

Abstract
Comparative study of STICS using HPLC-assisted experimental set-up clearly demonstrated benefits of using longer spacer-anchoring systems. The use of mixed self-assembled monolayers helps to provide the required space for glycosylation reaction around the immobilized glycosyl acceptor. Both extension of the spacer length and using mixed self-assembled monolayers help to promote reaction and the beneficial effects may include moving the glycosyl acceptor further out into solution and providing additional conformational flexibility. It is possible that surface-immobilized glycosyl acceptors with a longer spacer (C8-O-C8)-lipoic acid have a higher tendency to mimic a solution-phase reaction environment than that of acceptors with shorter spacers.
Keywords: glycosylation, solid-phase synthesis, carbohydrates, oligosaccharides, self-assembled monolayers, nanoporous gold
Introduction
Organic synthesis on solid phases represents an active area of research.1,2 The end of the last century has witnessed heated interest in the area of solid phase-supported synthesis of carbohydrates and, as a result, dramatic improvements have emerged. Early efforts to synthesize oligosaccharides and glycoconjugates using a manual approach3–7 have evolved into the development of an automated oligosaccharide synthesizer by Seeberger et al.8–16 Both manual and automated polymer supported synthesis is very attractive because it allows for the rapid synthesis of oligosaccharide sequences without the necessity of purifying (and characterizing) the intermediates. Another important advantage of oligosaccharide synthesis on solid phase support is the ease of excess reagent removal (usually achieved by filtration).17,18 In spite of remarkable progress, solid phase techniques still suffer from significant limitations: long reaction times, large reagent excess, limited use of molecular sieves, large volumes of waste solvent, cumbersome analysis of intermediates, lower stereoselectivity, loss and poisoning of resin, reagent trapping, etc.
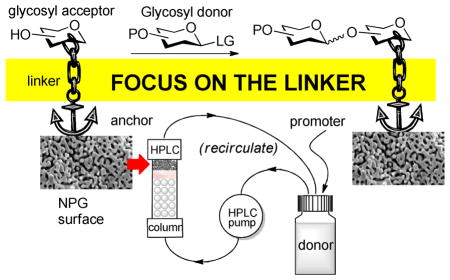
To investigate whether these limitations could be addressed by using a nanoporous gold surface instead of polymer support, recently we developed a new technique that we named STICS (surface-tethered iterative carbohydrate synthesis).19 As illustrated in Scheme 1, at the basis of the concept is a surface functionalized ‘stick’ made of chemically stable high surface area material that would simplify the transformation of the solid support-bound molecules between the reaction vessels. The glycosyl acceptor-anchored stick (A) is placed in the reaction vessel (B), containing sufficient quantities of the glycosyl donor, promoter and molecular sieves. Upon completion of the reaction, the stick, functionalized by a protected terminal sugar moiety, is transferred into another vessel (C) containing an appropriate solvent, wherein excess reagent is being rinsed off. Subsequently, the stick is dipped into the vessel (D) containing a certain reagent to remove a strategically placed temporary substituent P. This transformation results in the formation of the second-generation glycosyl acceptor. To conclude the cycle, the stick is then briefly placed in a thick-wall container connected to the vacuum (E). Upon completion of this sequence, again, a hydroxyl-modified stick is available for consequent glycosylation. To repeat the cycle, coupling-washing-deprotection-drying steps are performed once again. At the end of the synthesis, the oligosaccharide can be either cleaved off from the stick or deprotected directly on the stick to be used for immunoassay or molecular recognition studies.
Scheme 1.

STICS concept outline and focus of this study
Whereas it is common that reactions on solid supports are performed multiple times to ensure their completeness, we intentionally performed each reaction only once to elucidate the scope of the STICS approach. For instance, the lipoic acid anchor/linker-glycosyl acceptor scaffold provided the best yields (~60%) in comparison to those using other anchoring and glycosylation approaches used in our original STICS study.19 This result clearly illustrates the need of significant improvement for the STICS approach to become attractive on a practical basis. Although quantitative conversion yields can be achieved by repeating this reaction 2–3 times, this time and material investment would be hardly justified and would not serve the needs to reduce drawbacks of the solid-phase synthesis.
We hypothesized that one way to improve reaction yields is by lengthening the linker group20 that will extend the glycosyl acceptor further away from the nanoporous gold (NPG) surface and create more reaction space and the prospect for improved reactivity and higher yields. The distancing from the surface should be especially helpful for those molecules immobilized along the ligament sections of positive curvature as the carbohydrate groups become further apart as they project out into the solution. The typical diameters of the pores and ligaments of the NPG used in these studies falls in the range of 50 – 200 nm.21 This may help to reduce the gap between relatively slow surface or polymer-bound reactions and fast reactions in solution. In other words, the STICS would approach being solution-like in terms of efficiency, reactivity, and yields. Regions of negative curvature where ligaments meet are not as favorable as reaction spaces but overall account for a smaller amount of the available surface.
As the anchor we chose to continue with commercially available lipoic acid, as that gave the best results so far. In the previous study, lipoic acid was connected directly to the glycosyl acceptor. Lipoic acid was proven sufficient for the formation of a chemically resistant self-assembled monolayer (SAM) on the surface of the porous gold plates used in STICS. The latter conclusion was drawn from the fact that no desorption of the glycosyl acceptor from the surface was detected during either the glycosylation or deprotection steps. This application of lipoic acid-based anchor would help to avoid another potential drawback of polythiolated compounds, such as previously investigated tripod and adamantane anchors,19 in which about 30% of the mercapto groups may remain unbound to the surface and hence may potentially interact with thiophilic reagents used in STICS. While it is not guaranteed that all sulfurs of a lipoic acid anchor are involved in gold-thiolate bonds,22 with two points of attachment such accommodation is easier to achieve. All anchors bearing a cleavable lipoic acid ester should allow for convenient cleavage under basic conditions when needed.
As a part of ongoing research program in our laboratories, herein we report the synthesis and study of a series of lipoic acid anchor/extended linker/glycosyl acceptor scaffold molecules 1–3 depicted in Figure 1. The purpose of this study is to test our hypothesis that the spacer length has a significant effect on the outcome of glycosylation on the surface of the nanoporous gold. The spacer moieties were based on 1,4-butanediol, 1,8-octanediol and the dimer of the latter, 9-oxaheptadecane-1,17-diol. This will allow us to extend the acceptor molecule away from the surface by O(CH2)4 (in scaffold 1), O(CH2)8 (in scaffold 2), and O(CH2)8O(CH2)8 (in scaffold 3) in comparison to the original study wherein the acceptor was conjugated to lipoic acid directly.
Figure 1.
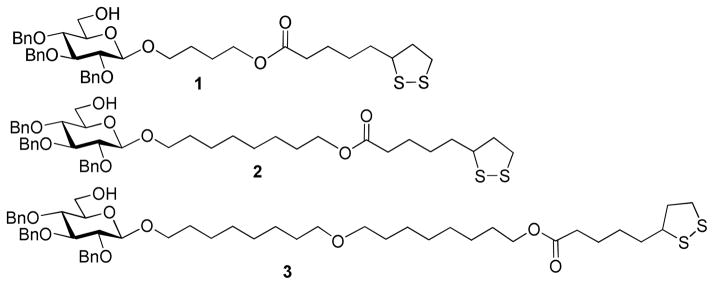
A series of lipoic acid anchor/extended linker/glycosyl acceptor scaffold molecules 1–3.
