

Abstract
The effects of standard adenosine receptor (AR) agonists and antagonists on the proliferation of human T lymphocytes, unstimulated and phytohemagglutinin-stimulated human peripheral blood lymphocytes (PBL), and Jurkat T cells were investigated. Real-time PCR measurements confirmed the presence of all four AR subtypes on the investigated cells, although at different expression levels. A2A ARs were predominantly expressed in PBL and further upregulated upon stimulation, while malignant Jurkat T cells showed high expression levels of A1, A2A, and A2B ARs. Cell proliferation was measured by [3H]-thymidine incorporation assays. Several ligands, including the subtype-selective agonists CPA (A1), BAY60-6583 (A2B), and IB-MECA (A3), and the antagonists PSB-36 (A1), MSX-2 (A2A), and PSB-10 (A3) significantly inhibited cell proliferation at micromolar concentrations, which were about three orders of magnitude higher than their AR affinities. In contrast, further investigated AR ligands, including the agonists NECA (nonselective) and CGS21680 (A2A), and the antagonists preladenant (SCH-420814, A2A), PSB-1115 (A2B), and PSB-603 (A2B) showed no or only minor effects on lymphocyte proliferation. The anti-proliferative effects of the AR agonists could not be blocked by the corresponding antagonists. The non-selective AR antagonist caffeine stimulated phytohemagglutinin-activated PBL with an EC50 value of 104 μM. This is the first study to compare a complete set of commonly used AR ligands for all subtypes on lymphocyte proliferation. Our results strongly suggest that these compounds induce an inhibition of lymphocyte proliferation and cell death through AR-independent mechanisms.
Keywords: Adenosine receptors, Real-time PCR, Human lymphocytes, Jurkat T cells, Proliferation, [3H]Thymidine incorporation, Receptor-independent mechanisms
Introduction
The endogenous nucleoside adenosine regulates many physiological processes through the activation of four specific cell surface receptors, referred to as A1, A2A, A2B, and A3 [1]. They belong to the family of G protein-coupled receptors characterized by seven transmembrane-spanning helical domains (seven TM receptors) [1]. While adenosine A2A and A2B receptors interact with Gs proteins to stimulate adenylate cyclase activity resulting in an increase in intracellular cAMP levels, A1 and A3 receptors are coupled to Gi proteins inhibiting adenylate cyclase activity and, therefore, cAMP production. Coupling to further second messenger systems has been described, e.g., stimulation of phospholipase C (A1, A2B, A3), activation of potassium and inhibition of calcium channels (A1), and mobilization of intracellular calcium by an as yet unknown mechanism (A2B) [1, 2]. Adenosine inhibits the release of excitatory neurotransmitters in the brain and leads to sedation, whereas in the periphery it shows negative inotropic, chronotropic, dromotropic, antihypertensive, and antidiuretic effects [3]. Adenosine receptors (ARs) are also found on cells of the immune system and have been recognized to play an important role in inflammatory conditions [4–7], in which adenosine can act as an anti-inflammatory and immunosuppressive mediator mainly via the activation of A2A receptors. It inhibits TNF-α production and has been implicated as a physiological signal in the apoptotic deletion of T cells during intrathymic cell selection, a process which functions to prevent autoimmunity [8–11]. The effects of adenosine on lymphocytes are of particular interest since the accumulation of extracellular and intracellular adenosine and 2′-deoxyadenosine in the absence of adenosine deaminase (ADA) activity is lymphotoxic and causes SCID [12, 13], which is characterized by a loss of function and depletion of T and B lymphocytes [14]. Part of this effect may be attributed to adenosine via the activation of ARs and sustained increase in cAMP levels [5]. Besides the activation of extracellular receptors [11, 15–17], direct intracellular toxicity (also by adenosine metabolites) has been described [18–21]. Furthermore, adenosine is able to stimulate cell growth and proliferation and/or to induce apoptosis in various normal and tumor cell lines. Proliferative or anti-proliferative effects appear to be dependent on the cell type, the distribution of adenosine receptor subtypes, and their expression on the cell surface [22–25]. A3 adenosine receptors seem to play an important role [26–28], but recent evidence accumulates that A2B ARs can be upregulated on malignant cells and are involved in the control of tumor growth and development [16, 24, 29–32]. In contrast to these findings, adenosine and various related AR agonists as well as antagonists can trigger apoptosis by receptor-independent mechanisms that require the entry of the compounds into the cells. A prominent representative is 2-chloro-2′-deoxyadenosine (cladribine) which is applied as a chemotherapeutic anticancer agent for the treatment of chronic lymphoid malignancies [10, 33, 34]. Further examples include 2-chloroadenosine (nonselective AR agonist) [10, 33], 1,3-dipropyl-8-cyclopentylxanthine (A1AR antagonist) [35], and 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (A3AR agonist) [36].
In the present study, we investigated the effects of standard adenosine receptor agonists and antagonists on the proliferation of human T lymphocytes. Initially, we evaluated the expression of adenosine receptor subtypes in unstimulated and phytohemagglutinin (PHA-)stimulated human peripheral blood lymphocytes (PBL) and Jurkat T cells, a human lymphocytic leukemia cell line, using real-time PCR. Subsequently, the effects of various nonselective and subtype-selective AR agonists as well as antagonists on the proliferation of these cells were determined. The rate of incorporation of [6-3H]-thymidine into DNA during the S-phase of the cell cycle was used as an indicator for the rate of cell proliferation. We provide evidence for an inhibitory effect of several AR agonists as well as antagonists on the proliferation of human lymphocytes through AR-independent mechanisms.
Materials and methods
Chemicals
DMSO and ADA were obtained from Fluka (St. Gallen, Switzerland), [3H]thymidine (20 Ci/mmol) was from Hartmann Analytic (Braunschweig, Germany), EDTA was from Roth (Karlsruhe, Germany), 5′-N-ethylcarboxamidoadenosine (NECA) from Applichem (Darmstadt, Germany), CGS21680 from Tocris (Ellisville, USA), and Pancoll from PANbiotech (Aidenbach, Germany). BAY60-6583 was kindly provided by Dr. Thomas Krahn, Bayer-Schering Pharma, Wuppertal, Germany. All other chemical reagents, cell culture materials, and AR ligands were obtained from Sigma (Munich, Germany) or synthesized in our laboratories.
