

Abstract
In follicular lymphoma, somatic hypermutation of the immunoglobulin heavy chain genes facilitates the identification of different lymphoma cell clones, and the construction of genealogical trees. To investigate the dissemination of lymphoma cells, and the role of bone marrow in disease progression, we simultaneously analyzed the somatic hypermutation patterns of lymph node and bone marrow specimens taken from three patients at onset and relapse of their disease. Immunoglobulin heavy chain genes were amplified by polymerase chain reaction, cloned and sequenced. Mutational pedigrees were constructed in a hierarchical order. When direct transition of one mutation pattern into that of a successor clones was not feasible, hypothetical predecessor clones were created, and a probability measurement calculation was introduced. Eighty-five sequenced clones were generated. The average mutation rates were 13.45% for the lymph node specimens, and 9.78% for the bone marrow ones. Forty-two hypothetical predecessor clones were introduced into inter-compartment pedigrees. The genealogical trees showed that early lymphoma clones with a low mutational load quickly migrate from lymph nodes into the bone marrow. Bi-directional lymphoma cell migration was detectable between the two compartments. In one case of follicular lymphoma, a clone identical to the initial lymph node clone was detected 2 years later in the bone marrow. The newly introduced algorithm allows the evaluation of both time and direction of follicular lymphoma cell migration. We found evidence that follicular lymphoma originates in the lymph node, and infiltrates the bone marrow early in the course of the disease. Moreover, inter-compartment migration between lymph nodes and bone marrow occurs in both directions.
Introduction
Follicular lymphoma (FL) accounts for 25%–40% of all B-cell non-Hodgkin’s lymphomas in Europe and the United States.1 Although often initially responsive to chemotherapy or radiotherapy, FL is characterized by relapses and progression to treatment-resistant disease or transformation to high-grade lymphoma.2 Almost all cases of FL harbor the t(14;18)(q32;q21) chromosomal translocation, resulting in overexpression of the oncogene BCL2. The frequent occurrence of the t(14;18)(q32;q21) translocation in B cells in the peripheral blood of healthy individuals3 as well as the occurrence of “FL in situ” with a low risk of progression demonstrate that the translocation alone is insufficient for a malignant phenotype.4,5 Specific attention has been focused on the microenvironment, especially of germinal centers of lymph nodes (LN).6–8 As the putative malignant counterpart of germinal center B cells, the (pre)-malignant FL cells are believed to overcome selective control mechanisms of the germinal center microenvironment by constitutive expression of the anti-apoptotic protein Bcl-2, allowing for secondary genetic aberrations leading to a fully malignant phenotype and progression of FL.9–12
The germinal center B-cell origin of FL is supported by ongoing somatic hypermutation of immunoglobulin heavy chain variable region (IgVH)-genes of t(14;18)-positive FL cells.13 The mutation patterns in the IgVH genes of FL have been found to be very similar to those in normal antigen-selected B cells.14 The active hypermutation machinery of FL cells results in an intraclonal sequence heterogeneity of neoplastic clones.15 During the course of the disease, the tumor cells disseminate to lymphatic organs, including the bone marrow (BM).16–18 It is now accepted, that somatic hypermutation of the original neoplastic clones is retained during the expansion and dissemination to adjacent germinal centers and distant LN.19,20 However, the molecular details of tumor cell dissemination into the BM are largely unknown.21,22
We conducted a simultaneous mutational analysis of the IgVH genes of LN and corresponding BM specimens from three patients with FL, to delineate the migration of FL cells between these two compartments on the basis of reconstructed temporal sequences of FL cell clones. We used a newly developed algorithm to describe clonal hierarchy and migration patterns more thoroughly.
Methods
Patients, histology, and immunohistochemistry
This study comprised three patients with synchronous LN and BM infiltration by FL at presentation. Biopsies were performed during the diagnostic and staging procedures. The selection criteria were the diagnosis of FL according to the fourth edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.1 Clinical information was obtained from the patients’ medical records. Material was collected from patients after their informed consent in accordance with the Declaration of Helsinki. The study was approved by the responsible institutional ethic boards. Further details are provided in the Online Supplementary Methods.
Sequence analysis of IgVH genes and definition of mutational patterns
DNA from the IgVH gene segments was extracted and amplified as described elsewhere.23,24 Cloning, sequencing, and the mutational analysis of the obtained segments are described in detail in the Online Supplementary Methods.
Delineation of tumor cell evolution by construction of pedigrees
For each patient and each compartment (LN and BM separately), the mutational patterns of IgVH gene segments were arranged in an ascending order of mutations to illustrate the mutational hierarchy of intraclonal sequence heterogeneity. Consequently, mutational patterns of early clones with few mutations had to be included in successor clones. When direct transition of one mutation pattern into that of successor clones with higher mutation loads was not observable, hypothetical predecessor clones (HPC) were introduced to retrace the evolution of sequenced clones back to the determined initial VHDJH gene rearrangement (wild-type sequence). Accordingly, compartment-specific pedigrees were constructed. Thereafter, a third “summary-pedigree” comprising all sequenced clones was constructed, to evaluate the possibility of inter-compartmental exchange between LN and BM.
