ |
| Thr rp | 5′TGTTGAATTCGATTGATCTTCGTTG3′ |
| metY fp (EcoRI) | 5′GCGTGAATTCTGCAGATTTTACGTCCCGTC3′ |
| metY rp (NcoI) | 5′GCTGCCATGGGCACTTTCCAGAAGGATTTT3′ |
| SSU9 | 5′CTCAAGTGUAGGCATGCAAGAGCT3′ |
Cloning of elongator tRNA genes into pACDH.
Elongator tRNA genes from the E. coli genome were amplified by PCR using flanking primers (Table 2): Asn fp, Asn rp for the gene asnT, Glu fp and Glu rp for the gene gltT, and so on for the genes serX, cysT, argU, proL, thrW, and glyU. The forward and reverse primers contained the restriction site for either EcoRI or NcoI. A 50-μl PCR mixture was set up using ∼200 ng genomic DNA from E. coli KL16, 200 μM dNTPs, 20 pmol forward primer, 20 pmol reverse primer, 1× Pfu enzyme buffer as supplied by the vendor, and 2 U of Pfu polymerase. The PCR conditions used were initial heating at 94°C for 3 min followed by 30 cycles at 94°C for 1 min, 50°C for 30 s, and 72°C for 1 min. The reaction mixtures were then held at 70°C for 10 min and then stored at 4°C. The genes were cloned into the vector pACDH (Tetr) as ∼300-bp-sized fragments (Table 1).
Immunoblot analysis.
Immunoblot analyses for β-lactamase were carried out using 15 μg of total protein from the cell extracts. The samples were fractionated by electrophoresis on a 12.5% SDS-PAGE gel along with size markers, and a section of the gel including a ∼32-kDa protein was then electroblotted onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked overnight using 5% nonfat milk powder in TBST (20 mM Tris-HCl [pH 7.4], 0.2% Tween 20, 150 mM NaCl). It was then washed three times for 10 min each time in TBST in a rocker and then incubated with polyclonal antiserum of 1:2,000 dilutions for 2 h at room temperature, along with 0.5% nonfat milk powder in TBST. The blot was again washed three times in a similar manner and treated with anti-rabbit goat IgG (secondary antibody) conjugated with alkaline phosphatase, at a dilution of 1:3,000 along with 0.5% nonfat milk powder in TBST for 2 h at room temperature. It was then washed again in TBST and incubated in Tris-HCl (pH 9.0) for 10 min. The blot was developed in 20 ml of Tris-HCl using 200 μl of 5 mg/ml of the compound 5-bromo-4-chloro-3′ indolyl phosphate (BCIP) and 200 μl of 30 mg/ml of the compound nitroblue tetrazolium chloride (NBT) as the substrate along with 80 μl of 2 M MgCl2.
Uracil DNA glycosylase assay from cell extracts.
Based on Bradford's protein estimation, 75 ng of the total protein from cell extracts of the strains under study grown to mid-log or stationary phase as indicated was used in the Ung assays. E. coli cells grown in 2 ml LB containing Amp and Tet to mid-log or stationary phase as indicated and processed to prepare cell extracts by sonication (8) were used for Ung assays. The cell extract was used to set up a reaction mixture of 15-μl volume carrying 10,000 cpm of γ-32P-labeled SSU9 DNA oligomer, 1× Ung buffer (diluted from 10× Ung buffer containing 200 mM Tris-HCl, pH 8.0, 10 mM dithiothreitol, 10 mM Na2EDTA, and 1 mg/ml of bovine serum albumin [BSA]), 0.5 pmol of unlabeled (cold) SSU9, and water. The mixture was incubated at 37°C for 20 min, following which freshly prepared 0.2 N NaOH was added to each of the samples to stop the reaction. It was then incubated for 15 min at 90°C, loaded onto a 15% acrylamide gel containing 8 M urea, and separated by gel electrophoresis. The gel was then exposed in a phosphorimager cassette, and an image of the extent of product formation was obtained. The pixel values were quantitated in the spots corresponding to the substrate (S) and product (P), using a BioImageAnalyzer (FLA5000; Fuji). The Ung activities were calculated as pmol of SSU9 oligomer converted to product per microgram total protein [by multiplying the P/(S+P) ratio with the total pmol of SSU9 taken in the reaction divided by the total protein in the cell extract]. Ung activities in the cell extracts were determined from range-finding experiments wherein dilutions of the extracts were reacted with uracil-containing DNA oligomer to determine the fraction of the substrate converted into product. If the chosen dilution resulted in limiting substrate, the extract was diluted further and/or the amount of DNA was increased (see Fig. S3, S5, and S7 in the supplemental material as representatives). Mean activities of the extracts from triplicate sets of cultures (see Fig. 2, 4 and 5) were plotted together with standard errors of the means (SEM).
Fig 2.