Results and discussion
Synthesis of glycosyl acceptors molecules 1 and 2 both followed the same pathway as depicted in Scheme 2. First, benzobromoglucose 423 was glycosidated with acceptors 5a24/5b25,26 under the Helferich conditions (Hg(CN)2/HgBr2) to afford the corresponding glycosides 6a/6b in 89%/92% yield, respectively. After that, compounds 6a/6b were sequentially deacylated under Zemplen conditions (MeONa), tritylated (TrCl/pyridine), and benzylated (BnBr/NaH) to afford compounds 7a/7b in 62%/74% yield over three steps. The latter compounds were desilylated (Bu4NF) to afford alcohols 8a27/8b in 93%/88% yields. The latter was esterified with lipoic acid in the presence of EDC and the resulting compounds were detritylated (dilute trifluoroacetic acid) to give the target glycosyl acceptors 1 and 2 in 96% and 75% yield, respectively.
Scheme 2.

Synthesis of lipoic acid derived glycosyl acceptors 1 and 2.
Conditions: a) Hg(CN)2/HgBr2, ClCH2CH2Cl, 4Å molec. sieves, rt, 36 h; b) NaOCH3/CH3OH (and THF for 6b), rt, 5 h; c) TrCl, pyridine, DMAP, 80 °C, 6 h; d) BnBr, NaH, DMF, 0 °C, 5 h; e) TBAF/THF, THF, rt, 3 h; f) lipoic acid, EDC, DMAP, CH2Cl2, rt, 8 h; g) 10% TFA/CH2Cl2, rt, 2 h.
The synthesis of glycosyl acceptor 3 began from the commercially available 1,8-octanediol 9 as depicted in Scheme 3. Monobenzylation of 9 (BnBr/NaH) afforded alcohol 1028 (53% yield), which was then treated with TsCl in pyridine to afford tosylate 1129 in 91% yield. Reaction of 10 with 11 in the presence of NaH in THF at reflux produced dimeric product 12 in 87% yield. The latter was then hydrogenated (H2, Pd/C) to afford diol 13 in 98% yield. Glycosylation of diol 13 with glycosyl donor 1430 was catalyzed by TMSOTf and afforded mono glycosylated product 15 in 70% yield. After that, glycoside 15 was silylated (TBDPSCl in pyridine) and the resulting product 16 (91%) was sequentially deacylated by Zemplen (NaOMe in MeOH), tritylated (TrCl, DMAP, pyridine), and benzylated (BnBr, NaH in DMF) to afford fully protected compound 17 in 82% yield over three steps. Then, compound 17 was desilylated (95%) and the liberated hydroxyl in compound 18 was coupled with lipoic acid followed by detritylation afforded the target glycosyl acceptor 3 in 88% yield over two steps.
Scheme 3.

Synthesis of lipoic acid derived acceptor 3.
Conditions: a) BnBr, NaH, THF/DMF, 0 °C→rt, 16 h; b) TsCl, pyridine, DMAP, 0 °C→rt, 16 h; c) NaH, THF, 60 °C, 24 h; d) H2, Pd/C, MeOH, rt, 8 h; e) TMSOTf, CH2Cl2, 3Å molec. sieves, rt, 2 h; f) TBDPSCl, pyridine, 50 °C, 6 h; g) NaOCH3/CH3OH, rt, 5 h; h) TrCl, DMAP, pyridine, 80 °C, 8 h; i) BnBr, NaH, DMF, 0 °C, 5 h; j) TBAF, THF, rt, 4 h; k) lipoic acid, EDC, DMAP, CH2Cl2, rt, 8 h; l) 10% TFA/CH2Cl2, rt, 2 h.
Having obtained different thiolated glycosyl acceptors 1–3, we turned our attention to studying their immobilization on the nanoporous gold chips using CH2Cl2 as solvent. The quantitative estimation of the amount of sugar molecules absorbed on the surface to form self-assembled monolayers (SAMs) was determined by weighing the NPG chips using a microbalance before and after immobilization. The loading and unloading study on the nanoporous gold surface was aimed at examining the ease of recovery of sugar from the gold chips. Accordingly, the loading and unloading of the sugar portion from the nanoporous surface was conducted and it was estimated to be quantitative recovery of the protected sugar used in monolayer formation. The average increase in mass upon loading of 1–3 on a NPG plate (8.0 mm × 8.0 mm × 0.25 mm) was found to be 0.75–0.80 mg. The mass of an NPG plate of these dimensions is typically 80 mg. Using a specific surface area of 8.2 m2 g−1 for 72 h dealloyed NPG material,31 this corresponds to a total estimated surface of 0.66 m2. The molar masses of 1–3 are 668.9, 725.0, and 853.2 g mol−1, respectively. The loaded masses of 0.75–0.80 mg for 1–3 correspond to a loading of 0.94–1.1 μmol per plate, in the same range as found in our prior reported study.19 The loading is equivalent to a surface coverage of 1.44–1.66 μmol m−2, or 0.87–1.00 × 1018 molecules m−2.
During our initial experimentation, glycosylation of pure monolayer of glycosyl acceptors 1–3 with imidate donor was not successful. At this point, we assumed that overcrowding of sugar molecules of same length in the monolayer prevents any glycosidation reaction to occur. Hence, we decided to test the possibility of forming mixed monolayers with a similarly structured compound i.e., the methyl ester of lipoic acid derivative 1932,33 as the additive. Our goal in using mixed SAMs was to achieve less steric crowding around the acceptor sugar and its bulky protecting groups and thereby facilitate the glycosylation reaction by allowing better access for the donor and promoter to the reactive hydroxyl group. It is also likely that in a mixed SAM, the conformation flexibility of the protected sugars would be increased and they would be more likely to display a reactive conformation. After comparative study of different monolayer ratios between acceptor 1–3 and additive 19 (1/1, 2/3, 1/4, and 4/1, mol/mol) we determined that the monolayer ratio of 2/3 gives comparatively better disaccharide formation than others.
Hence, for the present study, freshly prepared nanoporous gold chips were immersed in a solution containing the desired ratio of one of the glycosyl acceptors 1–3 (0.0422 mmol) and additive 19 (0.0636 mmol) in CH2Cl2 (10 mL) for 10 h. Resultantly, we obtained gold chips loaded with a SAM containing the glycosyl acceptor and the additive 1a–3a (Scheme 4). It is known that the surface composition of SAMs as prepared from solution does not necessarily match the composition of the solution from which they are prepared in terms of ratio of the two components; therefore, the 2:3 ratio is the solution molar ratio and may differ from that actually on the gold surface.34 The typical mass loading onto a 8.0 mm × 8.0 mm × 0.25 mm NPG plate for the mixed SAM of this ratio is 0.35–0.40 mg per plate, and this lower mass than found for the loading of the pure glycosyl acceptors is expected since 19 has a lower molar mass of 220.4 g mol−1. The lipoic ester 19 should occupy less area on the gold surface than 1–3, since it lacks the large protected sugar unit.
Scheme 4.

Loading mixed SAMs: synthesis of NPG-immobilized glycosyl acceptors 1a–3a
Modified NPG chips 1a–3a were used in glycosylations using our recently developed HPLC-assisted automated technology for oligosaccharide synthesis.27 This experimental setup based on an unmodified HPLC instrument was executed as follows. The Omnifit SolventPlus chromatography glass column was loaded with freshly activated molecular sieves (3Å beads) and gold chips modified by SAMs (approx. size 4.0 mm × 4.0 mm × 0.25 mm, quantity, ~0.20 mg each) separated by a pad of cotton (Scheme 5). The use of molecular sieves represents a deviation from the experimental set-up reported previously for polymer-supported synthesis. This is because we noticed that performing glycosylation using SAM-modified NPG under a strictly anhydrous condition has a dramatic effect on the yield of glycosylation. The loaded column was then connected to the HPLC and a solution of glycosyl donor in CH2Cl2 (78 mM) was then first circulated through the packed column containing molecular sieves and NPG chips for 30 min (flow rate 1.0 mL/min, 8 atm) in the absence of promoter. This step helps to remove traces of moisture in the stock solution of the glycosyl donor. After that, promoter was added to the stock solution. After 2 h of continuous recirculation of reagents, the system was purged with CH2Cl2 to wash off any impurities that may be remaining on the NPG surface. Then, NPG chips loaded with disaccharide derivatives were carefully removed from the column and the sugar content was cleaved off by soaking the plates in a 1 M solution of NaOMe in MeOH for 16 h.