Lymphocyte isolation and stimulation
Lymphocytes were isolated from buffy coats kindly provided by the Blood Bank of the University Hospital of Bonn, prepared from the peripheral blood of healthy volunteers. A buffy coat-PBS/2 mM EDTA mixture (1:2) was centrifuged on Ficoll-Hypaque density gradients after the method of Böyum [37]. The mononuclear leukocyte fractions isolated were removed from the Ficoll gradients and washed in PBS (pH 7.2) containing 2 mM EDTA. The final pellet was resuspended in VLE-RPMI 1640 medium supplemented with 10 % (v/v) fetal bovine serum, 100 U/ml of penicillin, and 0.1 mg/ml of streptomycin at a concentration of 3 × 106 cells ml−1. Monocytes, which adhere to plastic dishes, were removed after an adherence time of 1 h at 37 °C, 5 % CO2. The supernatant contained the resulting cell suspension of PBL, which was maintained in VLE-RPMI 1640 medium supplemented with 10 % (v/v) fetal bovine serum, 100 U/ml of penicillin, and 0.1 mg/ml of streptomycin at 37 °C with 5 % CO2 until use. Stimulation of the PBL was achieved by the addition of 10 μg/ml phytohemagglutinin (PHA-M).
Cell culture conditions
Human leukemia Jurkat T cells were grown in RPMI 1640 medium supplemented with 10 % (v/v) fetal bovine serum, 2 mM l-glutamine, and antibiotics (100 U/ml of penicillin and 0.1 mg/ml of streptomycin) at 37 °C with 5 % CO2.
RNA isolation and real-time PCR
The total RNA from PBL or Jurkat T cells was isolated using the NucleoSpin®RNAII kit (Macherey-Nagel, Düren, Germany), and the cDNA was subsequently prepared with the iScriptTMcDNA Synthesis kit (BioRad, Munich, Germany), following the manufacturer’s instructions, to give a final concentration of 12.5 ng/μl cDNA. Quantitative PCR was performed using the iQTMSYBR® Green Supermix (BioRad). A typical reaction contained 12.5 μl of the kit reaction mixture, 7.5 μl of molecular biology grade water, 0.5 μl each of 10 μM primer stocks, and 4 μl of cDNA. Standard curves were produced using diluted cDNA. Real-time PCR monitoring was performed using an iCycler IQ Real-Time PCR detection system (BioRad). The thermal cycler tracks fluorescence levels over 40 amplification cycles: 95 °C for 15 s (denaturation), 58 °C for 30 s (annealing), and 72 °C for 10 s (polymerization). A melt curve was performed at the end of each run to verify that there was a single amplification product and a lack of primer dimers. All samples were normalized to the amount of ß-actin mRNA present in the sample. The transcript levels and fold change in mRNA were determined by analyzing the standard curves and/or using the ΔΔCT method as described previously [38]. Primer sets for the following genes (sense and antisense sequence, respectively) were used: A1 AR (5′-TGC ACT GGC CTG TTC TGT AG-3′, 5′-CTG CCT CTC CCA CGT ACA AT-3′); A2A AR (5′-GGA GTT TGC CCC TTC CTA AG-3′, 5′-CTG CTT CCT CAG AAC CCA AG-3′); A2B AR (5′-ATC TCC AGG TAT CTT CTC-3′, 5′-GTT GGC ATA ATC CAC ACA G-3′); A3 AR (5′-CCT TCT CGC GTG TCC TGA CT-3′, 5′-CTC TGA CTA CCG CCG TTG CT-3′); and human ß-actin (5′-GGT GGC TTT TAG GAT GGC AAG-3′, 5′-ACT GGA ACG GTG AAG GTG ACA G-3′) in identical reactions.
Cell proliferation assay—[3H]thymidine incorporation
Cell suspensions were distributed at 1 × 105 cells per vial in a vial containing a final volume of 1 ml (PBL) or 0.5 ml (Jurkat T) of the medium (PBL: VLE RPMI + FCS + antibiotics; Jurkat T: RPMI 1640 + FCS + 2 mM l-glutamine + antibiotics) and the test compounds (concentrations stated in the text). Cells were grown for 48 h, after which time 10 μl of [6-3H]thymidine (0.5 μCi per vial) in the medium was added and the cells were allowed to grow for a further 24 h. At the end of this period, cells were harvested through a GF/B glass-fiber filter using a Brandel cell harvester and washed several times with ice-cold water. The punched-out wet filters were transferred to mini-vials and incubated with 2.5 ml of Ready SafeTM scintillation cocktail (Beckman Coulter) for 9 h before counting in a liquid scintillation counter. Data were analyzed using Graphpad Prism® (San Diego, CA, USA), version 4.0. IC50/EC50 values were determined by fitting the data to a sigmoidal curve with variable slope.
Results
Adenosine receptor transcripts
To confirm the presence of different AR subtypes in human lymphocytes, the mRNA expression levels of A1AR, A2AAR, A2BAR, and A3AR were determined. Real-time PCR analysis was performed with RNA isolated from human PBL, unstimulated as well as phytohemagglutinin (PHA-)stimulated, and Jurkat T cells. Primers were used to amplify specifically a fragment of the human AR cDNAs. As shown in Fig. 1, in PBL as well as in Jurkat T cells, the mRNA for all AR subtypes was detectable.
Fig. 1.

Results from real-time PCR experiments determining adenosine receptor transcripts in PBL and Jurkat T cells. Data were calculated relative to the internal housekeeping gene (ß-actin) and are expressed as percentage of control mRNA expression ± SEM, n = 3. ß-Actin mRNA expression was not significantly different between the tested cells allowing a direct comparison (data not shown)
In PBL, the highest mRNA expression was found for A2AARs, which was further upregulated upon stimulation of the cells with PHA, followed by mRNA expression of A1ARs and A3ARs. The lowest expression levels were found for A2BARs, which—together with A3ARs—appeared to be somewhat downregulated during stimulation. In Jurkat T cells, mRNA expression for A1AR, A2AAR, and A2BAR was nearly the same; only the expression of A3AR was significantly lower as compared to that of the other AR subtypes.
Effects of adenosine deaminase on lymphocyte cell proliferation
The effect of the enzyme ADA (EC 3.5.4.4), which converts adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine, respectively, on the proliferation of resting, unstimulated and PHA-stimulated PBL as well as on Jurkat T cells, was investigated. The results are shown in Fig. 2.
Fig. 2.

a Effect of adenosine deaminase on [3H]thymidine incorporation in resting and PHA-stimulated PBL as well as Jurkat T cells. Cells were cultured in the presence of the indicated amounts of enzyme. Results are expressed as percentage of untreated control (=100 %) [3H]thymidine uptake. b Dose–response curve obtained from [3H]thymidine incorporation studies in PHA-stimulated PBL. Each bar/data point represents the mean of a minimum of three separate experiments carried out in triplicate ± SEM
In the presence of ADA, resting PBL displayed higher DNA synthesis and proliferation than parallel control cultures, as measured by the incorporation of [3H]thymidine. The effect of different doses of ADA is shown in Fig. 2a. Proliferation increased up to a concentration of 1 IU/ml. An EC50 value of 0.0927 IU/ml was calculated and a maximum effect of 55 ± 15 % stimulation was observed (Fig. 2b). At higher concentrations, the proliferation of resting PBL returned to background levels. ADA had no effect on the proliferative capacity of PHA-stimulated PBL and Jurkat T cells (Fig. 2a).