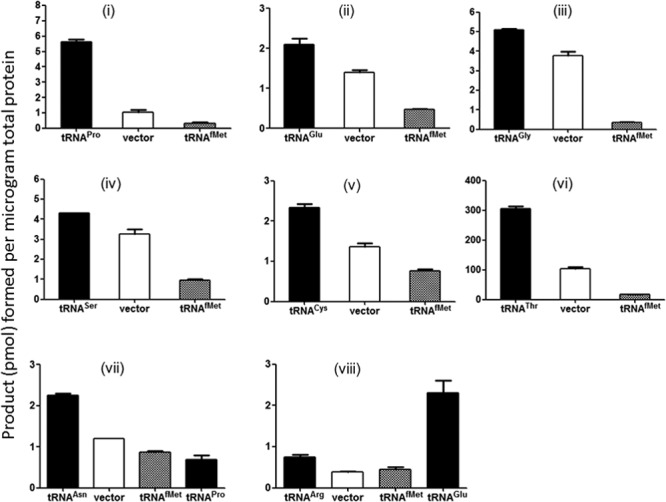
Initiation with elongator tRNAs is uncovered when the initiator tRNA content of the cell is decreased. (i to viii) Initiation activity measured from various start codons using Ung enzyme activity as readout. Three independent colonies were grown to mid-log phase (optical density at 600 nm [OD600], ∼0.6) in LB (Amp, Tet) and then used to prepare cell extracts. Means with standard errors are plotted. Cell extracts were prepared from strain TG1 Δung ΔmetZWV carrying plasmid pTrc-ung with various mutant start codons. In addition, the strain carries either pACDH vector alone (vector), pACDH carrying an elongator tRNA gene (as indicated by the relevant tRNA name), or pACDH carrying initiator tRNA (tRNAfMet). (iii) Results of overexpression of tRNAGly (CCC anticodon) with the reporter harboring the GGU (Gly) initiation codon. In panels vii and viii, a noncognate elongator tRNA has also been used as indicated.
Fig 4.
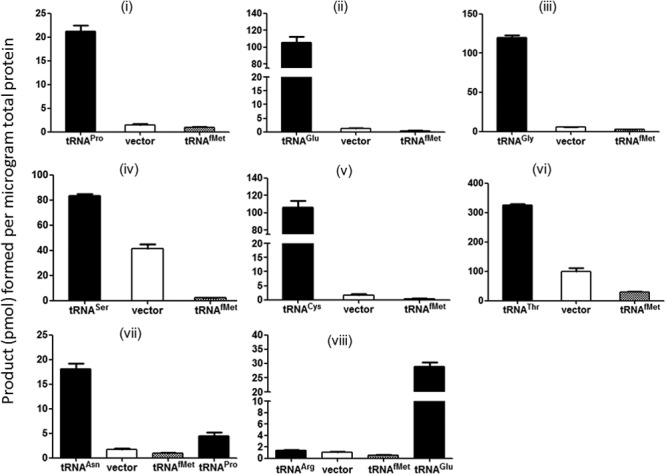
Initiation by elongator tRNAs in stationary phase. (i to viii) Initiation activity measured from various start codons using Ung enzyme activity as readout. Three independent colonies were allowed to reach saturation and then used to prepare cell extracts. Means with standard errors are plotted. Cell extracts were prepared from strain TG1 Δung ΔmetZWV carrying plasmid pTrc-ung with various mutant start codons. In addition, the strain carries either pACDH vector alone (vector), or pACDH carrying an elongator tRNA gene (as indicated by the relevant tRNA name), or pACDH carrying initiator tRNA (tRNAfMet). (iii) Results of overexpression of tRNAGly (CCC anticodon) with the reporter harboring the GGU (Gly) initiation codon. In panels vii and viii, a noncognate elongator tRNA has also been used as indicated.
Fig 5.

Ribosome recycling influences initiation with elongator tRNA. (A) Initiation activity recorded using Ung enzyme activity as a readout in strain LJ14, which carries a TS RRF allele. Three independent colonies of LJ14 harboring pTrc-ung (AAU) along with either pACDH, pACDH-Asn, or pACDH-metY were grown to mid-log phase at 30°C with shaking and then used to prepare cell extracts. Means with standard errors are plotted. (B) Growth analysis of the strains. Three independent colonies were grown as overnight cultures in LB (Amp Tet) at 30°C with shaking and diluted 100-fold in LB (Amp Tet), and growth was monitored using a Bioscreen C growth reader. Means with standard errors are plotted.
Preparation of ribosomes.
Ribosomes were isolated from E. coli KH-1 or E. coli KH-1 ΔmetZWV grown in LB to early log phase. Bacterial cells were collected by centrifugation in a Sorvall GS-3 rotor at 4,000 rpm and 4°C for 10 min and were resuspended in 1 ml lysis buffer (60 mM KCl, 60 mM NH4Cl, 50 mM Tris-HCl [pH 8], 6 mM MgCl2, 6 mM β-mercaptoethanol, 16% sucrose); lysozyme and DNase I (Amresco and GE Healthcare, respectively) were added to final concentrations of 1 mg/ml and 20 U/ml, respectively. The cells were incubated for 15 min at −70°C and then thawed in ice-cold water for 30 min. The freeze-thaw cycle was repeated twice, followed by centrifugation at 13,000 × g and 4°C for 20 min. The supernatant was diluted 2-fold with buffer A (60 mM KCl, 60 mM NH4Cl, 10 mM Tris-HCl [pH 8], 12 mM MgCl2, 6 mM β-mercaptoethanol). Lysate was first loaded onto a 30-ml, 10 to 25% (wt/wt) sucrose gradient prepared in buffer A and then centrifuged at 23,000 rpm in an SW28 rotor (Beckman) at 4°C for 13.5 h. “Heavy” ribosomes (Arg10:Lys8, CNLM-539, and CNLM-291; Cambridge Isotope Laboratories) used as standards in quantitative mass spectrometry (MS) experiments were a kind gift from T. Tammsalu (University of Tartu). Equal amounts of heavy and light ribosomes were mixed and digested with trypsin or doubly digested with LysC and trypsin according to the filter-aided proteome preparation (FASP) protocol (9), and the resulting peptides were fractionated using the SAX-C18 StageTip-based protocol (9) and analyzed using nano-liquid chromatography-coupled mass spectrometry (Nano-LC-MS/MS).
Quantitation of ribosomal proteins by Nano-LC-MS/MS.