Scheme 5.
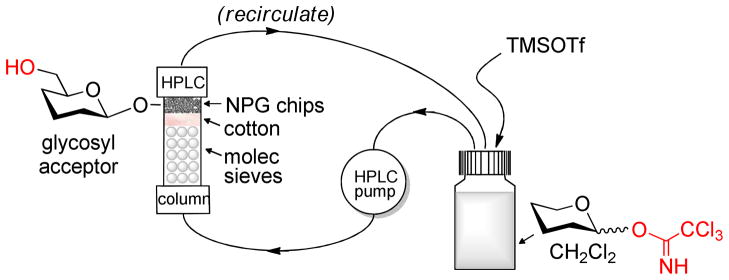
The outline of the HPLC-assisted experimental set-up for glycosylation of SAM-modified NPG chips.
Following this general experimental set-up, we conducted comparative studies of immobilized acceptors 1a–3a. Glycosylation with glycosyl donor 14 followed by deprotection/cleavage and acetylation afforded the corresponding disaccharides 20–22 in yields ranging between 60 and 90%. The yield calculation was based on TLC and NMR analysis of the oligosaccharide mixtures cleaved off from the surface. More precise calculations were deemed impractical given the use of a mixed SAM of glycosyl acceptor 1–3 and methyl lipoic ester 19 since this introduces some uncertainty as to the exact percent of the SAM composed of the glycosyl acceptor. More detailed studies seeking to estimate the surface bound ratios of glycosyl acceptor and additives such as the lipoic ester (19) on the NPG surface with aid of surface analytical methods are currently underway. If the exact number of immobilized acceptor sugar units on the NPG chips used could be determined, then yields could be based on the mass of disaccharide obtained. The best yield was obtained for the synthesis of disaccharide 22 from acceptor 3a equipped with the longest C8-O-C8 spacer. In this case, practically no monosaccharide derived from the immobilized glycosyl acceptor was detected. In contrast, in the case of the synthesis of disaccharides 20 (60%) and 21 (75%) a notable amount of monosaccharide was detected (30% and 15%, respectively).
Conclusions
As evidenced from comparative glycosylations, the glycosyl acceptor with longer spacer has a higher tendency to mimic a solution-phase reaction environment than that of acceptors with shorter spacers. The use of mixed SAMs helps to provide the required space for reaction around the immobilized acceptor. Extension of the spacer length could in principle have a number of effects to promote reaction with the donor, including moving the acceptor sugar further out into solution and providing additional conformational flexibility through which the acceptor sugar is more likely to adopt a reactive orientation. Another strategy other than the use of mixed SAMs that could be considered to potentially improve reactivity would be the use of a SAM anchor molecule which in itself has a larger surface footprint to accommodate the dimensions of the protected sugar. However, such a strategy may not significantly increase the ultimate amount of product recovered. In principle, the gold chip is loaded with the disaccharide derivative that can be subjected for further oligosaccharide elongation via alternating deprotection/glycosylation steps. Additionally, as shown in our previous study,27 all steps including glycosylation, deprotection, and cleavage from the solid support can be monitored using a standard HPLC detection system set to record changes in the UV absorbance of the solution eluting off the column.
Experimental part
4-[5-(1,2-Dithiolan-3-yl)pentanoyl]oxybut-1-yl 2,3,4-tri-O-benzyl-β-D-glucopyranoside (1)
Lipoic acid (0.048 g, 0.235 mmol) was added to a solution of compound 8a27 (0.180 g, 0.235 mmol), EDC hydrochloride (0.050 g, 0.259 mmol), and DMAP (0.014 g, 0.117 mmol) in CH2Cl2 (5.0 mL) and the resulting mixture was stirred under argon for 8 h at rt. After that, a 10% aq. TFA (5.0 mL) was added dropwise and the resulting mixture was stirred for 1 h at rt. The mixture was then diluted with CH2Cl2 (~50 mL) and washed with water (3 × 50 mL), 5% aq. NaHCO3 (50 mL), and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone – toluene gradient elution) to afford the title compound (0.160 g, 96%) as a white foam. Analytical data for 1: Rf = 0.50 (acetone/toluene, 1/5, v/v); 1H-NMR (300 MHz, CDCl3): δ, 1.41–1.50 (m, 2H, -CH2-), 1.62–1.80 (m, 9H, 4 × -CH2-, -CH-), 1.83–1.95 (m, 1H, -CH2-), 2.29 (t, 2H, J = 7.3 Hz, -CH2COO-), 2.39–2.50 (m, 1H, -CH2-), 3.06–3.21 (m, 2H, -CH2-), 3.33–3.38 (m, 1H, H-5), 3.40 (dd, 1H, J2,3 = 8.8 Hz, H-2), 3.51–3.62 (m, 3H, H-4, OCH2-, OH ), 3.66 (dd, 1H, J3,4 = 9.0 Hz, H-3), 3.71 (dd, 1H, J5,6a = 4.6 Hz, J6a,6b = 11.7 Hz, H-6a), 3.86 (dd, 1H, J5,6b = 2.6 Hz, H-6b), 3.93 (m, 1H, OCH2-), 4.09 (t, 2H, J = 5.9 Hz, -CH2OCO-), 4.43 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.75 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.83 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.86 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 7.24–7.31 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 24.6, 25.4, 26.2, 28.7, 34.0, 34.6, 38.4, 40.2, 56.3, 62.0, 63.9, 69.4, 74.9, 75.0 (x 2), 75.7, 77.5, 82.3, 84.5, 103.6, 127.6, 127.7, 127.8 (x 2), 127.9, 128.0 (x 2), 128.1 (x 2), 128.4 (x 4), 128.5 (x 2), 137.9, 138.3, 138.5, 173.5 ppm; HR-FAB MS [M+Na]+ calcd for C39H50O8S2Na 733.2845, found 733.2827.
8-(tert-Butyldiphenylsilyloxy)oct-1-yl 2,3,4,6-tetra-O-benzoyl-β-D-glucopyranoside (6b)
A solution of 2,3,4,6-tetra-O-benzoyl-α-D-glucopyranosyl bromide (4,23 2.0 g, 3.03 mmol) in ClCH2CH2Cl (10 mL) was added to a suspension of 8-(tert-butyldiphenylsilyloxy)octan-1-ol (5b,25,26 1.28 g, 3.33 mmol), Hg(CN)2 (0.766 g, 3.03 mmol), HgBr2 (0.546 g, 1.50 mmol), and molecular sieves (4 Å, 3.0 g) in ClCH2CH2Cl (20 mL) and the resulting mixture was stirred under argon for 36 h at rt. After that, the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~100 mL) was washed with 10% aq. Na2S2O3 (2 × 50 mL), 5% aq. NaHCO3 (50 mL), water (50 mL), and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to afford the title compound (2.67 g, 92%) as a white foam. Analytical data for 6b: Rf = 0.50 (ethyl acetate/hexane, 1/3, v/v); [α]D22 +13.4 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 0.97 (s, 9H, t-butyl), 1.01–1.14 (m, 8H, 4 × -CH2-), 1.33–1.49 (m, 4H, 2 × -CH2-), 3.45 (dt, 1H, J = 9.7, 6.2 Hz, OCH2a-), 3.52 (t, 2H, J = 6.6 Hz, -CH2OSi), 3.83 (m, 1H, J = 9.7, 6.2 Hz, OCH2b-), 4.05–4.10 (m, 1H, H-5), 4.43 (dd, 1H, J5,6a = 5.2 Hz, J6a,6b = 12.1 Hz, H-6a), 4.56 (dd, 1H, J5,6b = 3.3 Hz, H-6b), 4.75 (d, 1H, J1,2 = 7.9 Hz, H-1), 5.45 (dd, 1H, J2,3 = 9.7 Hz, H-2), 5.60 (dd, 1H, J4,5 = 9.7 Hz, H-4), 5.83 (dd, 1H, J3,4 = 9.6 Hz, H-3), 7.18–7.95 ppm (m, 30H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 19.2, 25.6, 25.7, 26.8 (x 3), 29.1, 29.2, 29.3, 32.5, 63.2, 63.9, 69.8, 70.3, 71.9, 72.1, 72.9, 101.3, 127.5 (x 5), 128.3 (x 6), 128.4 (x 2), 128.8 (x 2), 129.3, 129.5 (x 2), 129.6, 129.7 (x 5), 129.8 (x 2), 133.0, 133.1, 133.2, 133.4, 134.1, 135.5 (x 5), 165.0, 165.2, 165.8, 166.1 ppm; HR-FAB MS [M+Na]+ calcd for C58H62O11SiNa 985.3959, found 985.3961.
