

Abstract
The six elements commonly known as metalloids are boron, silicon, germanium, arsenic, antimony, and tellurium. Metalloid containing compounds have been used as antiprotozoal drugs. Boron-based drugs, the benzoxaboroles have been exploited as potential treatments for neglected tropical diseases. Arsenic has been used as a medicinal agent and arsphenamine was the main drug used to treat syphilis. Arsenic trioxide has been approved for the treatment of acute promyelocytic leukemia. Pentavalent antimonials have been the recommended drug for visceral leishmaniasis and cutaneous leishmaniasis. Tellurium (IV) compounds may have important roles in thiol redox biological activity in the human body, and ammonium trichloro (dioxoethylene-O, O’-)tellurate (AS101) may be a promising agent for the treatment of Parkinson’s disease. Organosilicon compounds have been shown to be effective in vitro multidrug-resistance reverting agents.
Keywords: Boron, Silicon, Germanium, Arsenic, Antimony, Metalloid
INTRODUCTION
The periodic table is divided into three types of elements: metals, non-metals and metalloids. The metalloids Boron (B), Silicon (Si), Germanium (Ge), Arsenic (As), Antimony (Sb) and Tellurium (Te) are located along a diagonal line separating metals from non-metals (Table 1). Metalloids have properties of both metals and non-metals, hence they are also known as semi metals. Metalloids react like non-metals when they react with metals and act like metals when they react with non-metals. Boron belongs to: Group 13, Period 2; Silicon: Group 14, Period 3; Germanium: Group 14, Period 4; Arsenic: Group 15, Period 4; Antimony: Group 15, Period 5; Tellurium: Group 16, Period 5 of the periodic table of elements (Table 1).
Table 1.
List of elements considered to be metalloids.

Metalloid compounds generate various biological effects on cells and tissues and their therapeutic and potential uses have been evolving over centuries, starting, for example, with the empiric use of arsenic in ancient times up to the current Food and Drug Administration approval of As2O3 for the treatment of acute promyelocytic leukemia in human. However, opportunities remain unrealised regarding therapeutic potential of metalloid compounds. Herein, an attempt has been made to review this topic due to awakening interest on metalloids for the treatment of human diseases.
BORON
In the quest for novel drug candidates, researchers have substituted boron for its neighbour carbon in numerous classes of drug molecules (1,2). The polyether-macrolide antibiotic, boromycin (C45H74BNO15, Fig. 1) containing the trace element boron, is a complex of boric acid with a tetradentate complexing agent. Boromycin was isolated as a potent anti-human immuno-deficiency virus (HIV) antibiotic from a fermentation broth of Streptomyces sp. A-3376 (3,4).
Fig. 1.
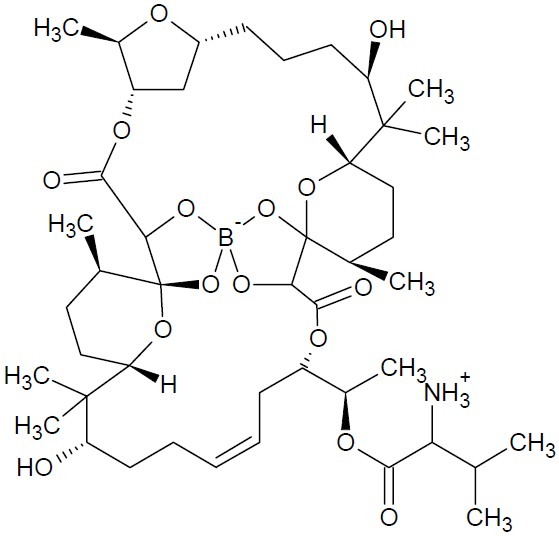
Boromycin
Boric acid is a weak acid and has antiseptic, antifungal, and antiviral properties (5,6). Mild solutions of boric acid have been used as eye antiseptics. Boron has a unique trigonal planar geometry and can form a dative bond under specific conditions through its empty p-orbital to generate tetrahedral structure. The exploit-tation of empty p-orbital expands drug design possibilities. Boron-based drugs have exhibited attractive properties and activities against a number of protozoans contributing to neglected tropical diseases. The current advances in discovery of potential treatments for human African trypanosomiasis, malaria and Chagas disease from a class of boron-containing drugs, the benzoxaboroles were reported (7). A unique geometry allows boron based drugs to have two distinct shapes. These shapes provide boron based drugs the ability to interact with biological targets in novel ways. In addition, boron’s reactivity allowed boron based drugs to interact with a biological target to create a change that is specific to a particular disease or condition (8). Design of boronic acid protease inhibitors initiated in 1990s (Fig. 2).
Fig. 2.
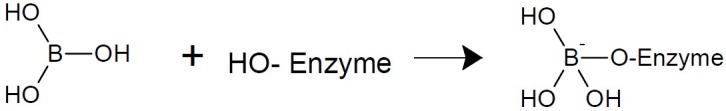
Formation of tetrahedral structure using an empty p-orbital on boron atom.
In this context, multiple disease targets have been pursued. Several proteasome inhibitors exert anti-tumour activity in vivo and potently induce apoptosis in tumour cells in vitro , including those resistant to conventional chemotherapeutic agents. Bortezomib (Mole-cular formula C19H25BN4O4, (Fig. 3) drug is an N-protected dipeptide and can be written as Pyz-Phe-boroLeu, which stands for pyrazinoic acid, phenylalanine and leucine with a boronic acid. Velcade (bortezomib), the first FDA approved therapeutic inhibitor of the 26S proteasome is an effective treatment for multiple myeloma and has reached FDA approval for treating relapsed multiple myeloma (the cancer of plasma cells) and mantle cell lymphoma. The boron atom in bortezomib molecule is a key substructure because through it certain proteasomes are blocked that would otherwise degrade proteins. The boron atom in bortezomib binds the catalytic site of the 26S proteasome (9) with high affinity and specificity. In normal cells, the proteasome regulates protein expression and function by degradation of ubiquitylated proteins, and also cleanses the cell of abnormal or misfolded proteins.
Fig. 3.
