

Abstract
MYO15A is located at the DFNB3 locus on chromosome 17p11.2, and encodes myosin-XV, an unconventional myosin critical for the formation of stereocilia in hair cells of cochlea. Recessive mutations in this gene lead to profound autosomal recessive nonsyndromic hearing loss (ARNSHL) in humans and the shaker2 (sh2) phenotype in mice. Here, we performed a study on 140 Iranian families in order to determine mutations causing ARNSHL. The families, who were negative for mutations in GJB2, were subjected to linkage analysis. Eight of these families showed linkage to the DFNB3 locus, suggesting a MYO15A mutation frequency of 5.71% in our cohort of Iranian population. Subsequent sequencing of the MYO15A gene led to identification of 7 previously unreported mutations, including 4 missense mutations, 1 nonsense mutation, and 2 deletions in different regions of the myosin-XV protein.
Keywords: autosomal recessive nonsyndromic hearing loss, DFNB3, Myo15A, Iran
INTRODUCTION
Nonsyndromic autosomal recessive deafness accounts for up to 80% of nonsyndromic hereditary hearing loss. To date, 95 loci responsible for this form of deafness have been mapped and 40 genes identified (Van Camp G, Smith RJH. 2011. http://www.hereditaryhearingloss.org). Autosomal recessive nonsyndromic hearing loss (ARNSHL) is generally congenital, prelingual, and severe-to-profound. In many different parts of the world, GJB2 mutations in the DFNB1 locus have been reported as the most common cause of nonsyndromic hearing loss [Nance et al., 2000].
The DFNB3 locus was initially identified in a population of 2,200 villagers of Bengkala; Bali. Homozygous individuals for DFNB3 are profoundly deaf from birth, with no other associated clinical features. This form of ARNSHL is caused by homozygous mutations in MYO15A, which encodes one of the unconventional myosins, myosin-XV [Friedman et al., 1995; Winata et al., 1995].
Myosins are a large family of actin-dependent molecular motors that bind and hydrolyze ATP to generate the force enabling the movement of actin filaments, which is necessary to drive a wide variety of cellular functions [Mooseker and Cheney, 1995; Krendel and Mooseker, 2005]. The myosin superfamily is subdivided according to sequence divergence of the motor domain and different tail domains. Conventional myosins belong to class II myosins and the other classes are referred as unconventional myosins [Friedman et al., 1999]. So far, about 20 classes of unconventional myosins have been identified [Krendel and Mooseker, 2005; Kalay et al., 2007].
Each myosin protein is composed of three evolutionarily conserved domains: the motor domain, neck region, and the tail domain. The motor domain is usually near the N-terminus and is highly conserved. One or more light chain binding IQ motifs are present in the neck region; the C-terminal tails are divergent in length and sequence. Therefore, it is assumed that these unique tail regions give specific binding properties for transport and association with membranes [Friedman et al., 1999].
Among 20 classes of unconventional myosins, five genes encoding myosin Ia, IIIa, VI, VIIa, XVa are necessary for auditory function. MYH9 and MYH14 have roles in normal auditory function and belong to conventional myosin genes [Kalay et al., 2007].
The MYO15A gene (OMIM #602666) is located on chromosome 17p11.2 and has 66 exons comprising 71,097 bp. The protein encoded by this gene, myosin-XV, is composed of 3,530 amino acids and is about 39.5 KDa in its longest form. (MYO15_HUMAN, Q9UKN7, and NP_057323.3). According to presence or lack of exon 2, there are two alternatively spliced transcripts; isoform classes I and 2 [Nal et al., 2007]. Thus, the long isoform of the human myosin XVA consists of an N-terminal domain (amino acids 1–1,223), a motor head domain (amino acids 1,224–1,899) which contains two binding sites for ATP and actin, the switches I and II helix, relay helix, SH3 helix, SH1/SH2 helix, and a converter domain. The neck region contains two light chain-binding motifs (IQ; amino acids 1,909–1,942). The tail region contains 2 MyTH4 (myosin tail homology 4) domains (amino acids 2,066–2,174 and 3,051–3,161), 2 FERM domains (amino acids 2,687–2,867 and 3,217–3,497) which have role in binding of cytoskeletal proteins to cytoplasmic domains of transmembrane proteins, a putative SH3 (src homology 3) domain (amino acids 2,865–2,959), and a C-terminal class I PDZ-binding motif [Garcia-Alvarez et al., 2003; Kalay et al., 2007] (Fig. 1).