8-(tert-Butyldiphenylsilyloxy)oct-1-yl 2,3,4-tri-O-benzyl-6-O-triphenylmethyl-β-D-glucopyranoside (7b)
NaOMe (1 mL, 0.5 M solution) was added to a solution of 6b (2.51 g, 2.61 mmol) in MeOH (30 mL) until pH ~9 and the resulting mixture was stirred under argon for 5 h at rt. After that, the reaction mixture was neutralized with Dowex (H+), the resin was filtered off and rinsed successively with MeOH. The combine filtrate was concentrated in vacuo and dried. The crude residue was dissolved in pyridine (15 mL), trityl chloride (2.17 g, 7.79 mmol) and DMAP (0.063 g, 0.519 mmol) were added, and the resulting mixture was stirred under argon for 10 h at 80 °C. After that, the volatiles were removed in vacuo, and the residue was co-evaporated with toluene (3 × 50 mL). The resulting residue was diluted with CH2Cl2 (~50 mL) and washed with water (3 × 50 mL) and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was filtered through a pad of silica gel (ethyl acetate – toluene elution) and concentrated in vacuo. The crude residue was dissolved in DMF (40 mL), NaH (60% dispersion in oil, 0.624 g, 15.6 mmol) was added portionwise, and the resulting mixture was stirred under argon for 30 min at 0 °C. Then, BnBr (1.11 mL, 9.35 mmol) was added and the resulting reaction mixture was stirred for 5 h at rt. The reaction mixture was neutralized with AcOH/MeOH (1/10, v/v), diluted with CH2Cl2 (80 mL), and washed with water (2 × 50 mL) and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to afford the title compound (2.04 g, 74%) as a pale-yellow foam. Analytical data for 7b: Rf = 0.50 (ethyl acetate/hexane, 1/9, v/v); [α]D22 +4.5 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 0.99 (s, 9H, t-butyl), 1.15–1.52 (m, 10H, 5 × -CH2-), 1.62–1.68 (m, 2H, -CH2-), 3.16 (dd, 1H, J5,6a = 3.9 Hx, J6a,6b = 10.1 Hz, H-6a), 3.31–3.35 (m, 1H, H-5), 3.44–3.59 (m, 6H, H-2, 3, 6b, OCH2a-, -CH2OSi), 3.73 (dd, 1H, J4,5 = 8.9 Hz, H-4), 3.98 (m, 1H, OCH2b-), 4.36 (d, 1H, J1,2 = 7.3 Hz, H-1), 4.45 (dd, 2H, 2J = 10.3 Hz, CH2Ph), 4.77 (dd, 2H, 2J = 10.8 Hz, CH2Ph), 4.81 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 6.77–7.62 ppm (m, 40H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 19.2, 25.7, 26.3, 26.5, 26.9 (x 3), 29.4, 29.5, 32.6, 62.4, 64.0, 69.8, 74.5, 74.8, 75.0, 75.9, 77.9, 82.5, 84.7, 86.3, 103.6, 126.9 (x 2), 127.5 (x 3), 127.6 (x 2), 127.7 (x 5), 127.8 (x 3), 128.0 (x 2), 128.1 (x 2), 128.2 (x 2), 128.3, 128.4 (x 2), 128.8 (x 3), 129.4, 129.6 (x 4), 134.1, 134.8 (x 4), 135.1 (x 2), 135.5 (x 3), 137.8, 138.6 (x 2), 143.9 (x 3) ppm; HR-FAB MS [M+Na]+ calcd for C70H78O7SiNa 1081.5415, found 1081.5392.
8-Hydroxyoct-1-yl 2,3,4-tri-O-benzyl-6-O-triphenylmethyl-β-D-glucopyranoside (8b)
TBAF (2.10 mL, 2.08 mmol) was added to a solution of 7b (1.10 g, 1.03 mmol) in THF (20 mL) and the resulting mixture was stirred under argon for 3 h at rt. The reaction mixture was then diluted with CH2Cl2 (~40 mL) and washed with water (50 mL) and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – toluene gradient elution) to afford the title compound (0.750 g, 88%) as a white foam. Analytical data for 8b: Rf = 0.50 (ethyl acetate/hexane, 1/4, v/v); [α]D24 +3.2 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.37 (s, 6H, 3 × -CH2-), 1.46–1.62 (m, 5H, 2 × -CH2-, OH), 1.74–1.83 (m, 2H, -CH2-), 3.27 (dd, 1H, J5,6a = 4.0, J6a,6b = 10.1 Hz, H-6a), 3.42–3.48 (m, 1H, H-5), 3.52–3.69 (m, 6H, H-2, 3, 6b, OCH2a-, -CH2OH), 3.84 (dd, 1H, J4,5 = 8.9 Hz, H-4), 4.10 (m, 1H, OCH2b-), 4.48 (d, 1H, J1,2 = 7.3 Hz, H-1), 4.56 (dd, 2H, 2J = 10.3 Hz, CH2Ph), 4.88 (dd, 2H, 2J = 10.8 Hz, CH2Ph), 4.92 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 6.87–7.56 ppm (m, 30H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 25.6, 26.2, 29.3, 29.4, 29.8, 32.7, 62.4, 62.9, 69.7, 74.5, 74.8, 75.0, 75.9, 77.9, 82.5, 84.7, 86.3, 103.6, 126.9 (x 2), 127.6 (x 2), 127.7 (x 6), 127.9 (x 2), 128.0 (x 2), 128.1 (x 3), 128.2 (x 3), 128.3 (x 4), 128.8 (x 6), 137.8, 138.5, 138.6, 143.9 (x 3) ppm; HR-FAB MS [M+Na]+ calcd for C54H60O7Na 843.4237, found 843.4250.