Generation of hypothetical predecessor clones and delineation of migration probability
For each sequenced FL population (i.e. LN, BM, and LN and BM together) the pool of possible HPC was derived from mutations shared by at least two sequenced clones. To select the most appropriate predecessor clones from the abundance of generated HPC, the probability measurement was introduced (Figure 1). Only HPC with the highest probability measurement values were introduced until the evolution of sequenced clones could be retraced to the “wild-type sequence”. Already established clone groups could not be disrupted by HPC with lower probability measurement values. These calculations resulted in a LN, a BM and an inter-compartment pedigree. If HPC of the inter-compartment pedigree displayed a higher probability measurement value than the corresponding LN or BM counterparts, inter-compartment migration was considered. The LN or BM allocation of these inter-compartment HPC was directed by the LN or BM affiliation of the majority of evolving clones (see Online Supplementary Methods).
Figure 1.
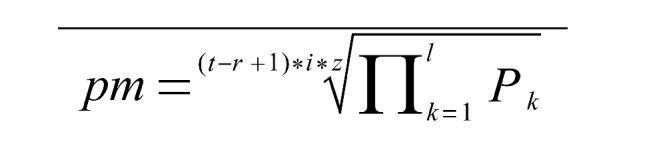
Formula for calculating the probability measure (pm). The pm-formula poses the pivotal part of hypothetical predecessor clone (HPC) stratification in the non-multiple sequence alignment-based algorithm “mutation evolution estimator” / “follicular lymphoma evolution reconstructor” created for this study to retrace follicular lymphoma evolution/dissemination: pm: probability measure of the considered HPC; t: total number of sequenced clones contained in the considered population [i.e. lymph node (LN)-, bone marrow (BM)- or LN+BM(inter-tissue)-population]; r: group strength; number of sequenced clones contained in the HPC-defining group of sequenced clones; i: number of mutations of the considered HPC; z: number of repeats of a HPC-sequence among the considered population (i.e. LN-, BM- or inter-tissue-population); l: number of nucleotide residues of the considered sequence; k: counter; starting at the first locus, being consecutively heightened by 1 and ending at the last locus of the considered sequence; P: relative frequency of a mutation at residue k among the considered population; nucleotide residues without mutations were given the value “1“.
Analysis of somatic hypermutation patterns
To evaluate the expression of functional B-cell receptors (BCR), sequences were evaluated for in-frame stop codons. Because preserved expression of complete BCR is considered mandatory for the survival of B cells, no further analysis was applied if an in-frame stop codon was detected.25 In cases without in-frame stop codons, the multinominally modified Chang and Casali formula for assessment of ongoing mutations was applied, using the www.stat.stanford.edu/immunoglobindatabase/software.26,27
Results
Clinical data
The first patient, a 35-year-old female, showed FL involvement of LN and BM. A regimen of conventional chemotherapy with rituximab was applied leading to clinical remission.
The second patient was a 44-year-old female with simultaneous FL involvement of LN and BM. Biopsies of each compartment were obtained in 2002 before therapy. The initial BM specimen from 2002 was not available, because of loss of material by sectioning. The patient received combined radio-chemotherapy and reached clinical remission. In 2005 another BM biopsy was taken for re-staging and showed infiltration by FL.
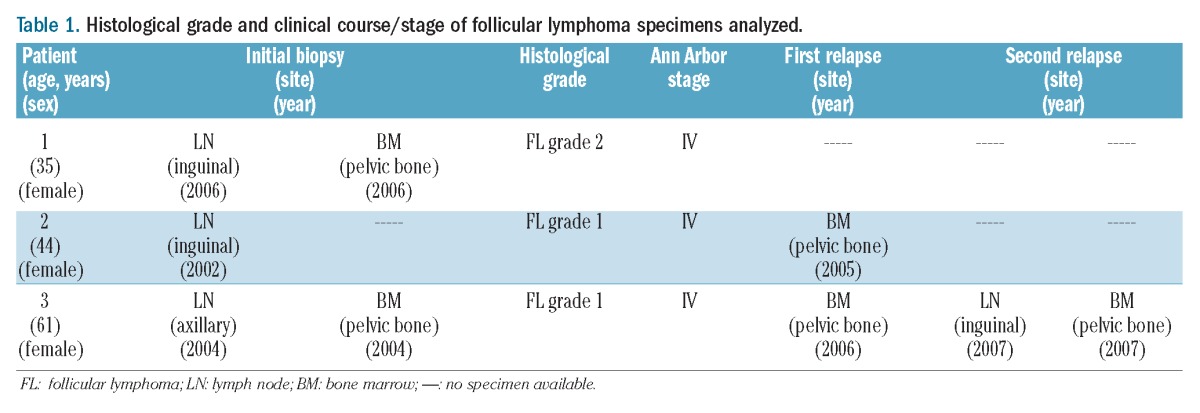
The third patient was a 61-year-old female with simultaneous FL involvement of the LN and BM. Pre-treatment biopsies of both tissues were taken in 2004. Chemotherapy was administered leading to a clinical remission. A BM specimen obtained in 2006 showed infiltration by FL. In 2007, a relapse prompted another pair of LN and BM biopsies showing infiltration of both compartments by FL. The patient received several regimens of chemotherapy with and without rituximab, and autologous stem cell transplantation in 2006 (Table 1).
Table 1.