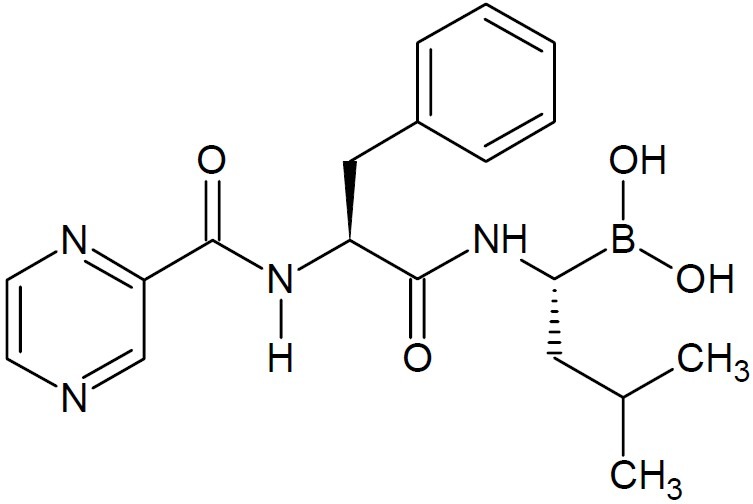
Structure of bortezomib
Anacor Pharmaceuticals has a rich pipeline of boron-containing therapeutics in the clinic. ABX (Fig. 4) inhibits bacterial Leucyl tRNA synthetase and represents a new class of Gram-negative antibacterial agents (10).
Fig. 4.
ABX
New series of dipeptidyl boronate inhibitors of 20S proteasome were identified to be highly potent drug-like candidates with IC50 values of 1.2 and 1.6 nM, respectively, which showed better activities than the drug bortezomib on the market (11,12). The potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[ [ (2S,3R)- 3-hydroxy-2-[ (6-phenylpyridine- 2-carbonyl) amino]-1 -oxobutyl] amino]- 3-methylbutyl] boronic acid (CEP-18770, (Fig. 5), has been reported (13).
Fig. 5.
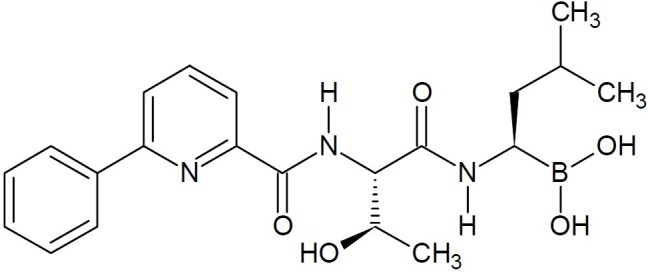
CEP-18770
Further, the anti-multiple myeloma protea-some inhibitor CEP-18770 enhanced the anti-myeloma activity of bortezomib and melphalan. The combination of anti-multiple myeloma proteasome inhibitor CEP-18770 intravenously and bortezomib exhibited complete regression of bortezomib-sensitive tumours. Moreover, this combination markedly delayed progression of bortezomib-resistant tumours compared to treatment with either agent alone (14). Structure-activity relationship study of 72 dipeptidyl boronic acid proteasome inhibitors constructed from β-amino acids revealed that bicyclic groups at the R1 position, 3-F substituents at the R2position, and bulky aliphatic groups at the R3position were favorable to the activities. Enzymatic screening results showed that compound (Fig. 6), comp-rising all of these features, was the most active inhibitor against the 20S human proteasome at less than a 2 nM level, as active as the marketed drug bortezomib (15).
Fig. 6.
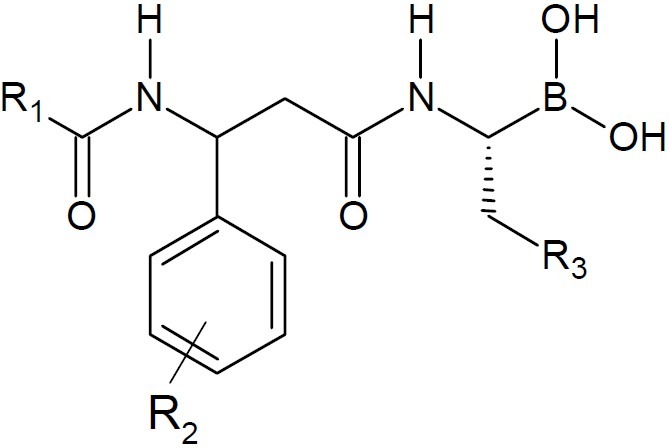
Dipeptidyl boronic acid proteasome inhibitors constructed from β-amino acids.
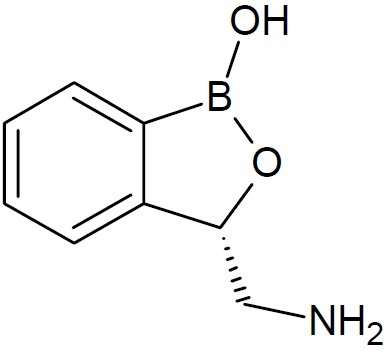
Interest in dipeptide boronic acids of the type H2N-X-Y-B(OH)2 as potent protease inhibitors for many diseases is growing. In this direction, dipeptides of boroLeu (Fig. 7) served as warheads in prodrugs as it was found to be adequately potent, cell-penetrating, cytotoxic, and stable to degradation by cellular peptidases (16). Anacor Pharmaceuticals, Inc., a biophar-maceutical company’s lead topical oxoborole product programs include AN2690 ((5-fluoro-1, 3-dihydro -1 -hydroxy -2, 1-benzoxaborole), (Fig. 8) candidate in Phase 3 clinical deve-lopment for the treatment of onychomycosis; AN2728(5-(4-cyanophenoxy) -1, 3-dihydro- 1- hydroxy- 2, 1-benzoxaborole, (Fig. 9), a topical anti-inflammatory PDE-4 inhibitor in Phase 2b clinical trial for the treatment of psoriasis and atopic dermatitis; and GSK 2251052, or GSK ’052, a systemic antibiotic in Phase 2 clinical trial for the treatment of infections caused by Gram-negative bacteria. It also engages in developing AN2718 (5-chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole) (Fig. 10), a topical antifungal product candidate which completed Phase 1 clinical trial for the treatment of onychomycosis and skin fungal infections (17,18). AN2690, a broad spectrum antifungal agent exhibited tremendous activity against yeast, molds and dermatophytes. It is a non-competitive inhibitor with ATP and leucine and inhibited protein synthesis in Saccharomyces cerevisiae targeting the edited domain of leucyl tRNA synthetase. No treatment-related systemic side effects have been observed in any of its clinical trials. AN2718 works similarly to AN2690. It targets common skin and fungal infectious agents including Trichophyton and Candida.