8-[5-(1,2-Dithiolan-3-yl)pentanoyl]oxyoct-1-yl 2,3,4-tri-O-benzyl-β-D-glucopyranoside (2)
The title compound was obtained from 8b in 75% yield (2 steps) as a white foam as described in the synthesis of compound 1. Analytical data for 2: Rf = 0.50 (acetone/toluene, 1/9, v/v); 1H-NMR (300 MHz, CDCl3): δ, 1.31 (s, 8H, 4 × -CH2-), 1.42–1.51 (m, 2H, -CH2-), 1.53–1.79 (m, 8H, 4 × -CH2-), 1.83–1.92 (m, 2H, -CH2-), 2.31 (t, 2H, J = 7.4 Hz, -CH2COO-), 2.40–2.51 (m, 1H, -CH2-), 3.06–3.22 (m, 2H, -CH2-), 3.31–3.39 (m, 1H, H-5), 3.41 (dd, 1H, J2,3 = 8.8 Hz, H-2), 3.49–3.62 (m, 3H, H-4, OCH2a-, OH ), 3.67 (dd, 1H, J3,4 = 9.0 Hz, H-3), 3.70 (dd, 1H, J5,6a = 4.6 Hz, J6a,6b = 11.5 Hz, H-6a), 3.86 (dd, 1H, J5,6b = 2.5 Hz, H-6b), 3.92 (m, 1H, OCH2b-), 4.04 (t, 2H, J = 6.7 Hz, -CH2OCO-), 4.43 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.67 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.75 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.80 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 7.25–7.35 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 24.9, 26.0, 26.2, 28.8, 28.9, 29.3, 29.4, 29.9, 34.3, 34.7, 38.6, 40.4, 56.5, 62.2, 64.6, 70.5, 75.0, 75.1, 75.3, 75.9, 77.8, 82.5, 84.6, 103.8, 127.8, 127.9, 128.0 (x 3), 128.1 (x 2), 128.2 (x 3), 128.3, 128.5 (x 2), 128.7 (x 2), 138.1, 138.5, 138.7, 173.8 ppm; HR-FAB MS [M+Na]+ calcd for C43H58O8S2Na 789.3471, found 789.3463.
1,17-Bisbenzyloxy-9-oxaheptadecane (12)
NaH (60% dispersion in mineral oil, 0.203 g, 5.08 mmol) was added to a solution of 8-benzyloxyoctan-1-ol (10,28 1.0 g, 4.23 mmol) in THF (10 mL) and the resulting mixture was stirred under argon for 30 min at 60 °C. Then, a solution of 8-(benzyloxy)oct-1-yl tosylate (11,29 1.98 g, 5.08 mmol) in THF (20 mL) was added dropwise and the resulting solution was stirred for 24 h at rt. After that, MeOH (~30 mL) and CH2Cl2 (~40 mL) were added and the resulting mixture was washed with water (2 × 30 mL) and brine (20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexanes gradient elution) to afford the title compound (2.0 g, 87%) as a colorless oil. Analytical data for 12: Rf = 0.50 (ethyl acetate/hexane, 1/9, v/v); [α]D21 -1.9 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.31 (s, 16H, 8 × -CH2-), 1.50–1.63 (m, 8H, 4 × -CH2-), 3.37 (t, 4H, 2 × OCH2-), 3.45 (t, 4H, 2 × OCH2-), 4.49 (s, 4H, 2 × CH2Ph), 7.23–7.33 ppm (m, 10H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 26.3 (x 4), 29.6 (x 4), 29.9 (x 4), 70.6 (x 2), 71.1 (x 2), 73.0 (x 2), 127.6 (x 3), 127.7 (x 3), 128.5 (x 4), 138.8 (x 2) ppm; HR-FAB MS [M+Na]+ calcd for C30H46O3Na 477.3345, found 477.3337.
9-Oxaheptadecan-1,17-diol (13)
Pd/C (10%, 0.060 g) was added to a solution of 12 (0.600 g, 1.32 mmol) in MeOH (15 mL) and the resulting mixture was stirred under a pressure of hydrogen gas for 8 h at rt. The solid was filtered off through a pad of Celite and washed successively with MeOH (5 × 10 mL). The combined filtrate was concentrated in vacuo to afford the title compound (0.355 g, 98%) as a white amorphous solid. Analytical data for 13: Rf = 0.50 (ethyl acetate/hexane, 3/2, v/v); [α]D21 -2.3 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3+D2O): δ, 1.32 (s, 16H, 8 × -CH2-), 1.51–1.59 (m, 8H, 4 × -CH2-), 3.39 (t, 4H, J = 6.7 Hz, 2 × OCH2-), 3.62 (t, 4H, 2 × -CH2OH); 13C-NMR (75 MHz, CDCl3): δ, 25.6 (x 2), 26.0 (x 2), 29.3 (x 4), 29.6 (x 2), 32.6 (x 2), 62.8 (x 2), 70.8 (x 2) ppm; HR-FAB MS [M+H]+ calcd for C16H35O3 275.2586, found 275.2582.
17-Hydroxy-9-oxaheptadec-1-yl 2,3,4,6-tetra-O-benzoyl-β-D-glucopyranoside (15)
A mixture of glycosyl trichloroacetimidate 1430 (2.70 g, 3.65 mmol), alcohol 13 (1.00 g, 3.65 mmol), and freshly activated molecular sieves (3 Å, 3.0 g) in CH2Cl2 (60 mL) was stirred under argon for 1 h at rt. TMSOTf (0.20 mL, 1.10 mmol) was added and the resulting mixture was stirred for 1 h at rt. After that, the solid was filtered off and the residue was rinsed with CH2Cl2 (4 × 20 mL). The combined filtrate (~140 mL) was washed with 20% aq. NaHCO3 (80 mL), water (80 mL), and brine (30 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – toluene gradient elution) to afford the title compound (2.17 g, 70%) as a white foam. Analytical data for 15: Rf = 0.50 (ethyl acetate/toluene, 1/3, v/v); [α]D21 +4.7 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.05–1.24 (m, 8H, 4 × -CH2-), 1.31 (s, 10H, 5 × -CH2-), 1.41–1.57 (m, 6H, 3 × -CH2-), 3.30–3.41 (m, 4H, 2 × -CH2O), 3.53 (dt, 1H, J = 9.7, 6.1 Hz, OCH2a-), 3.62 (t, 2H, J = 6.6 Hz, -CH2OH), 3.91 (m, 1H, OCH2b-), 4.12–4.18 (m, 1H, H-5), 4.50 (dd, 1H, J5,6a = 5.2 Hz, J6a,6b = 12.1, H-6a), 4.63 (dd, 1H, J5,6b = 3.3 Hz, H-6b), 4.83 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.99 (s, 1H, OH), 5.53 (dd, 1H, J2,3 = 7.9 Hz, H-2), 5.67 (dd, 1H, J4,5 = 9.7 Hz, H-4), 5.91 (dd, 1H, J3,4 = 9.6 Hz, H-3), 7.27–8.03 ppm (m, 20H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 25.6, 25.7, 25.9, 26.0, 29.1, 29.2, 29.3 (x 2), 29.4, 29.6, 29.7, 32.7, 62.9, 63.2, 69.8, 70.3, 70.9, 71.8, 72.0, 72.9, 79.1, 101.2, 128.2, 128.3 (x 3), 128.5 (x 3), 128.7 (x 3), 128.9, 129.3 (x 2), 129.5, 129.7 (x 4), 129.8 (x 2), 133.0, 133.1, 133.2, 133.4, 165.0, 165.2, 165.8, 166.1 ppm; HR-FAB MS [M+Na]+ calcd for C50H60O12Na 875.3982, found 875.3999.