Histological grade and clinical course/stage of follicular lymphoma specimens analyzed.
Histological analyses
The LN and BM tissues of the three patients showed FL involvement. All specimens from the three patients showed the typical immunophenotype of FL, with expression of CD20, CD10, Bcl6 and Bcl2 in LN and BM. Fluorescence in situ hybridization analysis revealed a break at the BCL2 locus at chromosome 18q21 in all three patients.
Analysis of IgVH-gene segments
Corresponding to the NCBI IgBLAST germline configuration with the most homologous sequence, one unmutated wild-type VHDJH-gene rearrangement could be determined for the sequenced clones of each patient. All sequences shared a variable number of single nucleotide mutations. No deletions, duplications or inversions were found. Nucleotide insertions were detected at the VH-D and the D-JH junctions.
Delineation of follicular lymphoma cell dissemination by pedigrees
A synopsis of the generation of FL pedigrees of LN and BM involvement is described for each patient, whereas a detailed description of the process is given in the Online Supplementary Results.
Patient 1
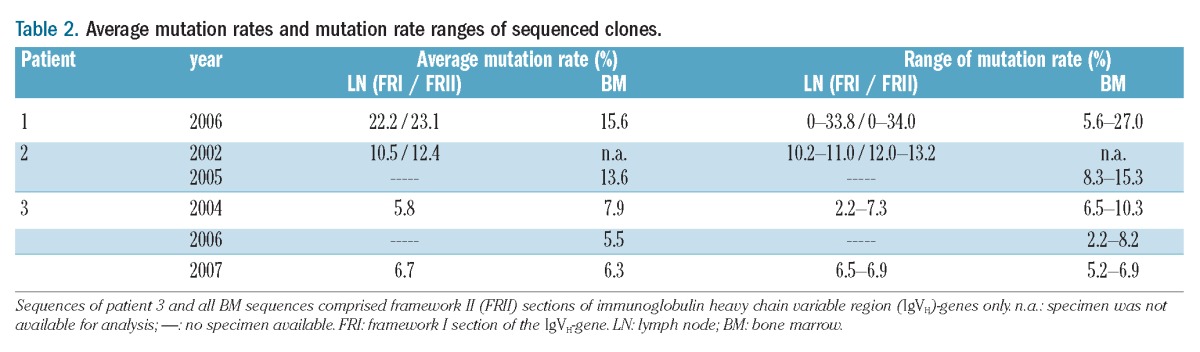
The IgVH germline gene with the highest homology was IGHV3-30*04. The VHDJH gene amplicons contained 349 basepairs (bp) in FR1 and 244 bp in FR2. The average mutation rate of the VHDJH gene sequences of the ten LN clones was 22.2% in FR1 and 23.1% in FR2, while that for the eight BM sequences was 15.6% (Table 2).
Table 2.
Average mutation rates and mutation rate ranges of sequenced clones.
Synopsis of the clonal evolution of the follicular lymphoma (Figure 2)
Figure 2.

Pedigree of the final FL cell evolution of patient 1 – synopsis of compartment-specific and inter-compartment pedigrees. The immediate hypothetical predecessor clones (HPC) of each sequenced clone (i.e. the compartment-specific HPC and the inter-compartment HPC) were compared to each other. Some shared an identical sequence, both in the compartment-specific and the inter-compartment pedigree (for example LN7 and LN+BM12 for a2, a7 and b1), some were different [for example LN9 (LN-specific pedigree) and LN+BM10 (inter-tissue pedigree) for clone b6]. In case of two different HPC, the respective probability measures (pm) were compared to decide whether evolution according to the compartment-specific or the inter-compartment FL pedigree was more likely. Consequently, sequenced BM clone k4 was allocated to compartment-specific evolution from the HPC BM7 with a higher pm value and this was, therefore, considered more likely than evolution from the inter-compartment HPC (LN+BM14). For sequenced clones b6, a8 and k5, inter-compartment evolution was considered more likely than compartment-specific evolution, according to pm-stratification. Blue lines represent putative clonal migratory events from the LN to the BM compartment, red lines represent migration in the opposite direction (i.e. putative clonal migratory events from the BM to the LN compartment).
The reader is encouraged to consult the figures while reading the descriptions of the pedigrees. One LN sequence (clone b4) was identical to the unmutated wild-type/germline sequence. Two LN sequences (clones a7 and b1) shared identical mutation patterns. Among the LN population, no direct transitions of one mutation pattern into another were observed. Consequently, nine HPC were introduced to reconstitute the most likely tumor cell evolution, according to the “probability measurement”-guided hierarchy. Since clone b4 represented the wild-type one, all other LN sequences and HPC were considered to be derived from clone b4. For the eight obtained BM sequences, no direct transitions of one sequence into another were observed. Subsequently, nine HPC were introduced to reconstitute the most likely tumor cell evolution in the BM.
For sequenced clones b6, a8, k4, and k5, a synthesis of the compartment-specific and the inter-compartment evolution served as the basis for deciding whether compartment-resident evolution or evolution by inter-compartment migration was more likely. Comparing the probability measurement values of the relevant immediate compartment-specific HPC with the corresponding inter-compartment HPC, BM-resident evolution was more likely for clone k4, whereas inter-compartment migration was obvious for LN-clones b6 and a8 and BM-clone k5.