Peptides were separated by reversed-phase chromatography using an Agilent 1200 series nanoflow system (Agilent Technologies) connected to an LTQ Orbitrap classic mass spectrometer (Thermo Electron, Bremen, Germany) equipped with a nanoelectrospray ion source (Proxeon, Odense, Denmark). Purified peptides were dissolved in 0.5% formic acid and loaded on a fused silica emitter (75 μm by 150 mm; Proxeon) packed in-house with Repropur-Sil C18-AQ 3-μm particles (Dr. Maisch, Germany) using a flow rate of 700 nl/min. Strong anion exchange (SAX)-fractionated stable isotope labeling with amino acids in culture (SILAC)-labeled peptides were separated with 240-min gradients as follows: pH 3 to 5 fraction, 8 to 36% B gradient; pH 6 fraction, 8 to 35% B gradient; pH 8 fraction, 5 to 33% B gradient; pH 11 fraction, 2 to 30% B gradient (A, 0.5% acetic acid; B, 0.5% acetic acid–80% acetonitrile) at a flow rate of 200 nl/min; isoelectric focusing (IEF)-fractionated SILAC-labeled peptides were separated with 120-min 3 to 40% B (A, 0.5% acetic acid; B, 0.5% acetic acid–80% acetonitrile) gradients at a flow rate of 200 nl/min. Eluted peptides were sprayed directly into an LTQ Orbitrap mass spectrometer operated at 180°C capillary temperature and 2.4-kV spray voltage. The LTQ Orbitrap was operated in the data-dependent mode with up to five MS/MS scans being recorded for each precursor ion scan. Precursor ion spectra were recorded in profile in the Orbitrap (m/z 300 to 1,900; R = 60 000; max injection time, 500 ms; max, 1,000,000 charges); data-dependent MS/MS spectra were acquired in centroid in the LTQ (max injection time, 150 ms; max, 5,000 charges; normalized CE 35%; wideband activation enabled). Monoisotopic precursor selection was enabled, singly charged ions and ions with an unassigned charge state were rejected, and each fragmented ion was dynamically excluded for 120 s. All measurements in the Orbitrap mass analyzer were performed with the lock-mass option enabled (lock-masses were m/z 445.12003 and 519.13882).
Data analysis.
Combined raw data files from SAX and IEF fractionations were analyzed with the MaxQuant software package, version 1.0.13.13 (10). Generated peak lists were searched with the Mascot search engine 2.2 against an E. coli protein sequence database (downloaded from www.ecogene.org on 22 September 2009) supplemented with common contaminants (e.g., human keratins or trypsin) and reversed sequences of all entries in order to estimate false-positive rates. Mascot searches were performed with full tryptic specificity (trypsin/P), a maximum of two missed cleavages, and a mass tolerance of 0.5 Da for fragment ions. Carbamidomethylation of cysteine was set as fixed, and methionine oxidation was set as variable modification. A maximum of three missed cleavages were allowed. In MaxQuant, false-discovery rate (FDR) thresholds were set to 1% at both peptide and protein levels, minimum required peptide length was set to six amino acids, maximum peptide phosphoenolpyruvate (PEP) was set to 0.005, and at least three peptides and two ratio counts were required for protein identification and quantitation.
RESULTS
Generation of the reporter assay system for initiation with the elongator tRNAs.
Uracil DNA glycosylase (Ung, encoded by ung) is a highly specific and efficient enzyme that excises uracil from DNA (11) and provides a sensitive reporter to study protein synthesis. Hence, we subjected the plasmid pTrc-ung, carrying the ung gene, to mutagenesis to generate a range of non-AUG triplets at the first codon position (Fig. 1A). These constructs were then introduced into E. coli TG1 Δung ΔmetZWV, lacking Ung protein and the initiator tRNA encoded by metZWV genes, to assess initiation from each of the constructs. Ung activity was estimated in vitro by supplying a radioactively labeled DNA oligomer carrying uracil. Excision of uracil followed by cleavage of the generated abasic site leads to production of two fragments, one of which is radioactively labeled (see Fig. S2 in the supplemental material). The radiolabeled uncleaved (leftover substrate) and the cleaved (product) DNAs are separated by denaturing gel, quantified by phosphorimager, and plotted as bar graphs (see Materials and Methods and Fig. S3, S5, and S7 in the supplemental material).
Fig 1.
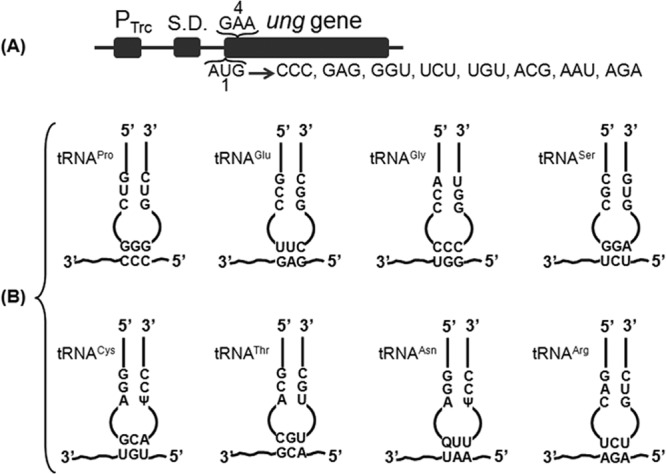
Assay system. (A) Schematic showing mutagenesis of the ATG start codon of the ung gene using the plasmid pTrc-ung as the template. The list shows the various combinations generated by random mutagenesis of the start ATG site. The first (AUG) and the fourth (GAA) codons in the ung gene are shown. The ung ORF was cloned into pTrc99c to generate pTrc-ung, and hence the mRNA bears the Shine-Dalgarno (S.D.) sequence of the expression vector. (B) Schematic representation of the elongator tRNAs used in this study and the changed start codons corresponding to them. The third schematic in the top row shows a mismatched pairing of tRNAGly (CCC anticodon) with the GGU codon for Gly. The anticodon and the region corresponding to the 3 G-C base pairs in the initiator tRNA anticodon stem are indicated.