Effects of adenosine receptor agonists on lymphocyte cell proliferation
As a next step, we investigated the effects of standard AR agonists on lymphocyte proliferation. The following agonists were studied: NECA (non-selective), CPA (A1), CGS21680 (A2A), BAY60-6583 (A2B), and N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (IB-MECA, A3). The structures and affinities of the compounds for the different AR subtypes are shown in Table 1.
Table 1.
Structures of investigated adenosine receptor agonists and their affinities or potencies at human adenosine receptor subtypes (data are taken from [1]
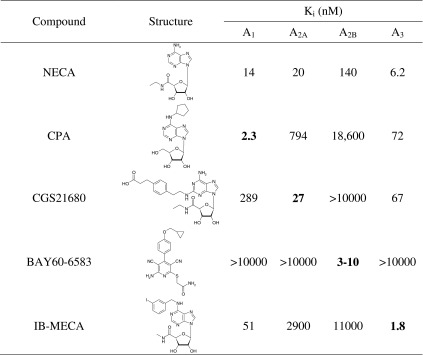
The nonselective agonist NECA and the A2AAR-selective CGS21680 showed no significant effect on the proliferation of unstimulated PBL. In contrast, the A1AR-selective CPA, the A2BAR-selective BAY60-6583, as well as the A3AR-selective agonist IB-MECA appeared to show a dose-dependent inhibition of [3H]thymidine uptake (Fig. 3a). For IB-MECA, the inhibition was statistically significant.
Fig. 3.

Effects of adenosine receptor agonists on [3H]-thymidine incorporation in unstimulated PBL (a), PHA-stimulated PBL (b), and Jurkat T cells (c). Cells were cultured in the presence of the compounds. Results are expressed as percentage of [3H]-thymidine uptake by untreated control cells (=100 %). Each bar represents the mean of a minimum of three separate experiments carried out in triplicate ± SEM
The effects were observed at relatively high concentrations in the micromolar range. Where possible, dose–response curves were determined and IC50 values were calculated: they ranged from 2.45 μM for CPA to 17.6 μM for IB-MECA (see Fig. 4a and Table 2). IB-MECA showed the highest maximum effect with an inhibition of the proliferation of 56 ± 6 %. CPA and BAY60-6583 inhibited the proliferation of unstimulated PBL by 41 ± 16 and 35 ± 11 %, respectively (Figs. 3a and 4a and Table 2). These effects were more pronounced in PHA-stimulated PBL: CPA, BAY60-6583, and IB-MECA showed a highly significant inhibition of the cell proliferation with a maximum effect of >95 % inhibition (CPA: 96 ± 1 % at 250 μM, BAY60-6583: 98 ± 1 % at 100 μM, IB-MECA: 96 ± 3 % at 100 μM; Figs. 3b and 4b). The effects were only observed at relatively high concentrations in the micromolar range (IC50 values: 12.7 μM for CPA, 7.10 μM for BAY60-6583, and 14.2 μM for IB-MECA). NECA and CGS21680 had no significant effects on the cell proliferation of PHA-stimulated PBL at concentrations up to 100 μM; only at a high concentration of 250 μM were both compounds weakly inhibitory.
Fig. 4.

Dose–response curves obtained with [3H]thymidine incorporation studies in unstimulated PBL (a) and PHA-stimulated PBL (b). Each data point represents the mean of a minimum of three separate experiments carried out in triplicate ± SEM (for IC50 values, see Table 2)
Table 2.
Anti-proliferative potency of standard adenosine receptor agonists and antagonists on peripheral blood lymphocytes and Jurkat T cell determined in [3H]thymidine uptake assays
| Compound | Unstimulated PBL | Stimulated PBL | Jurkat T | |||
|---|---|---|---|---|---|---|
| IC50 (μM) | % Inhibition | IC50 (μM) | % Inhibitiona | IC50 (μM) | % Inhibitionb | |
| CPA | 2.45 ± 2.62 | 41 ± 16 | 12.5 ± 1.2 | 96 ± 1 | >100 | 25 ± 3 |
| IB-MECA | 17.6 ± 1.9 | 56 ± 6 | 14.2 ± 1.4 | 96 ± 3 | >10 | 16 ± 2 |
| BAY60-6583 | 9.82 ± 2.58 | 35 ± 11 | 7.10 ± 1.25 | 98 ± 1 | >10 | 10 ± 12 |
| Caffeine | >100 | 3 ± 7 | EC50: 104 ± 1 | 26 ± 7c | >250 | 3 ± 0 |
| MSX-2 | 10.8 ± 1.7 | 63 ± 12 | 5.04 ± 1.63 | 76 ± 7 | 1.64 ± 1.29 | 83 ± 8a |
| PSB-10 | 4.61 ± 1.87 | 73 ± 11 | 14.4 ± 1.3 | 95 ± 1 | >10 | 41 ± 11 |
| PSB-36 | 8.19 ± 1.54 | 80 ± 11 | 7.31 ± 1.19 | 99 ± 0 | ∼100 | 55 ± 4 |
aMaximal inhibition, unless otherwise noted
bPercent inhibition at the indicated concentration, unless otherwise noted
cMaximal stimulation
In Jurkat T cells, CPA and IB-MECA also showed inhibitory effects on proliferation at a concentration of 10 μM, but not at 1 μM (Fig. 3c).
Effects of adenosine receptor antagonists on lymphocyte cell proliferation
The following antagonists were tested in this study: caffeine (A1, A2A, A2B); PSB-36 (1-butyl-8-(3-noradamantyl)-3-(3-hydroxypropyl)xanthine, A1); MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine, A2A); SCH420814 (preladenant, A2A); PSB-1115 (1-propyl-8-p-sulfophenylxanthine, A2B); PSB-603 (8-[4-[4-(4-chlorophenyl)piperazine-1-sulfonyl)phenyl]]-1-propylxanthine, A2B); and PSB-10 ((R)-8-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one, A3). The structures and affinities of the compounds for the different AR subtypes are shown in Table 3. Like AR agonists, several subtype-selective AR antagonists inhibited the proliferative capacity of human lymphocytes, as shown in Fig. 5.
Table 3.