Fig. 7.
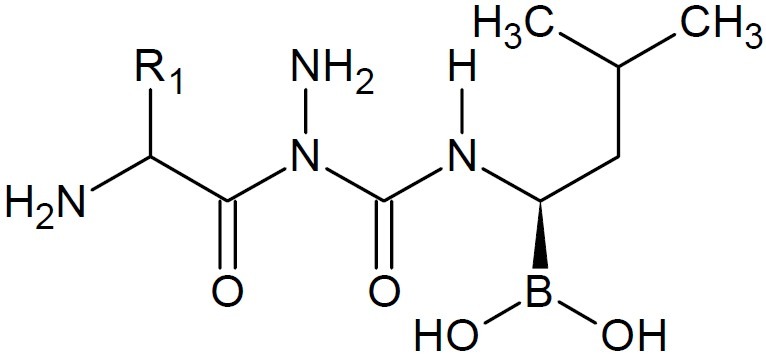
Dipeptides of boroLeu
Fig. 8.
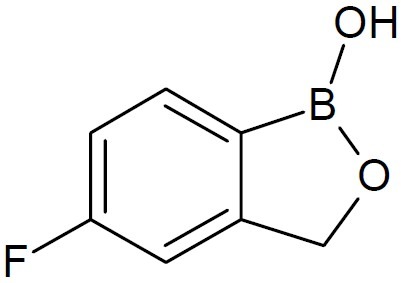
AN2690
Fig. 9.

AN 2728
Fig. 10.
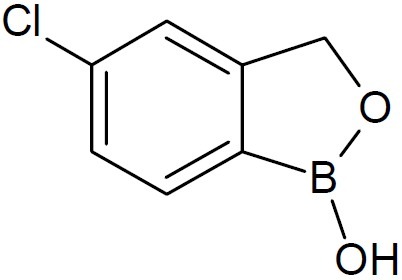
AN2718
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) (Fig. 11) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis. AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin. Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor (19). In July 2010, An3365 now GSK052 (Fig. 12) was developed for treatment of infections caused by Gram-negative bacteria (20).
Fig. 11.
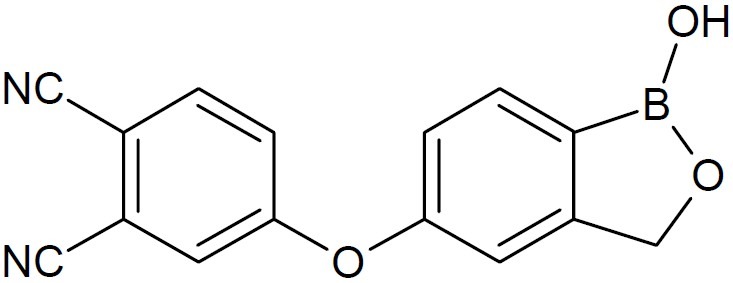
AN2898
Fig. 12.
GSK052
The presence of a boron atom in the heterocyclic core structure has been found essential for trypanocidal activity of orally active series of benzoxaborole-6-carboxamides in murine models of human African trypanosomiasis. SCYX-7158 (Fig. 13) has been identified as an effective, safe and orally active treatment for human African trypanoso-miasis to enter preclinical studies, with expected progression to phase 1 clinical trials in 2011 (21,22).
Fig. 13.
SCYX-7158
Recently, boron containing compounds have shown capability of acting as ligands on transmembrane receptors (ionotropic and metabotropic receptors) expressed in mamma-lians (23). Boron-based molecules can be used as new pharmacophores, or as markers of drugs in living tissue, and to improve long-hindered attempts to develop boron-neutron capture therapies to produce inhibiting agents for cancer treatment. Boron-10 (10B) isotope is good at capturing thermal neutrons and is used in boron neutron capturing medical therapy (BNCT). A number of potential boronated pharmaceuticals using boron-10 for use in boron neutron capture therapy and other inorganic medicinal agents have been reported (24–26). Biologically targeted BNCT treatment is based on producing radiation inside a tumour using 10B and thermal neutrons. 10B is introduced into cancer cells as borono-phenylalanine , while a neutron beam shines onto those cells, which causes the boron inside the cells to “fissle” into Lithium-7 and an alpha particle. This recoiling atom and alpha particle obliterate the cell, thus killing the cancer cells. Experts say that one to two BNCT treatment sessions may be sufficient to destroy a tumour. Moreover, BNCT treatment saves healthy tissue as the impact of radiation on surrounding healthy tissue has been reported to a minimum level (27,28).
Antimony
As therapeutic agents, antimony and its compounds have been mostly used for the treatment of two parasitic diseases (leishmaniasis and schistosomiasis) since their prescription by the alchemist John of Rupescissa in the 14th century (29). Leishma-niasis is a disease spread by the bite of the female sandfly. Schistosomiasis is disease of liver, gastrointestinal tract and bladder caused by schistosomes, trematode worms that parasitize people, and usually its source is from infested water.
Treatments chiefly involving antimony has been called antimonials. The treatment of leishmanial with potassiun antimony (III) tartrate (tartar-emetic) started in 1913. In potassiun antimony (III) tartrate, each Sb (III) coordinates to four oxygens from two tartrate with each in a bidentate mode. Potassium antimony tartrate has been used to treat cough for reducing the excretion of sputum as an effective ingredient in Compound Liquorice. Potassiun antimony (III) tartrate as an effective ingredient in compound Liquorice is used to induce sweating and also as an emetic (a drug that causes vomiting). With the introduction of the more efficacious, less toxic alternative, and subsequent larger use of praziquantel, the trivalent antimonials fell out of use from the treatment of schistosomiasis in the 1970s. Medicines called pentavalent antimony containing compounds like meglumine antimonite (Glucantime) (C7H18NO8Sb, (Fig. 14) and sodium stibogluconate(Pentostam) (C12H40Na3O26Sb2, (Fig. 15) have been used as first line drugs to treat cutaneous leishmaniasis for the past 50 years (30).