FIG. 1.
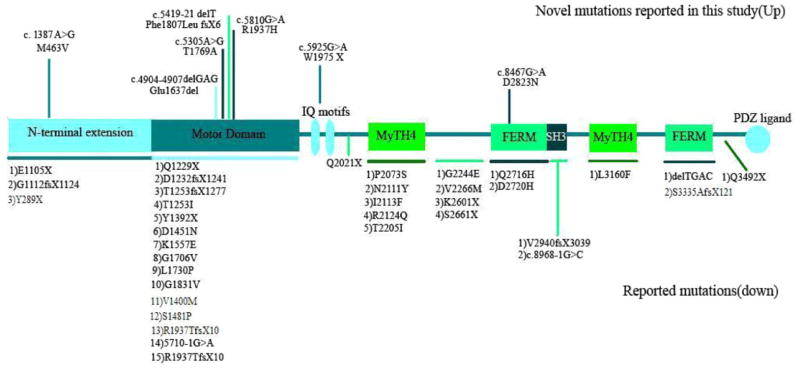
MYO15A gene structure, with novel mutations reported in this study indicated at the top of the figure and previously reported mutations listed at the bottom of the figure.
The myosin-XV protein has been shown to be integral for development and elongation of the stereocilia through delivery of whirlin to the tips of stereocilia. Whirlin binds to SH3-MYTH4-FERM-domain-containing region of the Myo15a protein and regulates actin filament elongation. Additionally, myosin-XV interaction with whirlin may play a role in cohesion of stereocilia [Wang et al., 1998; Belyantseva et al., 2005; Delprat et al., 2005; Kikkawa et al., 2005; Kalay et al., 2007].
There have been several reports of mutations in MYO15A causing hearing loss [Wang et al., 1998; Friedman et al., 1999; Liburd et al., 2001; Kalay et al., 2007; Nal et al., 2007; Lezirovitz et al., 2008; Belguith et al., 2009; Shearer et al., 2009; Cengiz et al., 2010]. Forty-three mutations have been previously reported in MYO15A, and primarily occur in the motor domain. In this study, we examined 140 Iranian families affected by deafness in order to determine the cause of deafness. We found eight families with genetic linkage to the DFNB3 locus and identified seven novel mutations in MYO15A (Fig. 1).
MATERIALS AND METHODS
Patients
Patients were 140 mostly consanguineous families of Iranian origin segregating ARNSHL. At least two affected children were observed in each family. Clinical examination was performed and environmental factors were eliminated as the cause of deafness in all affected family members. Features suggestive of syndromic anomalies were not present. Also, audiologic profiling was performed for all the patients who suffered from severe-to-profound hearing loss which was congenital and prelingual.
Informed consent was obtained from all participating subjects and then blood samples were obtained from family members. The Genetics Research Center at the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran, approved all procedures.
Genotyping and Mutation Analysis
Genomic DNA was isolated from peripheral blood samples according to a standard procedure [Miller et al., 1988]. All of the 140 families were analyzed for allele segregation at the DFNB3 locus on chromosome 17p11.2, using microsatellite markers closely linked to its gene and homozygosity mapping was performed.
The studied subjects were genotyped for microsatellite markers flanking the MYO15A gene (18,012,020–18,083,116 bp). Micro-satellite markers D17S2196 (17264393–17264673 bp), D17S842 (20667275–20667641 bp), D17S2187 (20694827–20695353 bp), D17S975 (28099976–28100268 bp), and D17S1857 (16415217–16415593 bp) had been selected and used for genotyping at least two affected and one normal children of all 140 families at the first screening stage. These markers were amplified according to a standard method and analyzed by conventional polyacrylamide gel electrophoresis (8%) and silver staining. PCR reactions were optimized as following; initial denaturation (95°C for 4 min), Denaturation (94°C for 40 sec), annealing (55°C or 60°C for 30 sec), extension (72°C for 40 sec) followed by final extension at 72°C for 2 min. While observing homozygosity for the affected children, additional microsatellite analysis was performed for the core family and the other branches of the pedigree, to confirm the cosegregation of the homozygous pattern with the phenotype.