Structures of investigated adenosine receptor antagonists and their affinities at human adenosine receptor subtypes (data taken from [1])a
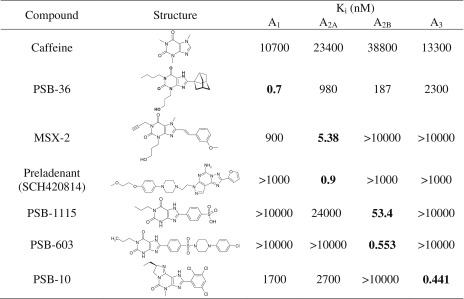
aunpublished data from our laboratory
Fig. 5.

Effects of adenosine receptor antagonists on [3H]thymidine incorporation in unstimulated PBL (a), PHA-stimulated PBL (b), and Jurkat T cells (c). Cells were cultured in the presence of the indicated compound concentrations. Results are expressed as percentage of untreated control (=100 %) [3H]thymidine incorporation. Each bar represents the mean of a minimum of three separate experiments carried out in triplicate ± SEM
The A1AR selective antagonist PSB-36, the A2AAR selective antagonist MSX-2, and the A3AR selective antagonist PSB-10 significantly inhibited the proliferation of unstimulated PBL in a concentration-dependent manner (Fig. 5a). Maximum effects of 80 ± 11, 63 ± 12, and 73 ± 11 % inhibition of cell proliferation at 250 μM, respectively, were observed. The plotted dose–response curves resulted in IC50 values of 8.19 μM for PSB-36, 10.8 μM for MSX-2, and 4.61 μM for PSB-10, indicating that the effects of the tested antagonists occurred at relatively high concentrations (Fig. 6a and Table 2).
Fig. 6.

Dose–response curves obtained from [3H]thymidine incorporation studies in unstimulated PBL (a), PHA-stimulated PBL (b), and Jurkat T cells (c). Each data point represents the mean of a minimum of three separate experiments carried out in triplicate ± SEM (for IC50 values, see Table 2)
Caffeine (nonselective), SCH-420814 (A2A-selective), and PSB-1115 and PSB-603 (both A2B-selective) showed no effects on the proliferation of unstimulated PBL. As observed for agonists, these results could be confirmed or were even more pronounced in PHA-stimulated PBL: PSB-36, MSX-2, and PSB-10 showed highly significant anti-proliferative properties with maximum effects and IC50 values of 99 ± 0 % and 7.31 μM for PSB-36, 76 ± 7 % and 5.04 μM for MSX-2, and 95 ± 1 % and 14.4 μM for PSB-10, indicating that the observed effects occurred at relatively high concentrations (Figs. 5b and 6b and Table 2). In addition, in PHA-stimulated PBL for SCH-420814 (A2A antagonist) and PSB-603 (A2B antagonist), anti-proliferative effects were observed at a concentration of 10 μM. PSB-1115 had no effect either on unstimulated or on PHA-stimulated PBL. Caffeine showed an additional small but significant stimulation of already PHA-stimulated PBL, with a maximum stimulation of 26 ± 7 % and an EC50 value of 104 μM (Figs. 5b and 6b and Table 2).
In Jurkat T cells, the A1 antagonist PSB-36 showed inhibitory effects on cell proliferation at 100 μM concentration and the A3 antagonist PSB-10 at 10 μM. MSX-2 exhibited the strongest inhibitory effects on the proliferative capacity of Jurkat T cells, with a maximum inhibition of 83 ± 8 % and an IC50 of 1.64 μM (Fig. 6c and Table 2). These results are in good agreement with the results obtained at unstimulated and PHA-stimulated PBL.
Further investigations of the anti-proliferative effects of adenosine receptor agonists and antagonists on lymphocyte cell proliferation
Additional experiments were performed to investigate whether the observed anti-proliferative effects of AR agonists and antagonists were receptor-dependent. Therefore, the effect of the co-administration of a subtype-selective AR agonist together with the respective subtype-selective AR antagonist on cell proliferation of resting and PHA-stimulated PBL was investigated. The results are shown in Fig. 7.
Fig. 7.

Effects of the combination of adenosine receptor agonists and antagonists on [3H]thymidine incorporation in resting PBL (a) and PHA-stimulated PBL (b). Cells were cultured in the presence of the indicated compound concentrations. Results are expressed as percentage of untreated control (=100 %) [3H]thymidine uptake. Each bar represents the mean of a minimum of two separate experiments carried out in triplicate ± SEM
As observed for all AR subtypes, the co-administration of a subtype-selective agonist and the corresponding subtype-selective antagonist did not abolish the anti-proliferative effects. For example, the co-administration of PSB-36 (10 μM) together with CPA (10 μM, A1AR) or the combination of IB-MECA (10 μM) and PSB-10 (10 μM, A3AR), all of which had shown inhibitory effects on cell proliferation of resting as well as PHA-stimulated PBL when applied as single compounds, increased the anti-proliferative effect as compared to the effects seen upon the administration of the single compounds (Fig. 7). PSB-36 inhibited cell proliferation with a maximum effect of 79 ± 1 % at unstimulated PBL and 78 ± 4 % at stimulated PBL and CPA with 57 ± 5 % (unstimulated) and 49 ± 4 % (PHA-stimulated), while the maximum effects after co-administration of the A1 antagonist PSB-36 and the A1 agonist CPA amounted to 90 ± 2 % (unstimulated) and 91 ± 1 % (PHA-stimulated; see Fig. 7a, b). IB-MECA (A3 agonist) inhibited cell proliferation with a maximum effect of 46 ± 7 % (unstimulated) and 48 ± 3 % (PHA-stimulated) and PSB-10 (A3 antagonist) with 54 ± 4 % (unstimulated) and 52 ± 4 % (PHA-stimulated), while the combination of both compounds showed a maximum effect of 90 ± 2 % (unstimulated) or 71 ± 3 % (PHA-stimulated), respectively. The combination of MSX-2 (A2A antagonist, 10 μM), exerting maximum inhibitory effects of 82 ± 19 % (unstimulated) and 80 ± 27 % (PHA-stimulated) when given as a single compound, in combination with CGS21680 (A2A agonist, 10 μM), which alone had no effect on the proliferation of PBL, revealed maximum inhibitory effects on the cell proliferation of 79 ± 23 % at unstimulated PBL and 82 ± 24 % at PHA-stimulated PBL. The administration of BAY60-6583 (agonist) and PSB-1115 (antagonist), ligands of the A2BAR, showed no effect on the proliferative capacity of PBL (resting and PHA-stimulated), either as single compounds or when applied in combination.
A second series of experiments was carried out to exclude the possibility that the observed inhibitory effects of the antagonists (PSB-36, MSX-2, and PSB-10) on cell proliferation were due to the upregulation of the receptors and subsequent stimulation by endogenously released adenosine. To ensure high levels of antagonists during the whole experiment, the compounds (10 μM) were added daily without changing the medium, resulting in a three times administration during the 3-day assay period. In addition, ADA (1 IU/ml) was added either only once, or daily, in order to remove endogenously released adenosine.