Fig. 14.
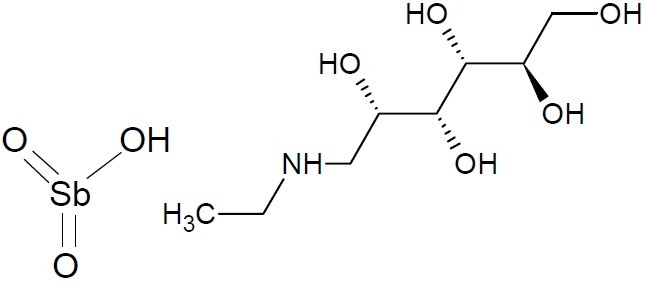
Meglumine antimonite
Fig. 15.

Sodium stibogluconate
In case of antimony treatment of schistosomiasis, antimony attached itself to sulphur atoms in trypanothione reductase (the putative enzyme targeted by antimonial compounds) which was used by both the parasites and human host. Based on the structural analysis of the trypanothione reductase complex with NADPH and Sb (III) in the reduced state, the trivalent Sb(III) ion inhibited the trypanothione reductase activity involving Cys52, Cys57 and His461’ of the two-fold symmetry related subunit in the catalytic cleft at the dimeric interface. Sb(III) coordination to Thr335 was also reported. Moreover, binding of the semimetal is only possible upon enzyme reduction because in the oxidized enzyme the two cysteine residues form a disulfur bridge (31). Antiomaline (Anthiomaline, Fig. 16), a brand of lithium antimony thiomalate has been successful in tropical nasal granuloma (Schistosomiasis) of cattle, and in veterinary practice as a skin conditioner.
Fig. 16.
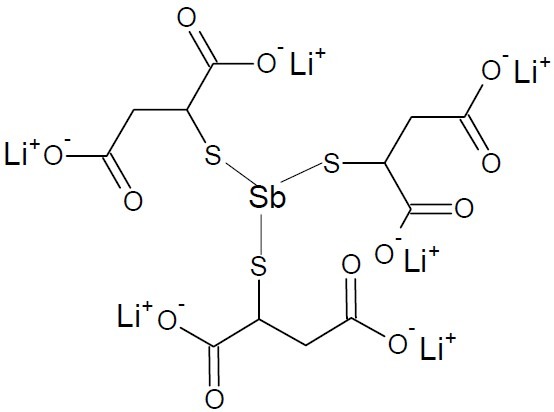
Anthiomaline
Sb5+ behaves as a prodrug and was converted to its active trivalent state (Sb3+) inside the cells by trypanothione forming an Sb(III)-trypanothione complex. Sodium stibogluconate is a potent protein tyrosine phosphatase inhibitor, and as an enhancer of cytokine signalling, appears to be a better inhibitor than suramin which is a compound known for its antineoplastic activity against several types of cancers (32). The formation of binary and ternary complexes due to the strong binding of antimony (III) at the thiolate group of Cys residue of glutathione, trypanothione, and nucleotides may assist antimony (III) to be transported in cells (33). Antimony compounds therapy exhibited toxicity (including cardiotoxicity and pancreatitis) which is quite common in HIV-associated leishmaniasis (34). Recently improvements have been achieved by combination therapy, reducing the time and cost of treatment. Recently, acquired resistance to antimonials has become a clinical threat during the last 16 years and this resistance has been found unique to L. donovani Researchers showed that some peroxovanadate complexes as antileishmanial agents have Sb(V)-resistance modifying ability in experimental infection with Sb(V) resistant Leishmania donovani isolates in murine model. These findings suggested thae use of vanadium compounds in combination with Sb(V) in the treatment of Sb(V) resistant cases of visceral leishmaniasis (35). In March 2010, the World Health Organization Expert Committee on the Control of Leishmaniases recommended sodium stibogluconate & paromomycin combination as first-line treatment for visceral leishmania sis in East Africa. It is already being used to treat patients in the countries such as Sudan and South Sudan (36,37).
Recently, several major intrinsic proteins (MIPs) comprising water-channelling aquaporins and glycerol-channelling aquaglyceroporins facilitate the diffusion of reduced and non-charged species of the metalloids silicon, boron, arsenic and antimony, thereby, suggesting their role as potential pharmacological targets (38). Researchers showed the presence of subcellular-aquaporins in L. donovani that are similar to tonoplast intrinsic protein of plants (39). Quantum dots consists of a metalloid core and a shell that sorrounds core and renders Quantum dots bioavailable (40).
Germanium
Salts and oxides lacking germanium carbon bond are categorized as inorganic forms. The most common inorganic form is Germanium dioxide (GeO2). Germanium sesquioxide (Organic Germanium-132) is a popular supplement used for cancer symptoms. The active compound, carboxylethyl germanium sesquioxide ((GeCH2CH2COOH)2O3, Fig. 17) is found must abundantly in the Reishi mushroom, as well as in Ginseng.
Fig. 17.
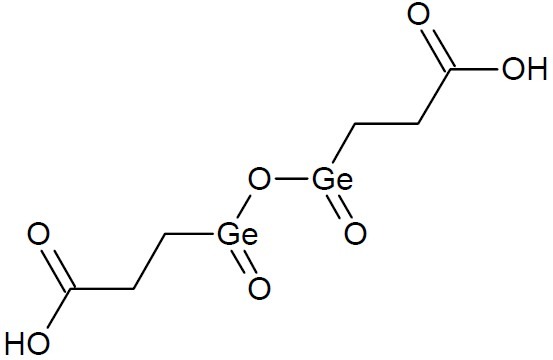
Carboxyethylgermanium sesquioxide
Germanium 132 having 3 atoms of oxygen is a very efficient donor of electrons. It merges with the free radicals and eliminates them from the organism via the excretion processes. Germanium sesquioxide aids the body natural defence against disease and aging. Its primary action is the restoration of the natural balance of cells in the immune system. Ge compounds were tested as chemotherapeutics agents (41) but no germanium compound has yet demonstrated a pharmaceutical use, as either an antibacterial or cancer chemotherapeutic agent. Propagermanium or organic germanium or beta-carboxyethylgermanium sesquioxide are used with complementary medicines and are helpful in boosting the immune system of cancer patients. It is also used to aid in treatment of AIDS, heart diseases and arthritis.