Elongator tRNAs initiate protein synthesis when the initiator tRNA content of the cell is decreased.
To see if depletion of initiator tRNA impacted the ability of elongator tRNAs to decode at the P site, the metZWV genes were knocked out of E. coli TG1, removing ∼75% of the initiator tRNA content of the cell (3). Figure 1B shows pairings of the mutant (non-AUG) initiation codons and the corresponding tRNA anticodons. Each of the Ung reporter plasmids (Fig. 1A) was introduced into E. coli TG1 Δung ΔmetZWV. A compatible plasmid (vector alone) or its derivative harboring the desired elongator tRNA gene was also introduced in the strain. When the corresponding elongator tRNAs were overexpressed, enzyme activities of the Ung reporters used went up compared with those of the vector-alone controls (Fig. 2i to viii). It may be mentioned that the initiation activities of the vector-alone controls are due to the cellular pools of respective elongator tRNAs. However, overexpression of initiator tRNA (instead of the cognate elongator tRNA) with the mutant Ung constructs resulted in substantially decreased Ung activities compared to the vector alone control, indicating that the initiator tRNA prevented the elongator tRNAs that decoded the changed start codons from gaining access to the P site (Fig. 2i to viii). As seen in Fig. 2iii, initiation from the GGU codon (Gly) using tRNAGly with the CCC anticodon having a mismatch at the third base of the codon resulted in only a small increase. Also, when we overexpressed a noncognate tRNAPro with the AAU start codon (Fig. 2vii), it did not lead to an increase in Ung activity (compare the AAU codon [Asn] with tRNAPro overexpression and the same codon with tRNAAsn overexpression and vector-alone activity), suggesting specificity of initiation with elongator tRNAs.
We noted that initiation from the AGA (Arg) construct was the lowest of all the constructs even upon overexpression of tRNAArg (Fig. 2viii) (see Discussion). Interestingly, when tRNAGlu (UUC anticodon) corresponding to the fourth codon (GAA) in the reporter (12) was overexpressed, it resulted in higher Ung activity (Fig. 2viii), suggesting that elongator tRNAs initiated even from a codon located suboptimally from the Shine-Dalgarno (SD) sequence. In some cases (tRNASer and tRNAGly), the activity from the native elongator tRNA (vector control) itself is high, so the increase upon overexpression of the cognate elongator tRNA is not as high as in other cases.
To gain a perspective on how strong these initiation activities are, we also recorded activity of the wild-type ung gene (with the AUG initiation codon), which showed a conversion of ∼80,000 pmol/μg total protein (see Fig. S3 in the supplemental material). The highest activity of an elongator here was by tRNAThr, which showed a conversion of ∼300 pmol/μg total protein (see Discussion).
Ribosomal particles of E. coli ΔmetZWV strains.
Correct ribosome subunit assembly depends upon production of stoichiometric amounts of ribosomal protein (13). Inhibition of protein synthesis often leads to ribosome assembly defects (14). Besides, ribosome assembly is temperature sensitive. Given that the E. coli strain with deletions of the metZWV genes is cold sensitive (2, 3), it was of interest to analyze its ribosome profile. As shown in Fig. 3, the ribosome profiles of the ΔmetZWV mutant and the wild-type strains grown at different temperatures of 25, 30, and 37°C were qualitatively similar at all temperatures. As even small ribosome assembly defects manifest as alteration of ribosome profile at lower temperatures (15), the ribosome profiles in Fig. 3 suggest that the reduced level of initiator tRNA does not cause any significant ribosome assembly defects. Interestingly, however, the relative amount of free ribosomal subunits is larger in the mutant strain than in the wild-type strain. This may be due to deficient translation initiation, although a deficiency at the later steps of ribosome assembly is not ruled out. Further, to see if the lower growth rate of the ΔmetZWV strains is correlated with cell size, we compared their cell sizes with that of the wild type and found that the two are near identical (see Fig. S4 in the supplemental material).
Fig 3.
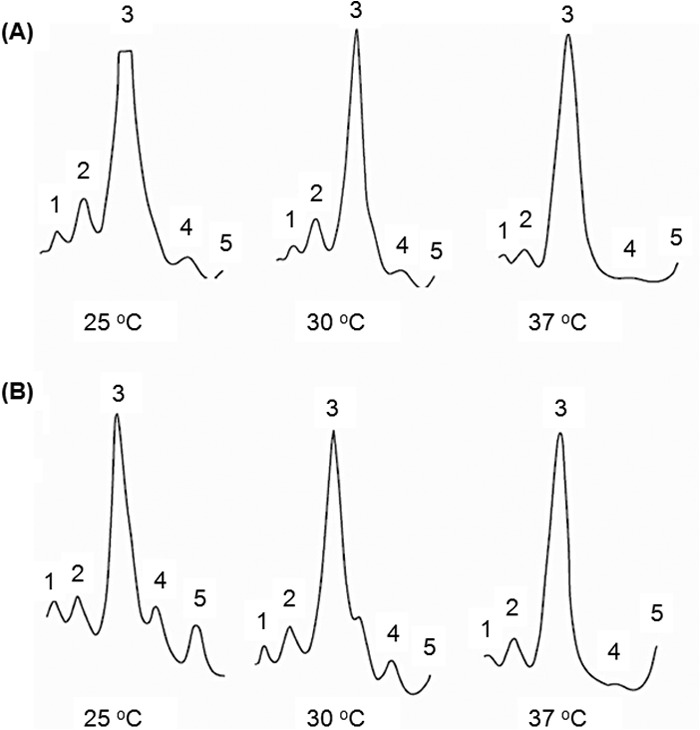
Ribosome profiles of wild-type E. coli and E. coli ΔmetZWV. Ribosome profiles of E. coli strain MG1655 (A) and E. coli strain MG1655 ΔmetZWV (B) at 25°C, 30°C, and 37°C are shown. The direction of sedimentation is from right to left. Peaks: 1, trisome; 2, disome; 3, 70S; 4, 50S; 5, 30S.