As shown in Fig. 8, the daily administration of the antagonists further increased the inhibitory effects on the proliferation of PBL, while the co-administration of ADA did not reduce the anti-proliferative effects of these compounds.
Fig. 8.

Effect of the daily administration of adenosine receptor antagonists (10 μM), ADA (1 IU/ml), or both on [3H]-thymidine incorporation in unstimulated PBL (a) and PHA-stimulated PBL (b). Cells were cultured in the presence of the indicated compound concentrations. Results are expressed as percentage of untreated control (=100 %) [3H]-thymidine uptake. Each bar represents the mean of a minimum of two separate experiments carried out in triplicate ± SEM
Discussion
Human lymphocytes are responsible for the adaptive cell-mediated immune response and are therefore of particular interest as immunomodulatory targets [4, 6, 9]. Inhibition of T lymphocyte proliferation and induction of apoptosis leads to immunosuppressive and anti-inflammatory effects. In the present study, we investigated the expression and function of AR subtypes in native human peripheral blood lymphocytes as well as in the human lymphoma cell line Jurkat T using real-time PCR. Moreover, we studied the effects of standard AR agonists and antagonists on lymphocyte proliferation. The results obtained from real-time PCR analysis showed expression for all four AR subtypes in the investigated cells, consistent with previous reports [22–24, 39]. The adenosine A2A receptor mRNA expression level was higher in PBL than in Jurkat T cells and was further upregulated after stimulation of the cells with PHA, as previously reported [40, 41]. The levels for A1AR mRNA did not change upon cell stimulation, while mRNA for the A2B and A3ARs appeared to be somewhat downregulated upon PHA stimulation. This result is in contrast to the results by Gessi et al. [42] who had observed an upregulation of A3ARs upon the activation of human lymphocytes. In comparison to the non-malignant peripheral blood lymphocytes, Jurkat T cells showed a much higher expression of A1AR and especially A2BAR, whereas the expression of A2AAR and A3AR was somewhat lower. This indicates that A2BAR may play a more important role than A3AR in this cancer cell line. Both receptor subtypes have previously been postulated to be associated with cancer cell proliferation [27, 29, 32, 42]. Our results indicate that all four AR subtypes are present on human lymphocytic cells, however at different expression levels, resulting in different receptor profiles. It should, however, be kept in mind that mRNA expression levels are not always well correlated with the protein levels expressed on the cell surface. The high expression levels of adenosine A2A receptors in native PBL and their upregulation after stimulation of the cells have previously been reported [40, 41]. These results underscore the importance of this receptor subtype as a mediator of anti-inflammatory effects triggering the emergency downregulation of overactive immune cells [43]. On the other hand, the relatively high expression levels of adenosine A2B receptors in cells of the malignant Jurkat T cell line compared to non-malignant PBL cells further indicate an important role of this subtype in malignant cell lines, as previously described [29, 32]. The adenosine A2B receptor may therefore serve as a target to control cell growth and proliferation in these cells. Previous investigations of the role of adenosine in the control of cell proliferation in a number of cell types had yielded conflicting results [24, 30, 44]. The next step of this study was therefore aimed at investigating the effects of several nonselective and subtype-selective AR agonists as well as antagonists on the proliferative capacity of these cells, as measured by [3H]thymidine uptake during DNA synthesis. In agreement with the reports showing that adenosine could impair cell proliferation [22, 24, 26], the present observations demonstrated an increased proliferation of human lymphocytes upon removal of endogenously released adenosine. The addition of ADA, which degrades adenosine to inosine, resulted in a significant stimulation of unstimulated PBL. No effect was seen upon treatment of cells, which were already strongly activated to grow, namely, PHA-stimulated PBL or Jurkat T cells, respectively. On the other hand, AR agonists, namely, the A1AR-selective CPA, the A2BAR-selective BAY60-6583, and the A3AR-selective IB-MECA, showed clear anti-proliferative properties in all investigated cells, stimulated and unstimulated PBL, and Jurkat T cells. However, for the observed effects, relatively high concentrations were required. All three agonists inhibited cell proliferation with IC50 values in the micromolar range despite low nanomolar affinities for these compounds, as previously demonstrated in radioligand binding and/or functional studies (see Table 1). However, there were some differences between resting and PHA-stimulated PBL concerning the maximum inhibitory effects: proliferation of unstimulated cells was inhibited to a lesser extent; maximum effects for the three compounds range from 35 to 56 % compared to proliferation inhibition rates of >95 % in stimulated PBL. The non-selective agonist NECA and the structurally related A2A-selective agonist CGS21680 showed no significant effect on T lymphocyte proliferation in our experiments. A very small A2AAR-dependent inhibition of thymocytes (7–15 % cell death) had been reported for CGS21680 [12].
Surprisingly, several AR antagonists, namely, the A1AR-selective PSB-36, the A2AAR-selective MSX-2, and the A3AR-selective PSB-10 also showed significant anti-proliferative properties on human lymphocytes. For these compounds, the same phenomena could be observed, as seen for AR agonists: the IC50 values determined for anti-proliferative activity were in the high micromolar range, while the previously determined AR affinities were about 1,000-fold lower (Ki values, see Table 3). The maximum inhibitory effects for the three compounds at resting PBL with 63–80 % inhibition of the proliferation were lower as compared to inhibitory rates of 76–99 % inhibition of cell proliferation of stimulated PBL. These results indicated that the observed inhibitory effects on cell proliferation by the agonists CPA, BAY60-6583, and IB-MECA and by the antagonists PSB-36, MSX-2, and PSB-10 may be caused by receptor-independent mechanisms. All of these anti-proliferative AR antagonists are xanthine derivatives related to caffeine. However, caffeine, and the A2B-selective xanthine derivative PSB-1115, did not show any anti-proliferative effect. Caffeine even showed a small stimulatory effect on the proliferation of PBL. Two other AR antagonists, the A2B-selective xanthine PSB-603 and the A2A-selective non-xanthine AR antagonist SCH420814, only showed a very small anti-proliferative effect on PHA-stimulated PBL at a high concentration of 10 μM. Thus, the xanthine structure does not appear to be responsible for the anti-proliferative effect of some of the AR antagonists.