Hence, all sequenced and hypothetical BM-clones evolved from LN-clones that migrated to the BM. Nodal clones b6 and a8 evolved from BM-derived clones that had previously evolved from the LN population. This represented multidirectional migration in the FL population of patient 1 emanating from the LN (see also Online Supplementary Results).
Patient 2
The common IgVH germline gene was IGHV3-23*01. The VHDJH gene amplicons contained 362 bp in FR1 and 244 bp in FR2. The average mutation rate of VHDJH gene sequences of the six LN clones (a2, a2, a3, a4, a5, a6) was 10.5% in FR1, 12.4% in FR2 and 13.6% for BM-derived clones (Table 2).
No LN or BM sequence was identical to the unmutated wild-type germline sequence. Within the 14 sequenced BM clones, the mutation pattern of clone k2 could be directly transferred into clone k6. Within the LN population, the HPC with the highest probability measurement value (HPC-LN1) included all following clones. The mutations of a1 and a6 were included in all successor clones. The HPC (HPC-LN3) was the predecessor of clones a3 and a5. Clone a2 evolved independently of HPC-LN3. A set of 18 BM-HPC was calculated. Five BM-HPC (HPC-BM1 –HPC-BM5) were introduced to reconstruct the probability measurement-guided clonal evolution of the BM. All BM clones shared 22 common mutations illustrated by HPC-BM1 (see also Online Supplementary Results).
Synopsis of the clonal evolution of the follicular lymphoma (Figure 3)
Figure 3.

Pedigree of the final FL cell evolution of patient 2 – synopsis of compartment-specific and inter-compartment pedigrees. Comparing the probability measure (pm) of the compartment-specific HPC to the inter-compartment HPC, a common HPC for all sequenced clones was determined (LN + BM1). Due to the time order of the obtained LN and BM specimens, this HPC was allocated to the LN compartment. By introduction of HPC LN + BM2 and LN + BM3, which constituted the following HPC among the clonal hierarchy of inter-comparment HPC, bidirectional clonal migration became evident. Thus, the LN population of year 2002 was composed of sequenced clones (a1, a6 and a4) that putatively had originally been derived from LN predecessors, and sequenced clones (a2, a5 and a3) that had putatively derived from BM resident HPC (see Online Supplementary Data). Thus contrary to MSA-based algorithms, the non-MSA-based algorithm “mutation evolution estimator” (MEE) / “follicular lymphoma evolution reconstructor” (FLER) renders a means of hypothetical backdating of clonal relations on the basis of mutational pattern-derived HPC creation (signified by the annotation ”postulated LN, year 2002”). Blue lines represents putative clonal migratory events from the LN to the BM compartment, red lines represent migration in the opposite direction (i.e. putative clonal migratory events from the BM to the LN compartment).
For the final pedigree, four HPC indicate two migration events between the LN and BM. Considering the synchronous involvement of LN and BM tissue at diagnosis, and the temporal discrepancy between the studied LN and BM specimens, the common HPC of all clones (HPC-LN+BM1) could not be allocated to a specific compartment. Whether an initial migration event was directed from the LN to the BM or in the opposite direction could not be determined. The hypothetical clones (LN+BM2), and (LN+BM3) were allocated to the BM compartment and proposed as clones that would have already existed in the BM at diagnosis in 2002, since all the sequenced BM clones could be derived from these two HPC.
In the synopsis of the compartment-specific and the inter-compartment pedigrees, LN clones a1, a6 and a4 evolved from precursor HPC-LN+BM1, whereas a2, a3 and a5 represented successor clones of BM-allocated HPC-LN+BM3, indicating a migration event from the BM to the LN. Whether BM clones of 2002 originated from LN-resident clones that had been infiltrating the BM prior to therapy could not be proven.
Patient 3
The common IgVH germline gene of the sequences of patient 3 was IGHV3-15*02. The average mutation rates for LN clones ranged from 2.2 to 7.3% and for BM sequences from 2.2 to 10.3% (Table 2).
Synopsis of the clonal evolution of the follicular lymphoma (Figure 4)
Figure 4.

Pedigree of the final FL cell evolution of patient 3 – synopsis of compartment-specific and inter-compartment pedigrees. Among the entire clonal population of patient 3 the least mutated sequenced clones were clone m2 of the initial lymph node (LN) population of 2004 and clone ×3 of the first relapse population of the bone marrow (BM) of year 2006. Interestingly both clones shared an identical mutation pattern suggesting a migratory link between the LN and the BM compartments. Considering the generated HPC, and the probability measure (pm)-values obtained for the compartment-specific and the inter-compartment populations, an initial inter-compartment migration in 2004 was a plausible option for clonal evolution, since the initial migration was directed from the LN to the BM where a HPC (BM1-04) included the mutation pattern of clone m2, which thus constituted the most likely “founder clone” of the clonal BM population. By detection of clone ×3 among the BM clones of the first relapse in 2006 showing identical mutations with the inital nodal clone m2, and the inclusion of this very mutation pattern in the HPC “founder clone” of the BM-population of 2004 (BM1 04) and the most likely HPC “founder clone” of the second BM relapse population of 2007 (BM1 07), BM persistence during therapy was evident. Chronological analysis of the different populations (not shown in detail), showed that the LN was reinfiltrated from the BM. Reinfiltration of the LN compartment of 2007 from the BM was more likely than nodal persistence during the interval from the 2004 to 2007 (Online Supplementary Tables S4F–H). However, whether reinfiltration took place in 2004, 2006 or 2007 could not be determined. Blue lines represent putative clonal migratory events from the LN to the BM compartment, red lines represent migration in the opposite direction (i.e. putative clonal migratory events from the BM to the LN compartment).