Bis (2-carboxyethylgermanium) sesquioxide (Ge-132), germanium oxide (GeO2) and germanium nanoparticles were tested as radiosensitizers. Nanometer-sized germanium particles have a similar radiosensitizing effect as that of GeO2. It was observed that inorganic but not organic germanium compounds exerted radiosensitizing effect in cells (42). More recently, in addition to the use of germanium as a dietary supplement, an elixir to cure diseases such as cancer and AIDS, germanium nanoparticles (GeCl4 and99m Tc-labeled organo-germanium nanoparticles), ranging in size from 60 to 80 nm, have been developed as a potential spleen imaging agent (43).
Arsenic
Arsenic is a metalloid or semi-metal and exists in two biologically important oxidation states, As (III) and As(V). Arsenic is considered to be a paradox in terms of its role both as a carcinogen and as a therapeutic agent and it has a fascinating history as a healer and killer. Early physicians, such as Hippocrates (c. 460 B.C -370 B.C.) and Paracelsus (1493-1541), recommended arsenic for the treatment of some diseases. In 1909, the first drug called Salvarsan (a compound of arsenic having chemical name – arsphenamine) was invented to cure syphilis, a sexually transmitted disease. The structure of this drug has only recently been determined almost 100 years after it was first used (44) (Fig. 18). It was the first important antisyphillitic, though was phased out in the 1930s by better arsenical compounds (neoarsphenamine), and eventually altogether by modern antibiotics.
Fig. 18.
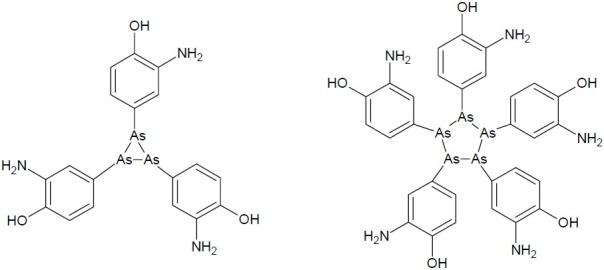
Two phenyl arsenic compounds found in the crystal structure of arsphenamine
As (III), in particular, reacts with closely spaced protein thiols, forming stable cyclic dithioarsinite complexes in which both sulfur atoms are bound to arsenic. The formation of cyclic dithioarsinite complexes is mostly responsible for arsenic cytotoxicity. Arsenic has been applied for the obstinate ulcers which sometimes occur at the roots of the nails. Arsenic trioxide was the main ingredient of the popular tonic of the 19th century, Fowler’s Solution or Liquor Arsenicalis (45)(a mixture of potassium arsenite and lavender water). Arsenic trioxide induces potent antitumor effects in vivo and in vitro and was a mainstay in the treatment against a certain form of blood cancer, acute promyelocytic leukemia (46). Arsenic induced complete remission when combined with all-trans retinoic acid and was effective in a high percentage of patients with acute promyelocytic leukemia, awakening interest in this metalloid for the treatment of human disease (47). Molecularly, arsenic induced the formation of reactive oxygen species through bonding with thiol residues, thereby, affecting numerous signaling pathways. In addition, arsenic directly binds to the C3HC4 zinc finger motif in the RING/B box/coiled-coil (RBCC) domain of promyelocytic leukemia protein (PML) and promyelocytic leukemia protein-retinoic acid receptor α (PML-RARα), induces their homodimerization and multi-merization, and enhanced their interaction with the SUMO E2 conjugase Ubc9 (The SUMO pathway parallels the classical ubiquitinylation pathway involving three steps including conjugation involving the E2 enzyme Ubc9 which plays an important role in the stability and packaging of gp120 into infectious HIV virions) facilitating subsequent sumoylation/ ubiquitination and proteasomal degradation (48). Based on clinical studies which showed a complete remission, especially in relapsed patient, the Food and Drug Administration (FDO) has approved the use of arsenic trioxide (Trisenox, Fig. 19) for the treatment of acute promyelocytic leukemia. (49).
Fig. 19.
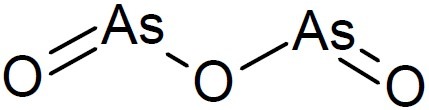
Structure of arsenic oxide
Mitochondria, a main source of ROS in cells, comprise the thioredoxin system which has been reported very sensitive to arsenic-based drugs. Major mitochondrial targets for binding of arsenic-containing compounds include many vicinal thiol-containing redox proteins. Such redox changes to vicinal thiols increased production of ROS and induction of apoptotic signalling pathways (50).
Melarsoprol (an organoarsenic compound introduced in 1949 as a very poorly tolerated non-aqueous solution, Arsobal(R) (Fig. 20), ability to cross the blood-brain barrier made it particularly effective against late-stage Gambian (or West African) sleeping sickness caused by T. brucei rhodesiense. Melarsoprol employed mainly in the treatment of Human African trypanosomiasis, has demonstrated an in vitro activity on myeloid and lymphoid leukemia derived cell lines.
Fig. 20.