Mutation analysis of MYO15A was performed for eight families who showed linkage to this locus. Conventional DNA sequencing of the coding exons and their splice junctions in addition to the 5′- and 3′-UTRs was performed for each proband of linked families, using Big Dye Terminators (Applied Biosystems 3130 Genetic Analyzer, Foster City, CA). Primers for sequencing (Supplementary Table I) were selected using bioinformatics softwares (Exon Primer, http://ihg.gsf.de/ihg/ExonPrimer.html; Primer 3, http://frodo.wi.mit.edu/primer3/; and Oligoanalyzer3.1, http://eu.idtdna.com/analyzer/applications/oligoanalyzer/).
Segregation study in family members was performed following the identification of mutations in probands. Additionally, 200 normal Iranian chromosomes were sequenced for each of the novel missense mutations.
The identified novel missense mutations in this study were analyzed by different softwares; Polyphen (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org), and ConSeq (http://conseq.tau.ac.il/).
RESULTS
In this study, homozygosity mapping of 140 Iranian families affected by severe-to-profound ARNSHL showed that eight families were linked to the DFNB3 locus. These data suggest the MYO15A mutation frequency of 5.71%, in our cohort of Iranian population. We also report on 7 novel MYO15A mutations including 4 missense mutations (1 of them is controversial), 1 nonsense mutation, and 2 deletions (Table I). All the mutations showed segregation with nonsyndromic hearing loss and were not found in 200 ethnicity-matched control chromosomes (Fig. 2). The results from one of these families have been previously reported [Shearer et al., 2009].
TABLE I.
Novel Mutations in MYO15A Identified in This Study
| Family code | Ethnicity | Type of mutation | Nucleotide change (NM_016239.3) | Amino acid change (NP_057323) | Location in protein | Polyphen prediction | SIFT prediction | Conseq prediction |
|---|---|---|---|---|---|---|---|---|
| L-3054 | Fars (Central Iran) | Missense | c.5305A>G; n.5643A>G | p.(Thr1769Ala) | Motor domain | Score = 0.999 probably damaging | Substitution is not tolerated | A predicted functional residue (highly conserved and exposed) Score in Conservation scale (1–9) = 9 |
| L-8800118 | Azeri (Northwestern Iran) | Nonsense | c.5925G>A; n.6263G>A | p.(Trp1975X) | IQ Motif | N/A | N/A | N/A |
| L-486 | Sistani (Southeastern Iran) | Deletion (1 nt) | c.5419-21delT; n.5759-61delT | p.(Phe1807Leu fsX6) | Motor domain | N/A | N/A | N/A |
| Missense | c. 1387A>G; n.1725A>G | p.(Met463Val) | N-terminal extension | Score = 0.099 benign | Substitution is not tolerated | Exposed amino acid Score in Conservation scale (1–9) = 5 | ||
| L-1828 | Fars (Central Iran) | Missense | c.8467G>A; n.8805G>A | p.(Asp2823Asn) | FERM domain | Score= 0.999 Probably damaging | Substitution is not tolerated | Exposed amino acid Score in Conservation scale (1–9) = 5 |
| L-8800094 | Azeri (Northwestern Iran) | Missense | c.5810G>A; n.6148G>A | p.(Arg1937His) | Motor domain | Score = 1.000 probably damaging | Substitution is not tolerated | Exposed amino acid Score in Conservation scale (1–9) = 6 |
| L-1406 | Kord (Western Iran) | Deletion (3 nt) | c.4904-4907delGAG | p.(Glu1637del) | Motor domain | N/A | N/A | Buried amino acid in the protein structure Score in Conservation scale (1–9) = 1 |
Note that the locations in the protein are predicted according to NP_057323.3.
FIG. 2.

Segregation study in five families. Abbreviated pedigrees are shown. A: Segregation of the c.A5305G (p.T1769A). B: Segregation of the c.4904–4907delGAG (p.E1637del). C: Segregation of the c.G8467A (p.D2823N). D: Segregation of the c.G5925A(p.W1975X). E: Segregation of the c.G5810A (p.R1937H).