For the entire evolution of the FL clones of patient 3, an inter-compartment population was created for sequenced LN and BM clones of 2004, which was then complemented by clones of the BM population of 2006. Subsequently, the inter-compartment population of sequenced LN and BM populations of 2007 were added. To assess the possibility of nodal persistence during disease progression from 2004 to 2007, both LN populations were analyzed as a separate inter-compartment collective.
The complete FL population of patient 3 was represented by 11 LN sequences from the year 2004, eight LN sequences from 2007, nine sequences for the BM compartment from the years 2004 and 2006, and ten sequences for the BM compartment from 2007. Two identical sequences were found in the LN population of 2004 (clones m1 and m4) and two lots of two identical sequences were found in the BM population of 2007 (clones s1 and s5; clones s3 and s7). Specifically, the LN clone m2 from year 2004 and the BM clone ×3 from year 2006 also had similar clones, showing identical mutation patterns among the sequences analyzed.
Since these mutations were detectable in all other sequenced clones, they were assumed to represent the “founder clones” of the FL of patient 3, and moreover indicated that the FL originated out of the LN sampled in 2004 (clone m2).
Consequently, by probability measurement-guided chronological analysis of the inter-compartment evolution of the five compartments, early migration of LN clone m2 into the BM and its persistence there was postulated for 2004, 2006 and 2007. This interpretation was supported by detecting clones with the identical mutation pattern among the first BM relapse population of 2006 (x3), in the LN tissue of 2004, and the BM and LN tissue of 2007 (HPC LN1 04). Hence, the clone survived the repeated therapy in the BM, giving rise to the second relapse in the BM.
According to chronological analysis of the different populations, it was most likely that the LN 2007 clone was a re-infiltration from the BM 2007 one, although theoretically re-infiltration by the clones (x3) from BM 2006 or LN1 04 from the BM 2004 cannot be entirely excluded (Figure 4).
Younger clones with a high mutational load were more susceptible to chemotherapy than older clones with only few mutations. (see also Online Supplementary Results)
Discussion
In this study, we analyzed the mutation patterns of the IgVH genes in LN and corresponding BM specimens from three patients with synchronous FL in both compartments. Based on our results, we conclude that FL originates in the LN and not in the BM, since the FL clone with the least mutated IgVH gene was found in the LN. We also found hints of early migration of LN clones with few IgVH gene mutations into the BM compartment. These early clones might represent “founder clones” of relapsed FL, possibly originating from the BM. This observation supports the theory of a BM niche for FL cells during the course of the disease and its treatment.28,29 We also showed that inter-compartment migration between LN and BM exists in both directions during disease.
Dissemination of FL clones among different lymph follicles and LN has been documented.16–19,21,30 Studies showing that FL cells colonize non-neoplastic germinal centers and that non-neoplastic germinal centers can serve as a niche for FL cells are further supported by the concept of “in situ” FL, which represents infiltration of pre-existing non-neoplastic germinal centers by FL cells, rather than de novo formation of malignant germinal centers.17,31–33 Our conclusion that FL originates in the LN is consistent with the hypothesis of a possible dormant FL tumor stem cell harboring an unmutated, rearranged IgVH/L gene as part of the t(14;18)(q32;q21) translocation, which is possibly exposed to the somatic hypermutation machinery by passing the germinal centers of LN, acquiring additional point mutations and progressing to clinically overt FL.34, 35
In the FL population of the first patient, we detected a putative naïve LN clone with an unmutated IgVH gene and consequently identified it as the founder clone of this FL. Unfortunately, no further naïve founder clones were found in this study, but clones with few mutations were detectable in patients 2 and 3. These naïve or less mutated clones might have migrated relatively early into the BM, where they persisted and presumably survived therapy. This interpretation is supported by the findings in the third patient, in whom FL clones with relatively high mutation loads disappeared during chemotherapy, whereas the clones with low mutation frequency reappeared in the BM during the first relapse. Specifically, one clone with the identical five mutations could be detected in the pre-treatment LN compartment and in the first post-treatment sample from the BM compartment. Interestingly, these sequences were the least mutated of the entire FL population and were detectable in all other clones. It is, therefore, possible that these early FL “founder” clones represent tumor stem cells, which survive in the BM niche and perhaps initiate the FL relapse either in the LN or in the BM. However, we cannot fully exclude that these “founder” clones have their niche in other peripheral lymphoid tissues from where they disseminate to the BM, for example, from in situ FL in other lymph nodes.