Proteomic analysis shows that strains lacking metZWV are deficient for protein S1 in their ribosomes.
Ribosomal particles were further characterized by a quantitative proteomic approach. To this end, 70S ribosomes of ΔmetZWV strains were mixed with equal amount of “heavy” standard ribosomes. Standard ribosomes contain heavy-isotope-labeled proteins (all Arg and Lys residues have molecular mass increments of +10 Da and +8 Da, respectively). Ribosomal proteins of the mixed ribosome population were digested with trypsin or LysC followed by quantitation of heavy and light peptides by high-pressure liquid chromatography (HPLC)-MS. The ratio of intensities of heavy and light peptides reflects the ratio of the corresponding proteins. All ribosomal proteins were found to be present in both types of ribosomes at the same level except the protein S1 (the relative ribosomal protein composition of 70S ribosomes in the wild-type versus ΔmetZWV strains according to quantitative HPLC-MS results for each r-protein was calculated as the R-protein average [without S1] of 1.0 ± 0.05, whereas for S1 protein it was 0.85 ± 0.02). Thus, the 70S ribosomes isolated from the initiator tRNA-deficient strain contain ∼15% less protein S1 than the parent strain. Protein S1 is known to affect mRNA binding to the ribosome (16). As binding of S1 to the small ribosome subunit is labile and is easily exchangeable (17), small changes such as those seen here may not be significant in general. However, given that the initiation activities of the elongator tRNAs are also low, we considered that such a small change in S1 on ribosomes, occurring as a consequence of deletion of metZWV, may contribute to the initiation activity of the elongator tRNAs.
Initiation by elongator tRNAs in stationary phase.
Ribosomal protein S1 has been implicated in the regulation of stationary-phase sigma factor RpoS (18) as well as in selective translation of mRNAs by bacterial ribosomes (19). Hence, in the light of the finding that the ribosomes in E. coli with deletion of metZWV were at least partially deficient in protein S1, we investigated if the efficiency of translational initiation by elongator tRNAs was affected during stationary phase. As shown in Fig. 4, translational initiation by all elongator tRNAs increased significantly during stationary phase. Interestingly, initiation increased even from the GGU (Gly) codon upon overproduction of tRNAGly with the CCC anticodon (Fig. 4iii) (see Discussion). As a control, tRNAfMet overproduction continued to result in suppression of initiation by elongator tRNAs (Fig. 4i to viii). In addition, as seen in Fig. 4vii, introduction of a plasmid version of the unrelated elongator tRNA led to only a minor increase in initiation, significantly less than that seen upon overexpression of the cognate tRNA. And, as seen with the log-phase cultures (Fig. 2viii), when tRNAGlu was overexpressed together with the AGA construct (Fig. 4viii), once again, it showed increased Ung activity. Furthermore, to ensure that the plasmid copy number itself was not increasing in stationary phase, we carried out immunoblot analysis for the plasmid-encoded β-lactamase protein and found that the levels remained the same across log and stationary phases in all cases (see Fig. S6 in the supplemental material). The wild-type ung gene again showed a conversion of ∼80,000 pmol/μg total protein (see Fig. S5 in the supplemental material), which is about the same as it did in the log phase.
Ribosome recycling influences initiation with elongator tRNA.
Initiation is closely linked with the step of ribosome recycling, as the free subunits generated in the last step are utilized for the next round of translation. Previous work indicated that a compromised recycling function could lead to loss of fidelity of initiation (20, 21). The rationale behind this idea was that, in addition to recycling posttermination complexes, ribosome recycling factor (RRF) could also dissociate initiation complexes carrying a wrong tRNA. Using our assay system, we carried out the same assays in a temperature-sensitive (TS) RRF background, to check if initiation with elongator tRNAs would now increase. As observed in Fig. 5A, compromising RRF function leads to an increase in the enzyme activity as seen with the start codon AAU upon overexpression of tRNAAsn. Again, this activity fell upon overexpression of initiator tRNA. It is important to note that this occurs even without the depletion of initiator tRNA. That is, compromising RRF function seems to have the same effect as depleting initiator tRNA. In fact, in the TS RRF background, we failed to delete the metZWV locus (perhaps due to high non-AUG initiation events). This would predict that the growth of a strain with compromised RRF function is retarded by events of wrongful initiation; i.e., the growth should improve upon overexpression of the wild-type initiator tRNA. And indeed, as is seen from Fig. 5B, overexpression of the wild-type initiator tRNA does improve growth at the permissive temperature of 30°C, while overexpression of the same initiator tRNA with a mutant anticodon CUA (such that it can only read a stop codon UAG at the first position and hence cannot decode cellular mRNAs) does not, indicating that restoration of initiation function is necessary for the toxicity to be offset.