17-tert-Butyldiphenylsilyloxy-9-oxaheptadec-1-yl 2,3,4,6-tetra-O-benzoyl-β-D-glucopyranoside (16)
TBDPSCl (0.61 mL, 2.344 mmol) was added to a solution of compound 15 (1.00 g, 1.77 mmol) in pyridine (4.0 mL) and the resulting mixture was stirred under argon for 7 h at 50 °C. After that, the reaction was diluted with CH2Cl2 (~30 mL), and washed with water (30 mL) and brine (10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – hexanes gradient elution) to afford the title compound (1.16 g, 91%) as a white foam. Analytical data for 16: Rf = 0.50 (ethyl acetate/hexanes, 1/5, v/v); [α]D21 +2.8 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.03 (s, 9H, t-butyl), 1.07–1.20 (m, 6H, 3 × -CH2-), 1.23–1.37 (br. s, 10H, 5 × -CH2-), 1.41–1.60 (m, 8H, 4 × -CH2-), 3.32 (t, 2H, ½ OCH2-), 3.37 (m, 2H, OCH2-), 3.53 (m, 1H, ½ OCH2-), 3.65 (t, 2H, -CH2OSi), 3.92 (m, 1H, ½ OCH2-), 4.12–4.18 (m, 1H, H-5), 4.50 (dd, 1H, J5,6a = 5.2 Hz, J6a,6b = 12.1 Hz, H-6a), 4.64 (dd, 1H, J5,6b = 3.3 Hz, H-6b), 4.83 (d, 1H, J1,2 = 7.8 Hz, H-1), 5.53 (dd, 1H, J2,3 = 7.9 Hz, H-2), 5.68 (dd, 1H, J4,5 = 9.7 Hz, H-4), 5.91 (dd, 1H, J3,4 = 9.6 Hz, H-3), 7.25–8.03 ppm (m, 30H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 19.2, 25.7 (x 2), 26.0, 26.1, 26.8 (x 3), 29.1, 29.2, 29.3 (x 2), 29.4, 29.7 (x 2), 32.5, 63.2, 63.9, 69.8, 70.3, 70.9 (x 2), 71.8, 72.1, 72.9, 101.3, 127.5 (x 5), 128.2 (x 2), 128.3 (x 3), 128.4 (x 2), 128.8 (x 2), 129.3, 129.4 (x 2), 129.5, 129.7 (x 6), 129.8 (x 2), 133.1 (x 2), 133.2, 133.4, 134.1, 135.5 (x 5), 165.0, 165.2, 165.8, 166.1 ppm; HR-FAB MS [M+Na]+ calcd for C66H78O12SiNa 1113.5160, found 1113.5194.
17-tert-Butyldiphenylsilyloxy-9-oxaheptadec-1-yl 2,3,4-tri-O-benzyl-6-Otriphenylmethyl-β-D-glucopyranoside (17)
The title compound was obtained from 16 in 82% yield (3 steps) as a white foam as described in the synthesis of compound 7b. Analytical data for 17: Rf = 0.50 (ethyl acetate/hexane, 1/9, v/v); [α]D21 +1.6 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.04 (s, 9H, t-butyl), 1.22–1.40 (m, 16H, 8 × -CH2-), 1.50–1.58 (m, 6H, 3 × -CH2-), 1.68–1.78 (m, 2H, -CH2-), 3.23 (dd, 1H, J5,6a = 3.9 Hz, J6a,6b = 10.1 Hz, H-6a), 3.37 (t, 4H, 2 × OCH2-), 3.38–3.43 (m, 1H, H-5), 3.51–3.60 (m, 4H, H-2, 3, 6b, OCH2a-), 3.64 (t, 2H, -CH2OSi), 3.81 (dd, 1H, J4,5 = 9.1 Hz, H-4), 4.05 (m, 1H, OCH2b-), 4.44 (d, 1H, J1,2 = 7.2 Hz, H-1), 4.52 (dd, 2H, 2J = 10.3 Hz, CH2Ph), 4.84 (dd, 2H, 2J = 10.8 Hz, CH2Ph), 4.89 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 6.84–7.68 ppm (m, 40H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 19.2, 25.7 (x 2), 26.1, 26.2, 26.3, 26.8 (x 3), 29.3 (x 2), 29.4, 29.5, 29.7, 29.8, 32.5, 62.4, 63.9, 69.8, 70.9 (x 2), 74.5, 74.9, 75.0, 75.9, 77.9, 82.7, 84.7, 86.3, 103.6, 126.9 (x 3), 127.5 (x 5), 127.6 (x 2), 127.7 (x 5), 128.0 (x 3), 128.1 (x 3), 128.2 (x 3), 128.3 (x 3), 128.4 (x 3), 128.8 (x 4), 129.4 (x 3), 134.1 (x 2), 135.5 (x 3), 137.8, 138.6 (x 2), 143.9 (x 3) ppm; HR-FAB MS [M+Na]+ calcd for C78H94O8SiNa 1209.6616, found 1209.6655.
17-Hydroxy-9-oxaheptadec-1-yl 2,3,4-tri-O-benzyl-6-O-triphenylmethyl-β-D-glucopyranoside (18)
The title compound was obtained from 17 in 95% yield as a white foam as described in the synthesis of compound 8b. Analytical data for 18: Rf = 0.50 (ethyl acetate/hexane, 1/3, v/v); [α]D21 +2.4 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.31 (s, 16H, 8 × -CH2-), 1.42–1.58 (m, 6H, 3 × -CH2-), 1.63–1.84 (m, 3H, -CH2-, OH), 3.23 (dd, 1H, J5,6a = 3.9 Hz, J6a,6b = 10.1 Hz, H-6a), 3.37 (t, 4H, J = 6.5 Hz, 2 × OCH2-), 3.41–3.47 (m, 1H, H-5), 3.52–3.65 (m, 6H, H-2, 3, 6b, OCH2a-, -CH2OH), 3.81 (dd, 1H, J4,5 = 9.0 Hz, H-4), 4.06 (dt, 1H, J = 9.4, 6.4 Hz, OCH2b-), 4.45 (d, 1H, J1,2 = 7.2 Hz, H-1), 4.53 (dd, 2H, 2J = 10.3 Hz, CH2Ph), 4.84 (dd, 2H, 2J = 10.8 Hz, CH2Ph), 4.90 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 6.84–7.50 ppm (m, 30H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 25.7, 26.2 (x 2), 26.4, 29.4, 29.5 (x 3), 29.8 (x 2), 29.9, 32.8, 62.5, 63.0 (x 2), 69.9, 70.9, 74.6, 74.9, 75.1, 76.0, 78.0, 82.6, 84.8, 86.4, 103.7, 127.0 (x 2), 127.7 (x 2), 127.8 (x 5), 127.9 (x 2), 128.1 (x 2), 128.2 (x 5), 128.3 (x 3), 128.4 (x 2), 128.5 (x 2), 128.9 (x 5), 137.9, 138.7 (x 2), 144.0 (x 3) ppm; HR-FAB MS [M+Na]+ calcd for C62H76O8Na 971.5438, found 971.5444.
17-[5-(1,2-Dithiolan-3-yl)pentanoyl]oxy-9-oxaheptadec-1-yl 2,3,4-tri-O-benzyl-β-Dglucopyranoside (3)
The title compound was obtained from 18 in 88% yield (2 steps) as a white foam as described in the synthesis of compound 1. Analytical data for 3: Rf = 0.50 (acetone/toluene, 1/9, v/v); 1H-NMR (300 MHz, CDCl3): δ, 1.25 (s, 16H, 8 × -CH2-), 1.35–1.68 (s, 14H, 7 × -CH2-), 1.77–1.88 (m, 2H, -CH2-), 2.23 (t, 2H, -CH2COO-), 2.33–2.43 (m, 1H, -CH2-), 2.98–3.14 (m, 2H, -CH2-), 3.26–3.37 (m, 6H, H-2, 5, 2 × OCH2-), 3.42–3.47 (m, 2H, OCH2a-, OH), 3.48 (dd, 1H, J4,5 = 9.3 Hz, H-4), 3.59 (dd, 1H, J3,4 = 9.0 Hz, H-3), 3.66 (dd, 1H, J5,6a = 4.3 Hz, J6a,6b = 8.9, H-6a), 3.77–3.88 (m, 2H, H-6b, OCH2b-), 3.98 (t, 2H, -CH2OCO-), 4.35 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.67 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.75 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.79 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 7.18–7.27 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 24.6, 25.8, 26.0, 26.1 (x 2), 28.6, 28.7, 29.2, 29.3 (x 2), 29.4 (x 2), 29.7 (x 2), 34.1, 34.5, 38.4, 40.2, 56.3, 62.0, 64.4, 70.3, 70.9 (x 2), 74.8, 74.9, 75.0, 75.6, 77.6, 82.3, 84.4, 103.7, 127.6 (x 2), 127.8 (x 2), 127.9, 128.0 (x 3), 128.3 (x 5), 128.4 (x 2), 137.9, 138.4, 138.5, 173.5 ppm; HR-FAB MS [M+Na]+ calcd for C51H74O9S2Na 917.4672, found 917.4674.