To further explore a possible AR-independent mechanism of the active AR antagonists, we tried to inhibit the agonist-induced inhibitory effects on lymphocyte proliferation by blocking the subtype-selective agonist effects with subtype-selective antagonists. As can be seen in Fig. 7, for none of the investigated AR agonists could the inhibition of cell proliferation be blocked by the addition of the corresponding AR antagonist, either in resting or in PHA-stimulated PBL. On the contrary, the inhibitory effects were retained or even increased for PSB-36 + CPA and IB-MECA + PSB-10, respectively. These results provide further evidence that the mechanism of the anti-proliferative action of AR agonist is independent of ARs. Another possible explanation for the fact that agonists as well as antagonists at ARs can cause a strong inhibition of the proliferation of resting and PHA-stimulated PBL and of Jurkat T cells could be an upregulation of ARs caused by the addition of antagonists, resulting in a subsequent increased stimulation by endogenous adenosine. To exclude this possibility, we chose an assay design in which it is guaranteed that the concentration of antagonist present is always high enough to block the receptors. This was achieved by a repeated administration of the antagonist during the incubation time. Additionally, we tested the effect of the presence of ADA in addition to the antagonist in order to remove endogenous adenosine. The results shown in Fig. 8 disproved the above hypothesis by showing that the observed inhibitory effects on cell proliferation could neither be diminished nor abolished by the repeated administration of the antagonist or by the presence of ADA. These findings exclude the possibility that AR antagonists inhibit lymphocyte cell proliferation by an upregulation of ARs.
Taken together, our results strongly indicate that the observed inhibitory effects of the subtype-selective AR agonists CPA, BAY60-6583, and IB-MECA, as well as those of the subtype-selective antagonists PSB-36, MSX-2, and PSB-10, on the proliferation of human lymphocytes are not mediated by AR-dependent mechanisms.
As can be seen in Fig. 6, caffeine was the only AR antagonist in this study which was able to stimulate the proliferation of already PHA-stimulated PBL. On the proliferation of resting PBL as well as on Jurkat T cells, caffeine had no measurable effect. The fact that the stimulating effect of caffeine only occurred in already stimulated PBL is indicative of an AR-independent mechanism. If it were a receptor-dependent mechanism, the effect should be observable also in resting PBL. For Jurkat T cells, one can argue that these cells—due to their malignancy—already proliferate at a maximum degree and cannot be further stimulated, a fact we also observed for stimulation with PHA, a strong T cell mitogen which was nevertheless unable to stimulate further proliferation in Jurkat T cells (data not shown). However, the stimulating effect of caffeine on the proliferation of already stimulated PBL can be explained by the reported ability of the methylxanthine to abolish cell cycle checkpoints [45]. As already mentioned, unstimulated PBL are resting in the G0 phase of the cell cycle after isolation and therefore show per se no mitotic and cell cycle activity. For these cells, a manipulation of the cell cycle exerts no effects, quite contrary to cells running through the cycle like PHA-stimulated PBL. In these cells, as observed in the present study, the abolishment of cell cycle checkpoints (e.g., through caffeine) results in an increased proliferation rate. The results of the present study indicate that the compounds probably do not only induce cell cycle arrest, as previously shown for certain AR ligands in some cancer cell lines [22, 36], but inhibit proliferation by additional mechanisms. An indication for this assumption is that unstimulated PBL resting in the G0 phase were significantly affected and [3H]thymidine uptake was potently reduced after the administration of CPA, BAY60-6583, and IB-MECA as well as PSB-36, MSX-2, and PSB-10.
The investigated AR agonists and antagonists can be structurally subdivided into several groups: (1) adenosine derivatives, (2) xanthine derivatives, and (3) non-nucleosidic adenine-like structures [46, 47]. Among the compounds that showed anti-proliferative activity, members of all three structural classes were represented. The mechanism of their anti-proliferative activity on lymphocytes is currently unknown. It might be speculated that adenosine derivatives, such as CPA, could serve as anti-metabolites of nucleic acid metabolism and thereby disrupt DNA or RNA synthesis. Another conceivable mechanism of the action of adenosine derivatives (CPA, IB-MECA), but also of the adenine-like compounds (BAY60-6583), and even of xanthine derivatives (PSB-36, MSX-2, PSB-10) might be due to kinase inhibition targeting the ATP co-substrate binding site [48–50]. However, further studies would be required to elucidate the mechanism of action for each of the compounds.
Conclusions
The first comprehensive study investigating the effects of standard agonists and antagonists for all four AR subtypes on the proliferation of unstimulated and PHA-stimulated PBL and on Jurkat T cells was performed. For several AR agonists and antagonists, anti-proliferative effects were determined. Several lines of evidence indicated that the effects found for the structurally diverse ligands—the agonists CPA, BAY60-6583 and IB-MECA and the antagonists PSB-36, MSX-2, and PSB-10—were AR-independent: (1) agonists as well as antagonists for the same AR subtype had the same effect (CPA and PSB-36; IB-MECA and PSB-10); (2) ligands for all four AR subtypes, some with opposing signaling pathways, were active; (3) subtype-selective antagonists did not block the effects of the agonists; and (4) the required concentrations for the anti-proliferative effects were about three orders of magnitude higher than those for binding to, blocking, or activating the ARs. The mechanisms of the anti-proliferative action on lymphocytes remain to be elucidated and can be expected to be different for each of the compounds.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (DFG) within the Graduiertenkolleg GRK 677 (S.K.L, C.E.M.) and the NRW-Forschungsschule Biotech Pharma (A.C.S., C.E.M.). We thank H. Eltzschig and S. Zug (University of Tübingen, Germany) and A. Bill (University of Bonn, Germany) for their support in real-time PCR experiments. The technical assistance of N. Florin is gratefully acknowledged.