DISCUSSION
Traditionally, the functions of initiator and elongator tRNAs are thought to be nonoverlapping. However, previous studies have shown that a depletion of the initiator tRNA content in the cell leads to enhanced initiation with mutant initiator tRNA molecules (3, 22). It was also found that an amber suppressor elongator tRNA shows initiation activity from a UAG start codon upon depletion of cellular initiator tRNA (3). This observation brought to light the importance of the relative concentrations of the two tRNA species in determining the fidelity of translation initiation. We designed an assay system to examine if native elongator tRNAs would initiate when competition from the initiator tRNA is reduced. We find that initiation with elongator tRNAs is indeed uncovered in vivo in such a scenario. Further weight is added to the observation by the fact that overexpression of initiator tRNA leads to a fall in reporter activity, indicating that the elongators are indeed initiating from the P site and prevented from doing so when the numbers of wild-type initiator tRNA molecules are increased. As the relative levels of initiator and elongator tRNAs and their aminoacylated forms are known to vary under different physiological conditions (23–25), our findings are biologically relevant. Earlier studies have mutagenized elongator tRNAs step by step, leading to gain of initiator function (7), and observed initiation from mutant initiator tRNAs upon depletion of the intracellular initiator tRNA content (22), but to the best of our knowledge this is the first time that initiation activity has been directly recorded from naturally occurring elongator tRNAs in vivo.
One of the crucial features of the initiator tRNAs is the presence of the three consecutive G-C base pairs in their anticodon stem (26). Some of the elongator tRNAs tested show significantly high initiation activity. Given the importance of the three G-C base pairs in determining the function of the initiator tRNA, we first looked at the status of the G-C base pairs in the anticodon stem. However, there is no apparent correlation between those tRNAs that bear two intact G-C base pairs and those that display initiation activity. tRNAAsn, which gives among the lowest increases in initiation, has the first two G-C base pairs intact, whereas tRNAGlu, which shows a higher initiation activity, does not. tRNASer, which shows activity comparable with that of tRNAGlu in our system, also has the first two G-C base pairs intact. Most of all, tRNAThr, which shows by far the highest increase in initiation, has only one G-C base pair, the first. Hence, the role of the three G-C pairs in determining which elongators can initiate appears unclear. It may well be that their role is context dependent, involving other features of the elongator tRNA. Another possibility, given that the whole phenomenon is determined by relative numbers, is that the most abundant tRNAs are the ones that initiate the best. However, upon examination of documented amounts of each species of tRNA in the E. coli cell (27), this appears to not be the ruling factor. Although tRNAGlu is among the most abundant tRNAs (∼7%), neither tRNAThr (∼1.4%) nor tRNAPro (∼1.1%), both among the strongest initiators in our assay, falls in the abundant category. A pairwise sequence comparison of initiator tRNA with elongator tRNAs revealed ∼47 to 59% sequence identity (metT, 51.1%; glyU, 54.4%; gltW, 57.8%; proL, 52.6%; thrW, 44.9%; asnU, 59.3%; serX, 50%; cysT, 47.4%; argU, 46.9%). tRNAAsn and tRNAArg show the highest similarities (50% and 42%), incidentally well above the similarity of tRNAMet to initiator tRNA (37%). Again this does not correlate, as tRNAArg in fact performs as the poorest initiator. However, it should also be said that at least the presence of Arg at the N terminus may severely shorten the half-life of the translated protein (28). Another possible explanation for the highest initiation activity of tRNAThr (among the ones tested here) could be that it is one of three codons (among the ones tested) that require no wobble pairing with the tRNA anticodon in the P site. The other two codons that also fall into this category are AGA (Arg) and CCC (Pro). While the lower efficiency of AGA (Arg) has been discussed above, lower initiation from CCC (Pro) could be due to its lower efficiency of peptide bond formation with the incoming amino acid in the A site (29–31). Other codons tested in this study are in fact decoded by tRNAs requiring wobble pairing with the third nucleotide of the codon, which are not normally favored for initiation (32). An unexpected observation was made with tRNAGly (CCC anticodon), whose overexpression resulted in a substantial increase in initiation from the GGU codon during the stationary phase in spite of the mismatched pairing (Fig. 4iii). Interestingly, this pairing, involving a mismatch at the third position of the codon, is reminiscent of the pairing of the CAU anticodon of tRNAfMet with the AUU initiation codon for translation of IF3 (33). Nevertheless, it should also be said that the reasons why some elongator tRNAs are able to function better than the others at the step of initiation remain unclear and need further investigation.
It was observed that the ribosomes in the strain deficient for tRNAfMet possessed substoichiometric amounts of S1. It is therefore possible that stoichiometric levels of S1 protein are important for preventing initiation with elongator tRNAs. It remains unclear, however, how deficiency of S1 may facilitate initiation with elongator tRNAs. This observation is of particular interest, as it is known to play a role in the selection of the start site on the mRNA (34–37). In addition, unlike with canonical mRNAs, it has been shown that leaderless mRNAs do not require the presence of S1 to bind the ribosome and be translated (38). This could provide a means for non-AUG initiation to occur, although it still begs the question as to why there should be smaller amounts of S1 in the ribosomes of a strain depleted of initiator tRNA content. An examination of the whole proteome in E. coli wild-type and ΔmetZWV strains yielded several minor differences, none of which involve translation-associated factors (data not shown). Thus, it remains unclear what factors may contribute to initiation with the elongator tRNAs. However, we noticed that E. coli strain LJ14, harboring a temperature-sensitive RRF, showed significant initiation with elongator tRNAs even when metZWV was not deleted, indicating that initiation with 70S ribosomes could contribute to the phenomenon. Furthermore, loss of fidelity check at the step of initiation due to RRF deficiency (21) could also have a role to play in initiation by elongator tRNAs, at least in E. coli LJ14.