We further demonstrated that FL cells migrate between the two compartments (LN and BM) in both directions during FL evolution. Such mutual trafficking between different compartments and concomitant exposure to different microenvironments might account for the observed molecular heterogeneity.6,10
Bognar et al. indicated that the BM niche plays a decisive role in FL, suggesting early infiltration events from the LN compartment.21 Applying the commonly used CLUSTAL-embedded multiple sequence alignment (MSA) algorithm, however, they were unable to delineate migration directions in relation to time.36,37 A basic feature of MSA algorithms is to calculate the evolutionary distance of operational taxonomic units (OTU), for instance of the genetic distance of nucleotide sequences, by a pairwise comparison of all units.38 Based on a received “guide tree”, the two units with the smallest distance are iteratively complemented by the remaining units, leading to a phylogenetic tree of relative genetic distance. As the MSA-based methods create phylogenies/pedigrees directed by the minimal genetic distances among sequences, they adhere to the principle of maximum sequence homology, which is appropriate for establishing relationships among potential members of an alleged species or of a protein family. Such methods do not, however, account directly for the mutational load of a particular sequence, the homology of mutation patterns of a collective of OTU, or for the frequency of equally mutated nucleotide residues. Instead, focus is put on the homology of unmutated sequence pairs without rooting to an unmutated wild-type sequence. Our goal was a more differentiated interpretation of ongoing somatic hypermutation in FL. Thus, we devised an algorithm that gives proper priority on homology of mutation patterns and thus more closely mirrors mutational proximity.
The relation of OTU in our method is based on pedigrees of FL evolution independent of sequence alignments. The pedigrees comprise stratified HPC and, as somatic hypermutation is a progressive event in FL, they reflect the temporal course of the disease. Due to the calculation of HPC based on the relative frequency of mutations and mutational patterns, migration and migration directions can be evaluated when comparing different populations of mutation patterns of sequenced IgVH genes. Thus, unlike MSA-based models, this method allows evaluation of both the time and direction of possible migratory interchange among different populations. However, more recently developed next generation high throughput/deep sequencing methods will probably overcome the necessity of introducing HPC, since these techniques might allow sequencing of all existing FL clones.
In the light of growing evidence of quiescent qualities of the BM niche in several malignancies, we complemented our migration analyses with the evaluation of potentially functional BCR expression.29,39–41 Functionally intact BCR are thought to convey pro-survival and differentiation signals to B-lymphocytes, independently of antigen stimulation.42,43 We, therefore, interpreted in-frame stop codons in IgVH genes as liberation from BCR restriction, and as a marker for possible niche-induced quiescence. The pre-treatment LN clones studied, especially from patients 1 and 2, showed expression of a putative functional BCR. In contrast, associated BM clones were less dependent on such BCR expression, since stop codons were detected more often among their sequenced IgVH genes. By comparing BCR conditions among pre-and post-treatment clone populations of patient 3 (LN and BM together, respectively), a more than 30% reduction of potentially functional BCR expression was observed. This was also true for patient 2. We interpret this putative loss of functional BCR as an argument for post-treatment re-infiltration of the LN from the BM.
Activation-induced cytidine deaminase (AID) was discovered to be the B-cell specific factor for somatic hypermutation and isotype switching.44 It has been proposed that there is a positive correlation between AID expression and the somatic hypermutation load in FL.35 Somatic hypermutation is physiologically triggered by interactions between antigens presented on follicular dendritic cells and the germinal center B cell.45,46 Whether antigens play a similar triggering role in the development of FL is not clear.47 Based on our findings and the outlined concept of multi-step FL evolution, we think that pre-malignant t(14;18)(q32;q21)-positive B cells are highly dependent on germinal center-resident environmental factors for fully malignant transformation. As in normal germinal center B cells, aberrant AID activity would drive mutagenesis paralleling normal B-cell differentiation programs in FL cells. Some minor mutated LN clones might alter their homing restriction as an early result of aberrant AID activity. Thus, hijacking of the CXCL12-secreting BM niche would be an early event during the evolution of FL. Accordingly, we found early BM infiltration during FL evolution by clones that could be traced back to the LN. Some FL cells in BM biopsies have an aberrant phenotype with lack of expression of both CD10 and BCL6, and have a deficiency of follicular dendritic cell networks within the neoplastic infiltrates.48 Future research should analyze these cells to determine whether they represent such early clones.