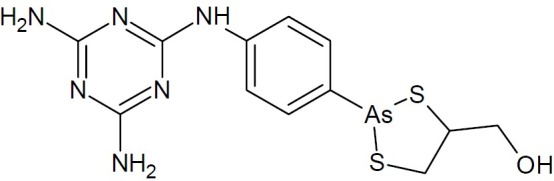
Melarsoprol
Complexed Melarsoprol (Melarsoprol hydroxypropyl-β-cyclodextrin and Melarsoprol randomly-methylated-β-cyclodextrin) has been employed as an oral treatment for CNS-stage human African trypanosomiasis, delivering con-siderable improvements over current parenteral chemotherapy (51). Inorganic arsenic com-pounds are suggested as useful agents for treatment of T-lymphoblastoid leukemia. In this context, As2O3 and arsenic acid inhibited proliferation and induced apoptosis in MOLT-4 and daunorubicine-resistant MOLT-4/DNR cells via glutathione-depletion and subsequent caspase-3/7 activation (52). Arsenic role as a therapeutic agent both as a single agent as well as in combination chemotherapy has been recently reported (53). Combination of arsenic trioxide and 5-fluorouracil increased cyto-toxicity. In this context, In vitro data showed that as a single agent, As2O3 down-regulated TS expression in HT29 cells. In vitro addition of 5-fluorouracil to these sensitized cells increased cytotoxicity (54). Arsenic trioxide has potential as a chemosensitizer in combi-nation therapy, especially to 5-fluorouracil. In this context, arsenic trioxide effect on cell proliferation of 5-fluorouracil-sensitive and -resistant HT29 colorectal cancer cells were dose dependent (54).
Tellurium
RT-01 (Empirical formula C13H22N+C3H3Cl4OTe-, (Fig. 21), an organotellurane compound, has been proved to be toxic against promastigotes and amastigotes (55).
Fig. 21.
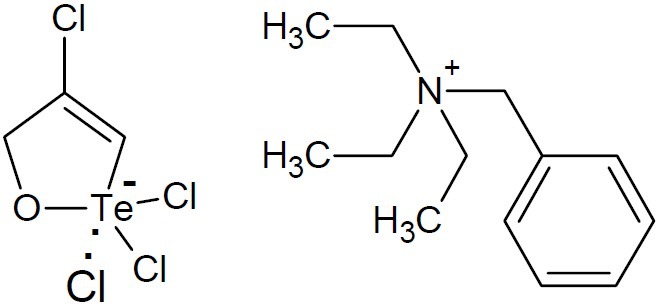
RT-01
Cadmium telluride nanoparticles are fluorescent and may be used as quantum dots in imaging and diagnosis. Some unsymmetrical 2- naphtyl diorganyltellurium dichlorides were most effective organotelluranes with gram-negative antibacterial effect(Fig. 22).
Fig. 22.
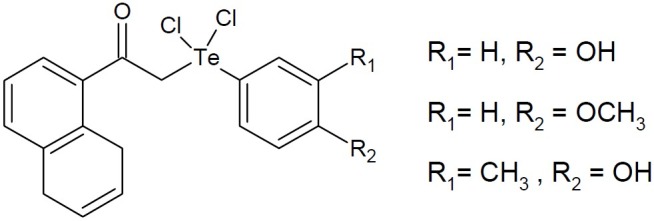
2- Naphtyl diorganyltellurium dichlorides
The antioxidant effects of organotellurides and diorganoditellurides, the immunomodulatory effects of the non-toxic inorganic tellurane, named AS-101 (Fig. 23), and organic telluranes (organotelluranes) as protease inhibitors were reported (56). The empirical formula of AS101 is C2H802NCl3Te and the chemical structure is shown below.
Fig. 23.
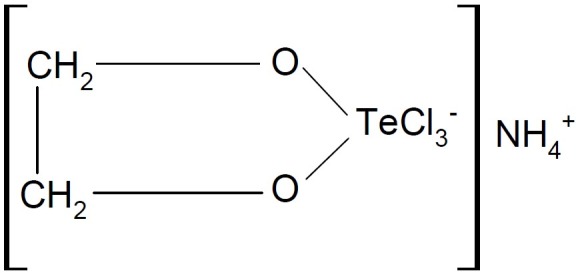
AS101
Te(IV) compounds influenced thiol redox biological activity, specifically inactivated cysteine proteases in the human body. The Te (IV)-thiol chemical bond formation or disulfide bond formation in a specific protein lead to conformational change, possibly resulting in the loss of its biological activity. Indeed, researchers demonstrated that AS101 and other Te (IV) compounds specifically inactivate cysteine proteases, while exhibiting no effect on the other families of serine, aspartic and metalloproteases, in good agreement with the predictions of their unique Te(IV)-thiol chemistry. The effects of two small inorganic tellurium complexes, ammonium small inorganic tellurium complexes, ammonium trichloro(dioxoethylene-O, O’)tellurate (AS101) and Octa-O-bis-(R,R)-tartarate ditellurane (SAS, Fig. 24) are primarily caused by their specific Te(IV) redox-modulating activities enabling the inactivation of cysteine proteases such as cathepsin B, inhibition of specific tumor survival proteins like survivin, or obstruction of tumor IL-10 production (57).
Fig. 24.
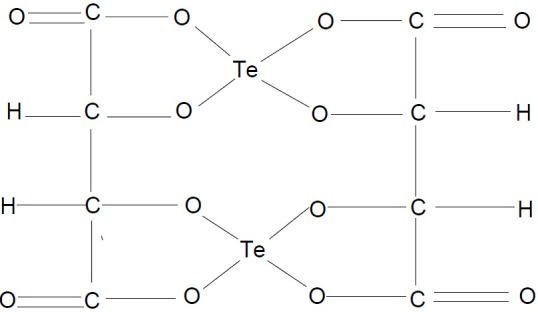
SAS
Multifunctional tellurium non-toxic molecule (ammonium trichloro(dioxoethylene-O, O’-) tellurate, AS101) protected dopaminergic neurons and improved motor function in animal models of Parkinson’s disease (58). AS101 induced PC12 differentiation and rescued the neurons from apoptotic death (59). AS101 has also been proved as a novel inhibitor of IL-1beta converting enzyme (60). Te (IV) compounds influenced thiol redox biological activity in the human body and represent a new class of anti-inflammatory compounds (61).
The specific TellurIV-thiol interaction enabled AS101 to interact with cysteine residues on both inflammatory and apoptotic caspases, resulting in their inactivation. AS101 downregulated IL-18 and IL-1beta serum levels in a mouse model of lipopolysaccharide (LPS)-induced sepsis, thereby, resulting in increased survival. Recent studies have emphasized the pathophysiologic role of IL-18 and IL-1beta in a variety of inflammatory diseases (62). The close chemical relationship between tellurium and sulfur also transcends into in vitro and in vivo situations and provided new impetus for the development of enzyme inhibitors and redox modulators, particularly, some of which may be of interest in the field of antibiotics and anticancer drug design.