Recent studies with an initiator tRNA-targeting enterotoxin produced by Salmonella and Shigella species called VapC showed that initiation from non-AUG codons could be uncovered when the toxin was overexpressed (39). Also, studies using eukaryotic antigen-presenting cells uncovered a novel alternate initiation strategy whereby the cell used either CTG (Leu) or the canonical ATG (Met) to generate different peptides for display to the immune system. However, a specific depletion of IF2-bound initiator tRNA did not lead to an increase in initiation with leucine elongator tRNA (40), indicating that other regulation was involved. Therefore, the selective alternate initiation using the CTG start site appears to be mechanistically different from the initiation by elongator tRNAs presented in this study.
The increasing number of examples of such promiscuous initiation by elongator tRNAs suggests that the phenomenon could well be exploited by the cell when the amount of initiator tRNA is reduced. Reduction in the amount of initiator tRNA by elimination of the metZWV genes leads to a distinct growth retardation as well as to cold sensitivity. Under such a stressful situation, the cell could potentially initiate at non-AUG sites along a canonical (or alternate) ORF, generating alternate peptides that could generate diversity in the peptide pool. Alternately, the elongator tRNAs capable of initiating could act at entirely new sites near SD-like sequences, generating entirely novel peptides. However, we speculate that if the S1 protein level is indeed low, it could even lead to initiation by elongator tRNAs independent of SD-like sequences. The fact that when tRNAGlu was overexpressed together with the AGA construct (Fig. 2 and 4viii) it effected initiation may also support this view. Initiation by the elongator tRNAs could be thought of as an SOS-like response mounted by the cell, but at the level of the proteome rather than at the level of the genome, incurring the risk of accumulating faulty peptides while generating some novel and potentially useful combinations. However, in view of the low levels of such activity (in comparison with that of the wild-type initiator tRNA) and the overwhelming background of initiation by initiator tRNA, it is also likely to be a rather weak phenomenon whose major role may be to signal a cellular strategy to offer the cell a fitness advantage under stressful conditions.
Supplementary Material
ACKNOWLEDGMENTS
We thank our laboratory colleagues and the anonymous reviewers for their suggestions on the manuscript.
This work was supported by grants from the Departments of Science and Technology (DST) and Biotechnology (DBT), New Delhi, India. U.V. is a J. C. Bose fellow of DST.
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00637-13.
REFERENCES
- 1.Kenri T, Imamoto F, Kano Y. 1994. Three tandemly repeated structural genes encoding tRNA(f1Met) in the metZ operon of Escherichia coli K-12. Gene 138:261–262 [DOI] [PubMed] [Google Scholar]
- 2.Kenri T, Kohno K, Goshima N, Imamoto F, Kano Y. 1991. Construction and characterization of an Escherichia coli mutant with a deletion of the metZ gene encoding tRNA (f1Met). Gene 103:31–36 [DOI] [PubMed] [Google Scholar]
- 3.Kapoor S, Das G, Varshney U. 2011. Crucial contribution of the multiple copies of the initiator tRNA genes in the fidelity of tRNA(fMet) selection on the ribosomal P-site in Escherichia coli. Nucleic Acids Res. 39:202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seong BL, RajBhandary UL. 1987. Mutants of Escherichia coli formylmethionine tRNA: a single base change enables initiator tRNA to act as an elongator in vitro. Proc. Natl. Acad. Sci. U. S. A. 84:8859–8863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seong BL, Lee CP, RajBhandary UL. 1989. Suppression of amber codons in vivo as evidence that mutants derived from Escherichia coli initiator tRNA can act at the step of elongation in protein synthesis. J. Biol. Chem. 264:6504–6508 [PubMed] [Google Scholar]
- 6.Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. 1998. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 26:148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varshney U, Lee CP, RajBhandary UL. 1993. From elongator tRNA to initiator tRNA. Proc. Natl. Acad. Sci. U. S. A. 90:2305–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor laboratory, Cold Spring Harbor, NY [Google Scholar]
- 9.Wisniewski JR, Zougman A, Mann M. 2009. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8:5674–5678 [DOI] [PubMed] [Google Scholar]
- 10.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 11.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. 1977. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 252:3286–3294 [PubMed] [Google Scholar]
- 12.Varshney U, Hutcheon T, van de Sande JH. 1988. Sequence analysis, expression, and conservation of Escherichia coli uracil DNA glycosylase and its gene (ung). J. Biol. Chem. 263:7776–7784 [PubMed] [Google Scholar]
- 13.Nomura M. 1984. The control of ribosome synthesis. Sci. Am. 250:102–114 [DOI] [PubMed] [Google Scholar]
- 14.Siibak T, Peil L, Donhofer A, Tats A, Remm M, Wilson DN, Tenson T, Remme J. 2011. Antibiotic-induced ribosomal assembly defects result from changes in the synthesis of ribosomal proteins. Mol. Microbiol. 80:54–67 [DOI] [PubMed] [Google Scholar]
- 15.Peil L, Virumae K, Remme J. 2008. Ribosome assembly in Escherichia coli strains lacking the RNA helicase DeaD/CsdA or DbpA. FEBS J. 275:3772–3782 [DOI] [PubMed] [Google Scholar]
- 16.Sorensen MA, Fricke J, Pedersen S. 1998. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo. J. Mol. Biol. 280:561–569 [DOI] [PubMed] [Google Scholar]