DISCUSSION
Here, we describe 8 families of 140 that show linkage to the DFNB3 locus, suggesting a MYO15A mutation frequency of 5.71% in Iranian patients with congenital severe-to-profound deafness. This study shows that MYO15A mutations are a relatively frequent cause of deafness in the cohort, which is highly genetically heterogeneous. The DFNB3 locus was first identified in residents of a village in Indonesia, Bengkala, with frequency of 2%. Further study on this population, showed a frequency of DFNB3 mutations of about 9.4% [Friedman et al., 1995; Winata et al., 1995].
In 2002, Friedman et al. reported that 5% responsible of hearing loss in Pakistan was associated with MYO15A. And recently, the frequency of MYO15A-related deafness in Turkey has been reported about 9.9% [Friedman et al., 2002; Duman et al., 2011]. Considering the relatively high frequency of MYO15A-related deafness in countries neighboring Iran (Pakistan and Turkey), the frequency of 5.71% that we report on here appears reasonable. In addition, because the families in our cohort were selected from different parts of Iran, this frequency is a good estimation for MYO15A-associated deafness in the aggregate Iranian population.
The audiologic profile of all affected members in these eight families showed the same down-sloping pattern. In this pattern, significant loss of hearing is observed in the higher frequencies more than in the lower frequencies (Fig. 3). Other studies on patients with MYO15A-associated deafness report a similar audioprofile, which leads us to conclude that this audioprofile is the common one in MYO15A-related recessive deafness [Nal et al., 2007; Lezirovitz et al., 2008; Shearer et al., 2009; Cengiz et al., 2010]. Because other studies have determined that DFNB3 deafness is one of the frequent causes of ARNSHL, especially in the Middle East, we can conclude that in patients for whom hearing loss is not due to GJB2, the MYO15A gene should be considered as the next choice for mutation analysis. This is especially true if the audioprofile is down-sloping [Friedman et al., 2002; Duman et al., 2011].
FIG. 3.

The typical down sloping audiogram in our patients.
In this study, we report on novel mutations in MYO15A gene, including 4 missense mutations (1 of which could not be resolved), 1 nonsense mutation, and 2 deletions in different regions of the myosin-XV protein. Two missense mutations, c.5810G>A (p.R1937H) and c.5305A>G (p.T1769A), 1 single-base pair deletion, c.5419-21delT(p.Phe1807LeufsX6), and 3 bp deletion c.4904–4907delGAG(E1637del) are located in the motor domain of the myosin-XV protein (Fig. 1). Considering that motor domain contains ATP- and actin-binding sites and produce force and move actin filaments in vitro [Reîdowicz, 1999], and high amount of reported mutations in this domain, we propose that these mutations most likely have a deleterious effect on protein function. Multiple alignment of human myosin-XV protein by SIFT and Conseq Server software showed high conservation of these two substitutions among different kinds of species (Table I). The threonine at position 1769 is highly conserved and about 89% of the sequences have this amino acid in this position. Also, arginine at position 1937 is highly conserved and is present in 95% of the sequences. Overall it seems that these amino acids may have an important role in the myosin-XV protein, and mutations at these sites result in pathogenicity and deafness.
The single-base pair deletion, c.5419-21delT, results in a frame shift followed by early stop codon. This deletion leads to loss the most part of the functional protein, presenting the pathogenic effect in hearing loss for this patient. However, sequencing of the other exons showed another change in this family, a missense mutation, c. 1387A>G or M463V, which occurs in the second exon of the gene and N-terminal extension of the protein. Because both mutations segregated with deafness in this family, we cannot specifically determine which mutation is causative in this family.
It should be noted that the methionine located in position 463 is exposed in protein structure, has an average conservation score and Polyphen software predicts benign effect on protein structure. Also, the biological function of N-terminal extension is still unknown. However, we could not observe this change in normal study on 200 ethnic specific samples. On the other hand, the single-base pair deletion causes a premature stop codon and therefore an abolishment of all protein function after this point. Therefore, we consider the single-base pair deletion more likely to be pathogenic (Fig. 4).
FIG. 4.