Silicon
Silicon forms four tetrahedral bonds just like carbon does. From a lipophilicity point of view, silicon containing analogues are more lipophilic than their carbon analogues. Simethicone [Poly(dimethylsiloxane, silicon dioxide)], (Fig. 25) is an oral anti-foaming agent used to reduce bloating, discomfort and pain caused by excess gas in the stomach or intestinal tract. It is a mixture of polydimethyl siloxane and hydrated silica gel.
Fig. 25.
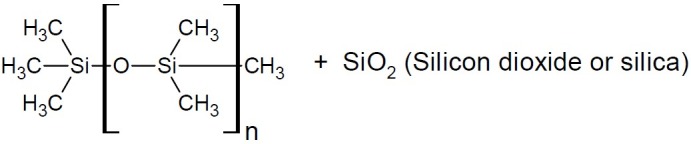
Dimethicone
Cisobitan, (Cyclic 2,6-cis-diphenylhexa methylcyclotetrasiloxane, (Fig. 26), is an organosilicon compound with estrogenic and antigonadotropic properties (63).
Fig. 26.
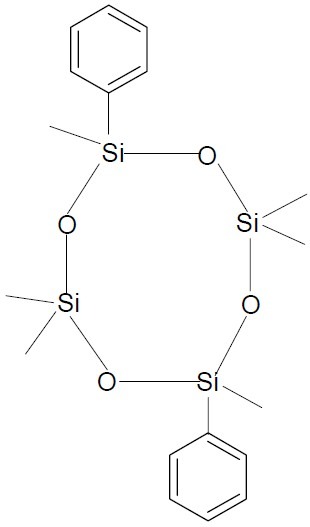
Cisobitan
Silicones are flexible compounds containing silicon-oxygen and silicon-carbon bonds and are widely used in applications such as artificial breast implants and contact lenses. ALIS 409 (1,3-dimethyl-1,3- p -fluorophenyl-1, 3(3- morfolinopropyl) -1, 3- disiloxan dihydrochloride, (Fig. 27) and ALIS 421 {1, 3-dimethyl -1, 3-(4- fluorophenyl) -1, 3[ 3(4-buthyl)- (1piperazinyl) -propyl] -1,3-disiloxan tetrahydrochloride} (Fig. 28), two water-soluble organosilicon compounds, have been shown to be effective in vitro multidrug-resistance reverting agents. The myorelaxing effect elicited either by ALIS 409 or by ALIS 421 was mainly due to the direct blockade of extracellular Ca2+influx. However, this effect was observed at concentrations much higher than those effective as modifiers of multidrug resistance in cancer cells (64,65).
Fig. 27.

ALIS 409
Fig. 28.
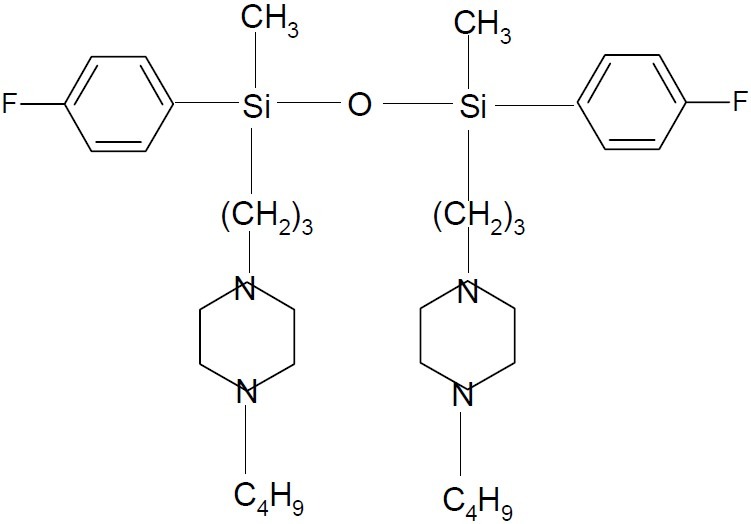
ALIS 421
The multidrug reversal effect of ALIS-409 was demonstrated in vivo without any apparent toxicity. In addition, it increased the apoptotic activity, exhibited some tumor growth delay, but did not affect the mitotic rate. This new organosilicon compound deserves further attention with a combination of multidrug-resistant substrate chemotherapeutic agents, especially in multidrug-resistant tumors (66).
The silicon switches haloperidol, fexofen-adine, bexarotene and venlafaxine drugs have been reported. The silicon-modified molecule exhibited far less toxicity than the all-carbon-based form of the drug (67). Mesoporous silicas was employed to fabricate three (3D)-dimensional scaffolds for bone tissue engineering. Further, peptides, hormones and growth factors were strongly grafted to the silica-based bioceramic matrix that acted as signals for bone cells and promoted bone regeneration process (68). Recently there have been some reports of organosilane-based protease inhibitors (69). A five-step methodology to prepare silanediol protease inhibitors (Fig. 29) has been reported (70).
Fig. 29.
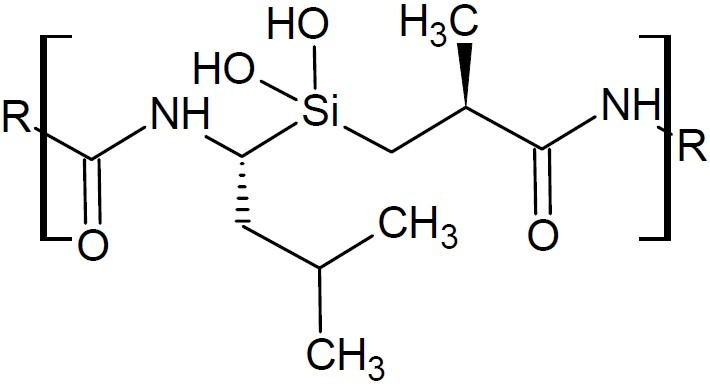
Silanediol protease inhibitor
applications of silica materials with regard to improving oral bioavailability and controlled drug delivery, multiparticulate systems for gastroretention or sustained release, composite xerogels, implant devices devoted to medical applications and in vivo biocompatibility have been reported (71–74). Nanostructures may be used to deliver drugs where they are required to avoid the harmful side effects. Preclinical and clinical data on nanostructured porous silicon provided evidence that such nanostructered particles deliver drugs at a prolonged controlled release to specific targets. Such studies offered new approaches to tackle hydrophobic and low bioavailable drugs that are being applied in a large range of healthcare settings (75).