An additional result of our study was the demonstration of the capacity of BM-resident FL cells to migrate back to the LN in a retrograde fashion, possibly inducing LN relapses. The abuse of quiescence-inducing qualities of the BM hematopoietic stem cell niche might account for the inability of conventional, anti-proliferative chemotherapeutic agents to cure FL. An explanation for why FL cells would suddenly be capable of leaving the hematopoietic stem cell niche remains elusive, and needs further investigation.28,29
In summary, our results suggest that FL originates from the LN, and that early derivates of the nodal clones migrate relatively soon into the BM, where somatic hypermutation is retained. These initial clones possibly represent “founder clones” of relapsed FL, possibly out of the BM. These observations support the theory of a BM niche for FL cells during the progression, persistence and relapse of disease. For the purposes of this study, we developed a new algorithm which allows a suitable analysis of tumor cell evolution and cell migration in FL. Further studies are necessary to investigate the potential role of the BM as a niche for progression, persistence and relapse of FL.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (4th ed). Lyon, France: IARC Press, 2008 [Google Scholar]
- 2.Horning SJ. Follicular lymphoma: have we made any progress? Ann Oncol. 2000;11 (Suppl 1):23–7 [PubMed] [Google Scholar]
- 3.Roulland S, Navarro JM, Grenot P, Milili M, Agopian J, Montpellier B, et al. Follicular lymphoma-like B cells in healthy individuals: a novel intermediate step in early lymphomagenesis. J Exp Med. 2006;203(11): 2425–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jegalian AG, Eberle FC, Pack SD, Mirvis M, Raffeld M, Pittaluga S, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118(11): 2976–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henopp T, Quintanilla-Martinez L, Fend F, Adam P. Prevalence of follicular lymphoma in situ in consecutively analysed reactive lymph nodes. Histopathology. 2011;59(1): 139–42 [DOI] [PubMed] [Google Scholar]
- 6.Lejeune M, Alvaro T. Clinicobiological, prognostic and therapeutic implications of the tumor microenvironment in follicular lymphoma. Haematologica. 2009;94(1):16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvaro T, Lejeune M, Salvado MT, Lopez C, Jaen J, Bosch R, et al. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J Clin Oncol. 2006;24(34): 5350–7 [DOI] [PubMed] [Google Scholar]
- 8.Carlotti E, Wrench D, Matthews J, Iqbal S, Davies A, Norton A, et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma may occur by divergent evolution from a common progenitor cell or by direct evolution from the follicular lymphoma clone. Blood. 2009;113(15): 3553–7 [DOI] [PubMed] [Google Scholar]
- 9.Bende RJ, Smit LA, van Noesel CJ. Molecular pathways in follicular lymphoma. Leukemia. 2007;21(1):18–29 [DOI] [PubMed] [Google Scholar]
- 10.de Jong D. Molecular pathogenesis of follicular lymphoma: a cross talk of genetic and immunologic factors. J Clin Oncol. 2005; 23(26):6358–63 [DOI] [PubMed] [Google Scholar]
- 11.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5(4):251–62 [DOI] [PubMed] [Google Scholar]
- 12.Stamatopoulos K, Kosmas C, Belessi C, Stavroyianni N, Kyriazopoulos P, Papadaki T. Molecular insights into the immunopathogenesis of follicular lymphoma. Immunol Today. 2000;21(6):298–305 [DOI] [PubMed] [Google Scholar]
- 13.Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341(20): 1520–9 [DOI] [PubMed] [Google Scholar]
- 14.Klein U, Goossens T, Fischer M, Kanzler H, Braeuninger A, Rajewsky K, et al. Somatic hypermutation in normal and transformed human B cells. Immunol Rev. 1998;162: 261–80 [DOI] [PubMed] [Google Scholar]
- 15.Aarts WM, Bende RJ, Steenbergen EJ, Kluin PM, Ooms EC, Pals ST, et al. Variable heavy chain gene analysis of follicular lymphomas: correlation between heavy chain isotype expression and somatic mutation load. Blood. 2000;95(9):2922–9 [PubMed] [Google Scholar]
- 16.Aarts WM, Bende RJ, Vaandrager JW, Kluin PM, Langerak AW, Pals ST, et al. In situ analysis of the variable heavy chain gene of an IgM/IgG-expressing follicular lymphoma: evidence for interfollicular trafficking of tumor cells. Am J Pathol. 2002;160 (3):883–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong P, Raffeld M, Teruya-Feldstein J, Sorbara L, Pittaluga S, Jaffe ES. In situ localization of follicular lymphoma: description and analysis by laser capture microdissection. Blood. 2002;99(9):3376–82 [DOI] [PubMed] [Google Scholar]
- 18.Oeschger S, Brauninger A, Kuppers R, Hansmann ML. Tumor cell dissemination in follicular lymphoma. Blood. 2002;99(6): 2192–8 [DOI] [PubMed] [Google Scholar]
- 19.Adam P, Schoof J, Hartmann M, Schwarz S, Puppe B, Ott M, et al. Cell migration patterns and ongoing somatic mutations in the progression of follicular lymphoma. Cytogenet Genome Res. 2007;118(2–4):328–36 [DOI] [PubMed] [Google Scholar]
- 20.Aarts WM, Bende RJ, Bossenbroek JG, Pals ST, van Noesel CJ. Variable heavy-chain gene analysis of follicular lymphomas: sub-clone selection rather than clonal evolution over time. Blood. 2001;98(1):238–40 [DOI] [PubMed] [Google Scholar]
- 21.Bognar A, Csernus B, Bodor C, Reiniger L, Szepesi A, Toth E, et al. Clonal selection in the bone marrow involvement of follicular lymphoma. Leukemia. 2005;19(9):1656–62 [DOI] [PubMed] [Google Scholar]