Abbreviations
- ADA
Adenosine deaminase
- AR
Adenosine receptor
- BAY60-6583
2-[6-Amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]pyridin-2-ylsulfanyl]acetamide
- cAMP
Cyclic AMP
- CGS-21680
(2-p-[2-Carboxyethyl]phenethylamino)-5′-N-ethylcarboxamido-adenosine
- CI-IB-MECA
2-Chloro-N6-(3-iodobenzyl)-9-[5-(methyl-carbamoyl)-β-d-ribofuranosyl]adenine
- CPA
N6-Cyclopentyladenosine
- DMSO
Dimethyl sulfoxide
- EDTA
Ethylenediaminetetraacetic acid
- FCS
Fetal calf serum
- GPCR(s)
G protein-coupled receptor(s)
- IB-MECA
N6-(3-Iodobenzyl)-5′-N-methylcarboxamidoadenosine
- MSX-2
3-(3-Hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine
- NECA
5′-N-Ethylcarboxamidoadenosine
- PBL
Peripheral blood lymphocytes
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PHA
Phytohemagglutinin
- PSB-10
(R)-8-Ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one
- PSB-36
1-Butyl-8-(3-noradamantanyl)-3-(3-hydroxypropyl)xanthine
- PSB-603
8-[4-[4-(4-Chlorophenyl)piperazine-1-sulfonyl)phenyl]]-1-propylxanthine
- PSB-1115
1-Propyl-8-p-sulfophenylxanthine
- SCID
Severe combined immunodeficiency
- SCH420814
Preladenant
- TNF
Tumor necrosis factor
Footnotes
Anke C. Schiedel and Svenja K. Lacher contributed equally.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63(1):1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mirabet M, Mallol J, Lluis C, Franco R. Calcium mobilization in Jurkat cells via A2b adenosine receptors. Br J Pharmacol. 1997;122:1075–1082. doi: 10.1038/sj.bjp.0701495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. 2010;185(4):1993–1998. doi: 10.4049/jimmunol.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linden J, Cekic C. Regulation of lymphocyte function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32(9):2097–2103. doi: 10.1161/ATVBAHA.111.226837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrero ME. Purinoceptors in inflammation: potential as anti-inflammatory therapeutic targets. Front Biosci. 2011;16:2172–2186. doi: 10.2741/3846. [DOI] [PubMed] [Google Scholar]
- 7.Burnstock G, Brouns I, Adriaensen D, Timmermans JP. Purinergic signaling in the airways. Pharmacol Rev. 2012;64(4):834–868. doi: 10.1124/pr.111.005389. [DOI] [PubMed] [Google Scholar]
- 8.Erdmann AA, Gao ZG, Jung U, Foley J, Borenstein T, Jacobson KA, Fowler DH. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707–4714. doi: 10.1182/blood-2004-04-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohta A, Kjaergaard J, Sharma S, Mohsin M, Goel N, Madasu M, Fradkov E, Sitkovsky M. In vitro induction of T cells that are resistant to A2 adenosine receptor-mediated immunosuppression. Br J Pharmacol. 2009;156:297–306. doi: 10.1111/j.1476-5381.2008.00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbieri D, Abbracchio MP, Salvioli S, Monti D, Cossarizza A, Ceruti S, Brambilla R, Cattabeni F, Jacobson KA, Franceschi C. Apoptosis by 2-chloro-2′-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem Int. 1998;32:493–504. doi: 10.1016/S0197-0186(97)00129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szondy Z. Adenosine stimulates DNA fragmentation in human thymocytes by Ca2+-mediated mechanisms. Biochem J. 1994;304:877–885. doi: 10.1042/bj3040877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apasov SG, Sitkovsky MV. The extracellular versus intracellular mechanisms of inhibition of TCR-triggered activation in thymocytes by adenosine under conditions of inhibited adenosine deaminase. Int Immunol. 1999;11:179–189. doi: 10.1093/intimm/11.2.179. [DOI] [PubMed] [Google Scholar]
- 13.Desrosiers MD, Cembrola KM, Fakir MJ, Stephens LA, Jama FM, Shameli A, Mehal WZ, Santamaria P, Shi Y. Adenosine deamination sustains dendritic cell activation in inflammation. J Immunol. 2007;179(3):1884–1892. doi: 10.4049/jimmunol.179.3.1884. [DOI] [PubMed] [Google Scholar]
- 14.Gelfand EW, Lee JJ, Dosch HM. Selective toxicity of purine deoxynucleosides for human lymphocyte growth and function. Proc Natl Acad Sci U S A. 1979;76:1998–2002. doi: 10.1073/pnas.76.4.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kizaki H, Suzuki K, Tadakuma T, Ishimura Y. Adenosine receptor-mediated accumulation of cyclic AMP-induced T-lymphocyte death through internucleosomal DNA cleavage. J Biol Chem. 1990;265:5280–5284. [PubMed] [Google Scholar]
- 16.Jackson EK, Gillespie DG, Dubey RK. 2′-AMP and 3′-AMP inhibit proliferation of preglomerular vascular smooth muscle cells and glomerular mesangial cells via A2B receptors. J Pharmacol Exp Ther. 2011;337(2):444–450. doi: 10.1124/jpet.110.178137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taliani S, La Motta C, Mugnaini L, Simorini F, Salerno S, Marini AM, Da Settimo F, Cosconati S, Cosimelli B, Greco G, Limongelli V, Marinelli L, Novellino E, Ciampi O, Daniele S, Trincavelli ML, Martini C. Novel N2-substituted pyrazolo[3,4-d]pyrimidine adenosine A3 receptor antagonists: inhibition of A3-mediated human glioblastoma cell proliferation. J Med Chem. 2010;53:3954–3963. doi: 10.1021/jm901785w. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka Y, Yoshihara K, Tsuyuki M, Kamiya T. Apoptosis induced by adenosine in human leukemia HL-60 cells. Exp Cell Res. 1994;213:242–252. doi: 10.1006/excr.1994.1196. [DOI] [PubMed] [Google Scholar]
- 19.Abbracchio MP. P1 and P2 receptors in cell growth and differentiation. Drug Dev Res. 1996;39:393–406. doi: 10.1002/(SICI)1098-2299(199611/12)39:3/4<393::AID-DDR21>3.0.CO;2-1. [DOI] [Google Scholar]
- 20.Jacobson KA, Hoffmann C, Cattabeni F, Abbracchio MP. Adenosine-induced cell death: evidence for receptor-mediated signalling. Apoptosis. 1999;4:197–211. doi: 10.1023/A:1009666707307. [DOI] [PubMed] [Google Scholar]
- 21.Mlejnek P, Dolezel P, Kosztyu P. P-glycoprotein mediates resistance to A3 adenosine receptor agonist 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-n-methyluronamide in human leukemia cells. J Cell Physiol. 2012;227:676–685. doi: 10.1002/jcp.22775. [DOI] [PubMed] [Google Scholar]
- 22.Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceschi C, Kim YC, Jacobson KA, Klotz KN, Lohse MJ, Abbracchio MP. Activation of the A3 adenosine receptor affects cell cycle progression and cell growth. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:225–234. doi: 10.1007/s002109900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, Leung E, Baraldi PG, Borea PA. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol. 2002;119:923–933. doi: 10.1046/j.1523-1747.2002.00111.x. [DOI] [PubMed] [Google Scholar]