General procedure for the preparation of NPG chips
The NPG plates were prepared by dealloying cut pieces (typical dimension 8 mm × 8 mm × 0.25 mm) of 10 carat yellow gold sheet (Hoover and Strong, Richmond, VA) in concentrated nitric acid for 72 h followed by successive rinsing with water and methanol and drying in vacuo.31 The pieces were then broken using the end of a spatula into 4 roughly equal fragments (ca. 4 mm × 4 mm × 0.25 mm) that could fit into the HPLC column.
General procedure for the formation of the mixed SAM
The freshly prepared nanoporous gold chips were allowed to stand in a solution of glycosyl acceptor (0.030 g, 0.0422 mmol) and methyl lipoic ester (0.014 g, 0.0636 mmol) in CH2Cl2 (10 mL) under argon for 10 h at rt. After that, NPG chips were carefully removed, washed with CH2Cl2 (4 × 5 mL), and dried in vacuo for 5 h. The loaded chips were then transferred into the Omnifit column that was then connected with HPLC system to be used in subsequent glycosidations.
General procedure for the HPLC-assisted glycosylation of 1a, 2a and 3a with glycosyl donor 14: synthesis of disaccharides 20–22
A solution of glycosyl donor 14 (Pump A, 78 mM soln, 0.175 g in 3.0 mL CH2Cl2), was passed through the column containing NPG-bound glycosyl acceptor 1a, or 2a or 3a and 3A molecular sieves in beads form) at the flow rate of 1.0 mL/min for 30 min with column output connects back to the same chamber A (i.e., recirculation mode). Then, after 30 min. TMSOTf (50 μL) was injected into the chamber A and continued circulation for 2 h at room temperature. Then the flow was switched to Pump B containing CH2Cl2 for 10 min (flow rate: 2 mL/min).
General procedure cleavage of sugar from NPG chip
A solution of NaOCH3 in CH3OH (0.1 M, 2.0 mL) was added to a reaction vessel containing NPG chips with disaccharides on it. The vessel was kept in a shaker for overnight, then the reaction mixture was neutralized with Dowex (H+) resin, filtered, and concentrated in vacuo to afford the corresponding deprotected disaccharide. The crude product was then acetylated as follows. To a stirred solution of crude disaccharide (~0.005 g, 0.0073 mmol) in pyridine (1 mL) Ac2O (0.5 mL) was added dropwise in the presence of catalytic DMAP. The reaction mixture was stirred under argon for 8 h at room temperature. The reaction mixture was quenched with CH3OH (1 mL) and the resulting mixture was concentrated under reduced pressure. The residue was diluted with CH2Cl2 (20 mL), and washed with 1N HCl (2 × 10 mL), water (20 mL), sat. aq. NaHCO3 (20 mL), and water (20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo to afford corresponding disaccharides 20–22 (60–90%, TLC estimation).
4-Acetoxybut-1-yl O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-β-D-glucopyranoside (20)
The title compound was synthesized from glycosyl acceptor 1 and glycosyl donor 14 in 60% yield (TLC estimation). Analytical data for 20: Rf = 0.40 (acetone/toluene, 1/9, v/v); [α]D21 -5.4 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.70–1.79 (m, 4H, 2 × -CH2-), 1.99, 2.00, 2.03, 2.04, 2.05 (5 s, 15H, 5 × -COCH3), 3.35–3.49 (m, 3H, H-2, 3, 5), 3.50–3.59 (m, 1H, OCH2a-), 3.61–3.69 (m, 3H, H-4, 5′, 6a), 3.99 (m, 1H, OCH2b-), 4.05–4.15 (m, 4H, H-6b, 6′a, -CH2OAc), 4.24 (dd, 1H, J5′,6′b = 4.6 Hz, J6′a,6′b = 12.3 Hz, H-6′b), 4.36 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.62 (d, 1H, J1′,2′ = 7.9 Hz, H-1′), 4.68 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.82 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.88 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.99 (dd, 1H, J2′,3′ = 9.2 Hz, H-2′), 5.08 (dd, 1H, J4′,5′ = 9.5 Hz, H-4′), 5.16 (dd, 1H, J3′,4′ = 9.3 Hz, H-3′), 7.23–7.33 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 20.6 (x 2), 20.7 (x 2), 20.9, 25.4, 26.2, 61.9, 64.1, 68.3, 68.4, 69.3, 71.2, 71.8, 72.9, 74.8, 74.9, 75.7, 77.2, 77.9, 82.2, 84.5, 100.8, 103.4, 127.7 (x 2), 127.9 (x 3), 128.0 (x 4), 128.4 (x 5), 128.5, 137.8, 138.3, 138.4, 169.0, 169.4, 170.3, 170.7, 171.1 ppm; HR-FAB MS [M+Na]+ calcd for C47H58O17Na 917.3572, found 917.3601.
8-Acetoxyoct-1-yl O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-β-D-glucopyranoside (21)
The title compound was synthesized from glycosyl donor 14 and glycosyl acceptor 2 in 75% yield (TLC estimation). Analytical data for 21: Rf = 0.50 (acetone/toluene, 1/4, v/v); [α]D21 -6.2 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.21–1.37 (m, 8H, 4 × -CH2-), 1.48–1.64 (m, 4H, 2 × -CH2-), 1.92, 1.92, 1.95 1.96, 1.98 (5 s, 15H, 5 × -COCH3), 3.26–3.43 (m, 3H, H-2, 3, 5), 3.44 (m, 1H, OCH2a-), 3.53–3.61 (m, 3H, H 4, 5′, 6a), 3.88 (m, 1H, OCH2b-), 3.96 (t, 2H, J = 6.7 Hz, -CH2OAc), 3.97–4.01 (m, 1H, H-6′a), 4.05 (dd, 1H, J5,6b = 2.3 Hz, J6a,6b = 12.5, H-6b), 4.17 (dd, 1H, J5′,6′b = 4.6 Hz, J6′a,6′b = 12.3 Hz, H-6′b), 4.29 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.55 (d, 1H, J1′,2′ = 7.9 Hz, H-1′), 4.61 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.75 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.77 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.95 (dd, 1H, J2′,3′ = 9.3 Hz, H-2′), 5.00 (dd, 1H, J4′,5′ = 8.1 Hz, H-4′), 5.10 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 7.15–7.26 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 20.6 (x 2), 20.7 (x 2), 20.9, 25.8, 26.1, 28.5, 29.2, 29.3, 29.7, 61.9, 64.5, 68.3, 68.4, 70.1, 71.2, 71.8, 72.9, 74.8, 74.9, 75.7, 77.2, 77.9, 82.1, 84.5, 100.7, 103.5, 127.6, 127.7, 127.8 (x 2), 127.9 (x 3), 128.0 (x 3), 128.3 (x 3), 128.5 (x 2), 137.8, 138.3, 138.4, 169.0, 169.3, 170.2, 170.6, 171.2 ppm; HR-FAB MS [M+Na]+ calcd for C51H66O17Na 973.4198, found 973.4187.