- 22.Ame-Thomas P, Maby-El Hajjami H, Monvoisin C, Jean R, Monnier D, Caulet-Maugendre S, et al. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood. 2007; 109(2):693–702 [DOI] [PubMed] [Google Scholar]
- 23.Kremer M, Spitzer M, Mandl-Weber S, Stecker K, Schmidt B, Hofler H, et al. Discordant bone marrow involvement in diffuse large B-cell lymphoma: comparative molecular analysis reveals a heterogeneous group of disorders. Lab Invest. 2003;83(1): 107–14 [DOI] [PubMed] [Google Scholar]
- 24.Segal GH, Jorgensen T, Scott M, Braylan RC. Optimal primer selection for clonality assessment by polymerase chain reaction analysis: II. Follicular lymphomas. Hum Pathol. 1994;25(12):1276–82 [DOI] [PubMed] [Google Scholar]
- 25.Zuckerman NS, McCann KJ, Ottensmeier CH, Barak M, Shahaf G, Edelman H, et al. Ig gene diversification and selection in follicular lymphoma, diffuse large B cell lymphoma and primary central nervous system lymphoma revealed by lineage tree and mutation analyses. Int Immunol. 2010;22 (11):875–87 [DOI] [PubMed] [Google Scholar]
- 26.Lossos IS, Tibshirani R, Narasimhan B, Levy R. The inference of antigen selection on Ig genes. J Immunol. 2000;165(9):5122–6 [DOI] [PubMed] [Google Scholar]
- 27.Chang B, Casali P. The CDR1 sequences of a major proportion of human germline Ig VH genes are inherently susceptible to amino acid replacement. Immunol Today. 1994;15(8):367–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66(9):4553–7 [DOI] [PubMed] [Google Scholar]
- 29.Shiozawa Y, Havens AM, Pienta KJ, Taichman RS. The bone marrow niche: habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia. 2008;22(5):941–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ottensmeier CH, Thompsett AR, Zhu D, Wilkins BS, Sweetenham JW, Stevenson FK. Analysis of VH genes in follicular and diffuse lymphoma shows ongoing somatic mutation and multiple isotype transcripts in early disease with changes during disease progression. Blood. 1998;91(11):4292–9 [PubMed] [Google Scholar]
- 31.Thomazy VA, Vega F, Medeiros LJ, Davies PJ, Jones D. Phenotypic modulation of the stromal reticular network in normal and neoplastic lymph nodes: tissue transglutaminase reveals coordinate regulation of multiple cell types. Am J Pathol. 2003;163 (1):165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheung MC, Bailey D, Pennell N, Imrie KR, Berinstein NL, Amato D, et al. In situ localization of follicular lymphoma: evidence for subclinical systemic disease with detection of an identical BCL-2/IGH fusion gene in blood and lymph node. Leukemia. 2009;23(6):1176–9 [DOI] [PubMed] [Google Scholar]
- 33.Corcione A, Ottonello L, Tortolina G, Facchetti P, Airoldi I, Guglielmino R, et al. Stromal cell-derived factor-1 as a chemoat-tractant for follicular center lymphoma B cells. J Natl Cancer Inst. 2000;92(8):628–35 [DOI] [PubMed] [Google Scholar]
- 34.Perez-Duran P, de Yebenes VG, Ramiro AR. Oncogenic events triggered by AID, the adverse effect of antibody diversification. Carcinogenesis. 2007;28(12):2427–33 [DOI] [PubMed] [Google Scholar]
- 35.Hardianti MS, Tatsumi E, Syampurnawati M, Furuta K, Saigo K, Nakamachi Y, et al. Activation-induced cytidine deaminase expression in follicular lymphoma: association between AID expression and ongoing mutation in FL. Leukemia. 2004;18(4):826–31 [DOI] [PubMed] [Google Scholar]
- 36.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003; 31(13):3497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23(10):403–5 [DOI] [PubMed] [Google Scholar]
- 38.Gotoh O. Multiple sequence alignment: algorithms and applications. Adv Biophys. 1999;36:159–206 [DOI] [PubMed] [Google Scholar]
- 39.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–21 [DOI] [PubMed] [Google Scholar]
- 40.Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007;21(2):304–10 [DOI] [PubMed] [Google Scholar]
- 41.Braun S, Kentenich C, Janni W, Hepp F, de Waal J, Willgeroth F, et al. Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol. 2000;18(1):80–6 [DOI] [PubMed] [Google Scholar]
- 42.Monroe JG. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat Rev Immunol. 2006;6(4):283–94 [DOI] [PubMed] [Google Scholar]
- 43.Corcos D. Ligand-independent activity of the B cell antigen receptor in physiology and pathology. Arch Immunol Ther Exp (Warsz). 2007;55(2):77–82 [DOI] [PubMed] [Google Scholar]
- 44.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–63 [DOI] [PubMed] [Google Scholar]
- 45.Choe J, Li L, Zhang X, Gregory CD, Choi YS. Distinct role of follicular dendritic cells and T cells in the proliferation, differentiation, and apoptosis of a centroblast cell line, L3055. J Immunol. 2000;164(1):56–63 [DOI] [PubMed] [Google Scholar]
- 46.Tew JG, Wu J, Fakher M, Szakal AK, Qin D. Follicular dendritic cells: beyond the necessity of T-cell help. Trends Immunol. 2001;22(7):361–7 [DOI] [PubMed] [Google Scholar]
- 47.Chang KC, Huang X, Medeiros LJ, Jones D. Germinal centre-like versus undifferentiated stromal immunophenotypes in follicular lymphoma. J Pathol. 2003;201(3):404–12 [DOI] [PubMed] [Google Scholar]
- 48.West RB, Warnke RA, Natkunam Y. The usefulness of immunohistochemistry in the diagnosis of follicular lymphoma in bone marrow biopsy specimens. Am J Clin Pathol. 2002;117(4):636–43 [DOI] [PubMed] [Google Scholar]