- 24.Panjehpour M, Karami-Tehrani F. Adenosine modulates cell growth in the human breast cancer cells via adenosine receptors. Oncol Res. 2007;16:575–585. doi: 10.3727/000000007783629981. [DOI] [PubMed] [Google Scholar]
- 25.Schneider C, Wiendl H, Ogilvie A. Biphasic cytotoxic mechanism of extracellular ATP on U-937 human histiocytic leukemia cells: involvement of adenosine generation. Biochim Biophys Acta. 2001;1538:190–205. doi: 10.1016/S0167-4889(01)00069-6. [DOI] [PubMed] [Google Scholar]
- 26.Fishman P, Bar-Yehuda S, Barer F, Madi L, Multani AS, Pathak S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp Cell Res. 2001;269:230–236. doi: 10.1006/excr.2001.5327. [DOI] [PubMed] [Google Scholar]
- 27.Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. 2005;280:19516–19526. doi: 10.1074/jbc.M413772200. [DOI] [PubMed] [Google Scholar]
- 28.Blad CC, von Frijtag Drabbe Kunzel JK, de Vries H, Mulder-Krieger T, Bar-Yehuda S, Fishman P, IJzerman AP. Putative role of the adenosine A3 receptor in the antiproliferative action of N6-(2-isopentenyl)adenosine. Purinergic Signal. 2011;7:453–462. doi: 10.1007/s11302-011-9244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panjehpour M, Castro M, Klotz KN. Human breast cancer cell line MDA-MB-231 expresses endogenous A2B adenosine receptors mediating a Ca2+ signal. Br J Pharmacol. 2005;145:211–218. doi: 10.1038/sj.bjp.0706180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson EK, Ren J, Gillespie DG. 2′,3′-cAMP, 3′-AMP, and 2′-AMP inhibit human aortic and coronary vascular smooth muscle cell proliferation via A2B receptors. Am J Physiol Heart Circ Physiol. 2011;301(2):H391–401. doi: 10.1152/ajpheart.00336.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Simioni C, Leung E, Maclennan S, Baraldi PG, Borea PA. Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1a, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol Pharmacol. 2007;72(2):395–406. doi: 10.1124/mol.106.032920. [DOI] [PubMed] [Google Scholar]
- 32.Ma DF, Kondo T, Nakazawa T, Niu DF, Mochizuki K, Kawasaki T, Yamane T, Katoh R. Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Hum Pathol. 2010;41(11):1550–1557. doi: 10.1016/j.humpath.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Ceruti S, Franceschi C, Barbieri D, Malorni W, Camurri A, Giammarioli AM, Ambrosini A, Racagni G, Cattabeni F, Abbracchio MP. Apoptosis induced by 2-chloro-adenosine and 2-chloro-2′-deoxy-adenosine in a human astrocytoma cell line: differential mechanisms and possible clinical relevance. J Neurosci Res. 2000;60:388–400. doi: 10.1002/(SICI)1097-4547(20000501)60:3<388::AID-JNR14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 34.Minelli A, Bellezza I, Tucci A, Rambotti MG, Conte C, Culig Z. Differential involvement of reactive oxygen species and nucleoside transporters in cytotoxicity induced by two adenosine analogues in human prostate cancer cells. Prostate. 2009;69:538–547. doi: 10.1002/pros.20900. [DOI] [PubMed] [Google Scholar]
- 35.Mirabet M, Mallol J, Lluis C, Franco R. Dipropylcyclopentylxanthine triggers apoptosis in Jurkat T cells by a receptor-independent mechanism. Cell Death Differ. 1997;4:639–646. doi: 10.1038/sj.cdd.4400291. [DOI] [PubMed] [Google Scholar]
- 36.Morello S, Petrella A, Festa M, Popolo A, Monaco M, Vuttariello E, Chiappetta G, Parente L, Pinto A. Cl-IB-MECA inhibits human thyroid cancer cell proliferation independently of A3 adenosine receptor activation. Cancer Biol Ther. 2008;7:278–284. doi: 10.4161/cbt.7.2.5301. [DOI] [PubMed] [Google Scholar]
- 37.Boyum A. Separation of leukocytes from blood and bone marrow. Introduction. Scand J Clin Lab Invest Suppl. 1968;97:7. [PubMed] [Google Scholar]
- 38.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohta A, Sitkovsky M. Methylxanthines, inflammation, and cancer: fundamental mechanisms. Handb Exp Pharmacol. 2011;200:469–481. doi: 10.1007/978-3-642-13443-2_19. [DOI] [PubMed] [Google Scholar]
- 40.Himer L, Csoka B, Selmeczy Z, Koscso B, Pocza T, Pacher P, Nemeth ZH, Deitch EA, Vizi ES, Cronstein BN, Hasko G. Adenosine A2A receptor activation protects CD4+ T lymphocytes against activation-induced cell death. FASEB J. 2010;24(8):2631–2640. doi: 10.1096/fj.10-155192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mills JH, Kim DG, Krenz A, Chen JF, Bynoe MS. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol. 2012;188(11):5713–5722. doi: 10.4049/jimmunol.1200545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gessi S, Varani K, Merighi S, Cattabriga E, Avitabile A, Gavioli R, Fortini C, Leung E, Mac Lennan S, Borea PA. Expression of A3 adenosine receptors in human lymphocytes: up-regulation in T cell activation. Mol Pharmacol. 2004;65(3):711–719. doi: 10.1124/mol.65.3.711. [DOI] [PubMed] [Google Scholar]
- 43.Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. doi: 10.1016/j.it.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Panjehpour M, Karami-Tehrani F. An adenosine analog (IB-MECA) inhibits anchorage-dependent cell growth of various human breast cancer cell lines. Int J Biochem Cell Biol. 2004;36:1502–1509. doi: 10.1016/j.biocel.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Feng Y, Wu J, Feng X, Tao D, Hu J, Qin J, Li X, Xiao W, Gardner K, Judge SI, Li QQ, Gong J. Timing of apoptosis onset depends on cell cycle progression in peripheral blood lymphocytes and lymphocytic leukemia cells. Oncol Rep. 2007;17:1437–1444. [PubMed] [Google Scholar]
- 46.Müller CE, Ferre S. Blocking striatal adenosine A2A receptors: a new strategy for basal ganglia disorders. Recent Pat CNS Drug Discov. 2007;2(1):1–21. doi: 10.2174/157488907779561772. [DOI] [PubMed] [Google Scholar]
- 47.Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta. 2011;1808(5):1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shelton JR, Cutler CE, Oliveira M, Balzarini J, Peterson MA. Synthesis, SAR, and preliminary mechanistic evaluation of novel antiproliferative N6,5′-bis-ureido- and 5′-carbamoyl-N6-ureidoadenosine derivatives. Bioorg Med Chem. 2012;20(2):1008–1019. doi: 10.1016/j.bmc.2011.11.043. [DOI] [PubMed] [Google Scholar]
- 49.Böhm L, Roos WP, Serafin AM. Inhibition of DNA repair by Pentoxifylline and related methylxanthine derivatives. Toxicology. 2003;193(1–2):153–160. doi: 10.1016/S0300-483X(03)00294-4. [DOI] [PubMed] [Google Scholar]
- 50.Garuti L, Roberti M, Bottegoni G. Small molecule aurora kinases inhibitors. Curr Med Chem. 2009;16(16):1949–1963. doi: 10.2174/092986709788682227. [DOI] [PubMed] [Google Scholar]