- 17.Pulk A, Liiv A, Peil L, Maivali U, Nierhaus K, Remme J. 2010. Ribosome reactivation by replacement of damaged proteins. Mol. Microbiol. 75:801–814 [DOI] [PubMed] [Google Scholar]
- 18.Sevo M, Buratti E, Venturi V. 2004. Ribosomal protein S1 specifically binds to the 5′ untranslated region of the Pseudomonas aeruginosa stationary-phase sigma factor rpoS mRNA in the logarithmic phase of growth. J. Bacteriol. 186:4903–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moll I, Grill S, Grundling A, Blasi U. 2002. Effects of ribosomal proteins S1, S2 and the DeaD/CsdA DEAD-box helicase on translation of leaderless and canonical mRNAs in Escherichia coli. Mol. Microbiol. 44:1387–1396 [DOI] [PubMed] [Google Scholar]
- 20.Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. 2006. How initiation factors maximize the accuracy of tRNA selection in initiation of bacterial protein synthesis. Mol. Cell 23:183–193 [DOI] [PubMed] [Google Scholar]
- 21.Seshadri A, Dubey B, Weber MH, Varshney U. 2009. Impact of rRNA methylations on ribosome recycling and fidelity of initiation in Escherichia coli. Mol. Microbiol. 72:795–808 [DOI] [PubMed] [Google Scholar]
- 22.Samhita L, Shetty S, Varshney U. 2012. Unconventional initiator tRNAs sustain Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 109:13058–13063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanduc D. 1997. Changes of tRNA population during compensatory cell proliferation: differential expression of methionine-tRNA species. Arch. Biochem. Biophys. 342:1–5 [DOI] [PubMed] [Google Scholar]
- 24.Elf J, Nilsson D, Tenson T, Ehrenberg M. 2003. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300:1718–1722 [DOI] [PubMed] [Google Scholar]
- 25.Dittmar KA, Sorensen MA, Elf J, Ehrenberg M, Pan T. 2005. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seong BL, RajBhandary UL. 1987. Escherichia coli formylmethionine tRNA: mutations in GGGCCC sequence conserved in anticodon stem of initiator tRNAs affect initiation of protein synthesis and conformation of anticodon loop. Proc. Natl. Acad. Sci. U. S. A. 84:334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong H, Nilsson L, Kurland CG. 1996. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 260:649–663 [DOI] [PubMed] [Google Scholar]
- 28.Tobias JW, Shrader TE, Rocap G, Varshavsky A. 1991. The N-end rule in bacteria. Science 254:1374–1377 [DOI] [PubMed] [Google Scholar]
- 29.Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339:85–88 [DOI] [PubMed] [Google Scholar]
- 30.Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K. 2013. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339:82–85 [DOI] [PubMed] [Google Scholar]
- 31.Bullwinkle TJ, Zou SB, Rajkovic A, Hersch SJ, Elgamal S, Robinson N, Smil D, Bolshan Y, Navarre WW, Ibba M. 2013. (R)-beta-lysine-modified elongation factor P functions in translation elongation. J. Biol. Chem. 288:4416–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arora S, Bhamidimarri SP, Bhattacharyya M, Govindan A, Weber MH, Vishveshwara S, Varshney U. 2013. Distinctive contributions of the ribosomal P-site elements m2G966, m5C967 and the C-terminal tail of the S9 protein in the fidelity of initiation of translation in Escherichia coli. Nucleic Acids Res. 41:4963–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gold L. 1988. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu. Rev. Biochem. 57:199–233 [DOI] [PubMed] [Google Scholar]
- 34.Suryanarayana T, Subramanian AR. 1983. An essential function of ribosomal protein S1 in messenger ribonucleic acid translation. Biochemistry 22:2715–2719 [DOI] [PubMed] [Google Scholar]
- 35.Thomas JO, Szer W. 1982. RNA-helix-destabilizing proteins. Prog. Nucleic Acid Res. Mol. Biol. 27:157–187 [DOI] [PubMed] [Google Scholar]
- 36.Hartz D, McPheeters DS, Gold L. 1991. Influence of mRNA determinants on translation initiation in Escherichia coli. J. Mol. Biol. 218:83–97 [DOI] [PubMed] [Google Scholar]
- 37.Roberts MW, Rabinowitz JC. 1989. The effect of Escherichia coli ribosomal protein S1 on the translational specificity of bacterial ribosomes. J. Biol. Chem. 264:2228–2235 [PubMed] [Google Scholar]
- 38.Tedin K, Resch A, Blasi U. 1997. Requirements for ribosomal protein S1 for translation initiation of mRNAs with and without a 5′ leader sequence. Mol. Microbiol. 25:189–199 [DOI] [PubMed] [Google Scholar]
- 39.Winther KS, Gerdes K. 2011. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U. S. A. 108:7403–7407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starck SR, Jiang V, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. 2012. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 336:1719–1723 [DOI] [PubMed] [Google Scholar]
- 41.Low B. 1968. Formation of merodiploids in matings with a class of Rec- recipient strains of Escherichia coli K12. Proc. Natl. Acad. Sci. U. S. A. 60:160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning. a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 43.Janosi L, Mottagui-Tabar S, Isaksson LA, Sekine Y, Ohtsubo E, Zhang S, Goon S, Nelken S, Shuda M, Kaji A. 1998. Evidence for in vivo ribosome recycling, the fourth step in protein biosynthesis. EMBO J. 17:1141–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bharti SK, Varshney U. 2010. Analysis of the impact of a uracil DNA glycosylase attenuated in AP-DNA binding in maintenance of the genomic integrity in Escherichia coli. Nucleic Acids Res. 38:2291–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao AR, Varshney U. 2001. Specific interaction between the ribosome recycling factor and the elongation factor G from Mycobacterium tuberculosis mediates peptidyl-tRNA release and ribosome recycling in Escherichia coli. EMBO J. 20:2977–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.