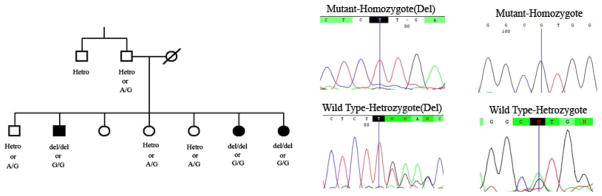
The core family of the L-486 pedigree has been shown here and segregation of the c.5419-21delT and (p. F1807L fsX6) c. 1387A>G (p.M463.V) in this family can be observed.
Another deletion in the motor domain, c.4904-4907delGAG, results in deletion of one amino acid in the myosin-XV protein. The deleted amino acid is glutamic acid at position 1637, which is buried in the structure of the protein and not conserved. However, according to the multiple alignment using SIFT, this amino acid is at least conserved in Rhesus monkey, giant panda, Sumatran Orangutan, Common Marmoset, Dog, Bovine, house mouse, brown rat, implicating a specific role for this position. So, considering normal sequence in all other exons, it seems that this deletion can be a reason for hearing loss in this patient.
The c.5925G>A (p.W1975X) mutation, truncates the protein and as a result of this truncation, myosin-XV function may be impaired, leading to nonsyndromic hearing loss in this patient. This substitution is located in the IQ motif of the myosin-XV protein. IQ motifs are highly conserved throughout myosins. They are basic amphiphilic helixes, usually present in 1–7 copies and follow the conserved motor domain sequence [Bähler and Rhoads, 2002]. In fact, this motif is a recognition site for calmodulin (CaM) binding. CaM is recognized as a major calcium sensor in the cell and regulator for the cell cycle as well as cytoskeletal organization. CaM can act in Ca+-independent manner too, for example, with unconventional myosins lacking Ca+ [Rhoads and Friedberg, 1997]. We predict that this nonsense mutation, which is predicted to lead loss of half myosin-XV, has a destructive effect on protein function.
The c.8467G>A (p.D2823N) mutation is located in the first FERM domain of myosin-XV. The FERM domain (F, 4.1 protein; E, ezrin; R, radixin; M, moesin) localizes proteins to the plasma membrane and can be observed in cytoskeletal-associated proteins, such as unconventional myosins X, VIIa, and XV that associate with various proteins at the interface between the plasma membrane and the cytoskeleton (European Bioinformatics Institute: http://www.ebi.ac.uk/interpro/IEntry?ac=IPR000299). Delprat et al. proposed that the C-terminal MyTH4-FERM region of myosin XVa binds to the PDZ1 and PDZ2 domains of the long whirlin isoform. So, this interaction controls the elongation of stereocilia. On the other hand, the interaction between whirlin and NGL-1 might be involved in the stabilization of inter stereociliar links [Delprat et al., 2005]. Aspartic acid at position 2823 can be seen in about 47% of the sequences, however considering observation of no other changes in all other exons and bioinformatics predictions, this substitution can be an explanation for hearing loss in this family.
In conclusion, we found a MYO15A mutational frequency of 5.71% in our cohort of deaf Iranian individuals. Sequencing of the MYO15A gene led to identification of 7 previously unreported mutations, including 4 missense mutations, 1 nonsense mutation, and 2 deletions in different regions of the myosin-XV protein.