17-Acetoxy-9-oxaheptadec-1-yl O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-β-D-glucopyranoside (22)
The title compound was synthesized from glycosyl donor 14 and glycosyl acceptor 3 in 90% yield (TLC estimation). Analytical data for 22: Rf = 0.50 (acetone/toluene, 1/9, v/v); [α]D22 -2.6 (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3): δ, 1.24 (s, 16H, 8 × -CH2-), 1.42–1.58 (m, 8H, 4 × -CH2-), 1.92, 1.92, 1.95 1.97, 1.98 (5 s, 15H, 5 × -COCH3), 3.26–3.37 (m, 6H, H-2, 5, 2 × OCH2-), 3.38–3.49 (m, 2H, H-3, OCH2a-), 3.53–3.62 (m, 3H, H-4, 5′, 6a), 3.88 (m, 1H, OCH2b-), 3.94–3.96 (m, 1H, H-6b), 3.97 (t, 2H, J = 6.7 Hz, -CH2OCO-), 4.05 (dd, 1H, J5′,6′a = 2.3 Hz, J6′a,6′b = 12.2 Hz, H-6′a), 4.17 (dd, 1H, J5′,6′b = 4.6 Hz, H-6′b), 4.29 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.55 (d, 1H, J1,2 = 7.9 Hz, H-1′), 4.61 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.75 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 4.77 (dd, 2H, 2J = 11.0 Hz, CH2Ph), 4.95 (dd, 1H, J2′,3′ = 9.3 Hz, H-2′), 5.00 (dd, 1H, J4′,5′ = 9.6 Hz, H-4′), 5.06 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 7.16–7.26 ppm (m, 15H, aromatic); 13C-NMR (75 MHz, CDCl3): δ, 20.6 (x 2), 20.7 (x 2), 20.9, 25.8, 26.1 (x 2), 26.2, 28.5, 29.2, 29.3, 29.4 (x 2), 29.7 (x 3), 61.9, 64.6, 68.3, 68.4, 70.1, 70.9 (x 2), 71.2, 71.7, 72.9, 74.8, 74.9, 75.7, 77.2, 77.9, 82.1, 84.5, 100.7, 103.5, 127.6, 127.7, 127.8, 127.9 (x 4), 128.0, 128.1 (x 2), 128.3 (x 3), 128.5 (x 2), 137.8, 138.3, 138.4, 169.0, 169.4, 170.2, 170.7, 171.2 ppm; HR-FAB MS [M+Na]+ calcd for C59H82O18 Na 1101.5399, found 1101.5404.
Supplementary Material
Scheme 6.

Comparative glycosylations of immobilized glycosyl acceptors 1a–3a with donor 14 using HPLC-assisted experimental set-up
Acknowledgments
This work was supported by an award from the NIGMS (GM090254).
Supporting Information: general experimental and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org/.
Contributor Information
Keith J. Stine, Email: kstine@umsl.edu.
Alexei V. Demchenko, Email: demchenkoa@umsl.edu.
References
- 1.Toy PH, Lam Y, editors. Solid-Phase Organic Synthesis. John Wiley & Sons, Inc; Hoboken: 2012. [Google Scholar]
- 2.Babaev EV, Ermolat’ev DS. Russ J Gen Chem. 2010;80:2572–2589. [Google Scholar]
- 3.Osborn HMI, Khan TH. Tetrahedron. 1999;55:1807–1850. [Google Scholar]
- 4.Krepinsky JJ, Douglas SP. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, New York: 2000. pp. 239–265. [Google Scholar]
- 5.Seeberger PH, Haase WC. Chem Rev. 2000;100:4349–4393. doi: 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]
- 6.Kunz H, Schultz M. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, New York: 2000. pp. 267–304. [Google Scholar]
- 7.Kochetkov NK. Russ Chem Rev. 2000;69:795–820. [Google Scholar]
- 8.Walvoort MTC, van den Elst H, Plante OJ, Krock L, Seeberger PH, Overkleeft HS, van der Marel GA, Codee JDC. Angew Chem Int Ed. 2012;51:4393–4396. doi: 10.1002/anie.201108744. [DOI] [PubMed] [Google Scholar]
- 9.Krock L, Esposito D, Castagner B, Wang CC, Bindschadler P, Seeberger PH. Chem Sci. 2012;3:1617–1622. [Google Scholar]
- 10.Seeberger PH. Chem Soc Rev. 2008;37:19–28. doi: 10.1039/b511197h. [DOI] [PubMed] [Google Scholar]
- 11.Werz DB, Castagner B, Seeberger PH. J Am Chem Soc. 2007;129:2770–2771. doi: 10.1021/ja069218x. [DOI] [PubMed] [Google Scholar]
- 12.Seeberger PH, Werz DB. Nature Rev. 2005;4:751–763. doi: 10.1038/nrd1823. [DOI] [PubMed] [Google Scholar]
- 13.Seeberger PH. Chem Commun. 2003:115–1121. doi: 10.1039/b210230g. [DOI] [PubMed] [Google Scholar]
- 14.Plante OJ, Palmacci ER, Seeberger PH. Adv Carbohydr Chem Biochem. 2003;58:35–54. doi: 10.1016/s0065-2318(03)58002-7. [DOI] [PubMed] [Google Scholar]
- 15.Hewitt MC, Snyder DA, Seeberger PH. J Am Chem Soc. 2002;124:13434–13436. doi: 10.1021/ja027538k. [DOI] [PubMed] [Google Scholar]
- 16.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt RR, Jonke S, Liu K. In: ACS Symp Ser (Frontiers in Modern Carbohydrate Chemistry) Demchenko AV, editor. Vol. 960. Oxford Univ. Press; 2007. pp. 209–237. [Google Scholar]
- 18.Tanaka K, Fukase K. In: Solid-Phase Organic Synthesis. Toy PH, Lam Y, editors. John Wiley & Sons, Inc; Hoboken: 2012. pp. 489–530. [Google Scholar]
- 19.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem Commun. 2009:1834–1836. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 20.Scott PJH. In: Solid-Phase Organic Synthesis. Toy PH, Lam Y, editors. John Wiley & Sons, Inc; Hoboken: 2012. pp. 3–82. [Google Scholar]
- 21.Tan YH, Davis JA, Fujikawa K, Ganesh NV, Demchenko AV, Stine KJ. J Mater Chem. 2012;22:6733–6745. doi: 10.1039/C2JM16633J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong Y, Abaci S, Shannon C, Bozack MJ. Langmuir. 2003;19:8922–8926. [Google Scholar]
- 23.Lemieux RU. In: Methods in Carbohydrate Chemistry. Whistler RL, Wolform ML, editors. Vol. 2. Academic Press Inc; New York and London: 1963. pp. 226–228. [Google Scholar]
- 24.Kobayashi J, Hatakeyama A, Tsuda M. Tetrahedron. 1998;54:697–704. [Google Scholar]
- 25.Clausen MH, Madsen R. Carbohydr Res. 2004;339:2159–2169. doi: 10.1016/j.carres.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Khan AT, Ghosh S, Choudhury LH. Eur J Org Chem. 2004:2198–2204. [Google Scholar]
- 27.Vijaya Ganesh N, Fujikawa K, Tan YH, Stine KJ, Demchenko AV. Org Lett. 2012;14:3036–3039. doi: 10.1021/ol301105y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao X, Hall DG. J Am Chem Soc. 2005;127:1628–1629. doi: 10.1021/ja042827p. [DOI] [PubMed] [Google Scholar]
- 29.Czech BP, Bartach RA. J Org Chem. 1984;49:4076–4078. [Google Scholar]
- 30.Colonna B, Harding VD, Nepogodiev SA, Raymo FM, Spencer N, Stoddart JF. Chem Eur J. 1998;4:1244–1254. [Google Scholar]
- 31.Shulga OV, Jefferson K, Khan AR, D’Souza VT, Liu J, Demchenko AV, Stine KJ. Chem Mater. 2007;19:3902–3911. doi: 10.1021/cm070238n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huwe CM, Küzer H. Tetrahedron Lett. 1999;40:683–686. [Google Scholar]
- 33.Hassan HM, Maltman BA. Bioorg Chem. 2012;40:6–9. doi: 10.1016/j.bioorg.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Dhayal M, Ratner DM. Langmuir. 2009;25:2181–2187. doi: 10.1021/la8031122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.