Supplementary Material
Acknowledgments
We are grateful to our patients and their families for their participation in this study. This study was supported by the Iranian National Science Foundation, with grant number 85073/23.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Bähler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–113. doi: 10.1016/s0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- Belguith H, Aifa-Hmani M, Dhouib H, Said MB, Mosrati MA, Lahmar I, Moalla J, Charfeddine I, Driss N, Arab SB, Ghorbel A, Ayadi H, Masmoudi S. Screening of the DFNB3 locus: Identification of three novel mutations of MYO15A associated with hearing loss and further suggestion for two distinctive genes on this locus. Genet Test Mol Biomark. 2009;13:147–151. doi: 10.1089/gtmb.2008.0077. [DOI] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- Cengiz FB, Duman D, Sirmaci A, Tokgöz-Yilmaz S, Erbek S, Oztürkmen-Akay H, Incesulu A, Edwards YJ, Ozdag H, Liu XZ, Tekin M. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomark. 2010;14:543–550. doi: 10.1089/gtmb.2010.0039. [DOI] [PubMed] [Google Scholar]
- Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, Perfettini I, Legrain P, Richardson G, Hardelin JP, Petit C. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet. 2005;14:401–410. doi: 10.1093/hmg/ddi036. [DOI] [PubMed] [Google Scholar]
- Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomark. 2011;15:29–33. doi: 10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, Arhya IN, Asher JH., Jr A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet. 1995;9:86–91. doi: 10.1038/ng0195-86. [DOI] [PubMed] [Google Scholar]
- Friedman TB, Sellers JR, Avraham KB. Unconventional myosins and the genetics of hearing loss. Am J Med Genet. 1999;89:147–157. doi: 10.1002/(sici)1096-8628(19990924)89:3<147::aid-ajmg5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Friedman TB, Hinnant JT, Ghosh M, Boger ET, Riazuddin S, Lupski JR, Potocki L, Wilcox ER. DFNB3, spectrum of MYO15A recessive mutant alleles and an emerging genotype–phenotype correlation. Adv Otorhinolaryngol. 2002;61:124–130. doi: 10.1159/000066824. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. http://www.mrc-lmb.cam.ac.uk/myosin/Review/Reviewframeset.html. [DOI] [PubMed] [Google Scholar]
- Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet Part A. 2007;143A:2382–2389. doi: 10.1002/ajmg.a.31937. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y, Mburu P, Morse S, Kominami R, Townsend S, Brown SD. Mutant analysis reveals whirlin as a dynamic organizer in thegrowing hair cell stereocilium. Hum Mol Genet. 2005;14:391–400. doi: 10.1093/hmg/ddi035. [DOI] [PubMed] [Google Scholar]
- Krendel M, Mooseker MS. Myosins: Tails (and heads) of functional diversity. Physiology. 2005;20:239–251. doi: 10.1152/physiol.00014.2005. [DOI] [PubMed] [Google Scholar]
- Lezirovitz K, Pardono E, de Mello Auricchio MTB, de Carvalho e Silva FL, Lopes JJ, Abreu-Silva RS, Romanos J, Batissoco AC, Mingroni-Netto RC. Unexpected genetic heterogeneity in a large consanguineous Brazilian pedigree presenting deafness. Eur J Hum Genet. 2008;16:89–96. doi: 10.1038/sj.ejhg.5201917. [DOI] [PubMed] [Google Scholar]
- Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z, Riazuddin S, Liang Y, Menon PSN, Smith T, Smith ACM, Chen KS, Lupski JR, Wilcox ER, Potocki L, Friedman TB. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith–Magenis syndrome. Hum Genet. 2001;109:535–541. doi: 10.1007/s004390100604. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker MS, Cheney RE. Unconventional myosins. Annu Rev Cell Dev Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Nal N, Ahmed ZM, Erkal E, Alper OM. Mutational spectrum of MYO15A: The large N-terminal extension of myosin XVA is required for hearing. Hum Mut. 2007;28:1014–1019. doi: 10.1002/humu.20556. [DOI] [PubMed] [Google Scholar]
- Nance WE, Liu XZ, Pandya A. Relation between choice of partner and high frequency of connexin-26 deafness. Lancet. 2000;356:500–501. doi: 10.1016/S0140-6736(00)02565-4. [DOI] [PubMed] [Google Scholar]
- Reîdowicz MJ. Myosins and deafness. J Muscle Res Cell Motil. 1999;20:241–248. doi: 10.1023/a:1005403725521. [DOI] [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Shearer AE, Hildebrand MS, Webster JA, Kahrizi K, Meyer NC, Jalalvand K, Arzhanginy S, Kimberling WJ, Stephan D, Bahlo M, Smith RJH, Najmabadi H. Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope. 2009;119:727–733. doi: 10.1002/lary.20116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage (07/2006) 2011 http://webhost.ua.ac.be/hhh/
- Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Morton CC, Morell RJ, Noben-Trauth K, Camper SA, Friedman TB. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280:1447–1451. doi: 10.1126/science.280.5368.1447. [DOI] [PubMed] [Google Scholar]
- Winata S, Arhya IN, Moeljopawiro S, Hinnant JT, Liang Y, Friedman TB, Asher JH. Congenital non-syndromal autosomal recessive deafness in Bengkala, an isolated Balinese village. J Med Genet. 1995;32:336–343. doi: 10.1136/jmg.32.5.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.