The application of new and approved drugs to target validated pathways involves planned replacement of carbon with silicon within biologically prevalidated drug scaffolds. Such approach has generated focused libraries of pharmaceutically relevant agents with novel, durable and marketable intellectual property. In this context, in vitro testing against a human pancreatic cancer cell line, MiaPaCa-2, and a panel of 14 human multiple myeloma cell lines showed that silicon-indomethacin derivatives have demonstrated superior anticancer activity at clinically achievable concentrations (76).
CONCLUSION
Metalloids include a group of biologically important elements (boron, silicon, germanium, tellurium, arsenic and antimony) ranging from the essential to the highly toxic. Metalloid-containing drugs are used as chemotherapeutic agents to combat infectious diseases caused by pathogenic parasites as well as cancer, including acute promyelocytic leukemia. Metalloids accumulation in cells is required to work as a drug. Specific major intrinsic proteins could be potential pharmacological targets as these represent an ancient and indispensable transport mechanism for metalloids. It is essential to develop effective strategies to exclude toxic substances such as arsenic and antimony from the cell and to acquire their tolerance. In order to elucidate the mechanisms involved in metalloid tolerance in yeast and other organisms, S. cerevisiae AP-1-like proteins Yap1p and Yap8p mediated metalloid (arsenic and antimony) tolerance by activating transcription of distinct defense genes. Organ-osilicon compounds have a bright future as multidrug-resistance reverting agents and as protease inhibitors.
ACKNOWLEDGMENT
Support by PCTE Group of institutes is acknowledged.
REFERENCES
- 1.Hunter P. Not boring at all. Boron is the new carbon in the quest for novel drug candidates. EMBO Rep. 2009;10(2):125–128. doi: 10.1038/embor.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almond JB, Cohen GM. The proteasome: A novel target for cancer chemotherapy. Leukaemia. 2002;16:433–443. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]
- 3.Kohno J, Kawahata T, Otake T, Morimoto M, Mori H, Ueba N, et al. Boromycin, an anti-HIV antibiotic. Biosci Biotechnol Biochem. 1996;60:1036–1037. doi: 10.1271/bbb.60.1036. [DOI] [PubMed] [Google Scholar]
- 4.Dunitz JD, Hawley DM, Mikloš D, White DNJ, Berlin Y, Marušiæ R, et al. Structure of boromycin. Helvetica Chimica Acta. 1971;54:1709–1713. doi: 10.1002/hlca.19710540624. [DOI] [PubMed] [Google Scholar]
- 5.Abercrombie P. Vaginitis. In: Maizes V, Low Dog T, editors. Integrative Women's Health. New York, NY: Oxford University Press; 2010. p. 192. [Google Scholar]
- 6.Harvey SC. Antiseptics and disinfectants; Fungicides; ectoparasiticides. In: Gilman AG, Goodman LS, Gilman A, editors. Goodman and Gillman's: The Pharmacological Basis of Therapeutics. 6th ed. New York: McGraw-Hil; 1980. p. 971. [Google Scholar]
- 7.Jacobs RT, Plattner JJ, Keenan M. Boron-based drugs as antiprotozoals. Curr Opin Infect Dis. 2011;24:586–592. doi: 10.1097/QCO.0b013e32834c630e. [DOI] [PubMed] [Google Scholar]
- 8.Baker SJ, Ding CZ, Akama T, Zhang YK, Hernandez V, Xia Y. Therapeutic potential of boron-containing compounds. Future Med Chem. 2009 Oct;1(7):1275–1288. doi: 10.4155/fmc.09.71. [DOI] [PubMed] [Google Scholar]
- 9.Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21:838–842. doi: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez V. The Ritz Carlton. Philadelphia, PA: 2010. Jan 25-27, Structure-guided discovery of (S)-3-(aminomethyl) benzo [c] [1,2] oxaborol -1 (3H)- ol hydrochloride (ABX): A first in class gram-negative antibacterial; Anti-Infectives Summit. [Google Scholar]
- 11.Zhu Y, Zhao X, Zhu X, Wu G, Li Y, Ma Y, et al. Design, synthesis, biological evaluation, and structure−activity relationship (SAR) discussion of dipeptidyl boronate proteasome inhibitors, Part I: Comprehensive understanding of the SAR of á-amino acid boronates. J Med Chem. 2009;52:4192–4199. doi: 10.1021/jm9005093. [DOI] [PubMed] [Google Scholar]
- 12.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: A link to clinical adverse events. Clin Cancer Res. 2011;17:2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 13.Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem. 2008;51:1068–1072. doi: 10.1021/jm7010589. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez E, Li M, Steinberg JA, Wang C, Shen J, Bonavida B, et al. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib. Br J Haematol. 2010;148:569–581. doi: 10.1111/j.1365-2141.2009.08008.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Y, Wu G, Zhu X, Ma Y, Zhao X, Li Y, et al. Synthesis, in vitro and in vivo biological evaluation, and comprehensive understanding of structure-activity relationships of dipeptidyl boronic acid proteasome inhibitors constructed from â-amino Acids. J Med Chem. 2010;53:8619–8626. doi: 10.1021/jm1009742. [DOI] [PubMed] [Google Scholar]
- 16.Milo LR, Jr, Lai JH, Wu W, Liu Y, Maw H, Li Y, et al. Chemical and biological evaluation of dipeptidyl boronic acid proteasome inhibitors for use in prodrugs and pro-soft drugs targeting solid tumors. J Med Chem. 2011;54:4365–4377. doi: 10.1021/jm200460q. [DOI] [PubMed] [Google Scholar]
- 17.Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, et al. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole, for the potential treatment of onychomycosis. J Med Chem. 2006;49:4447–4450. doi: 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- 18.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crépin T, Zhou H, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 19.Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. [Google Scholar]
- 20.Baker SJ, Tomsho JW, Stephen J, Benkovic SJ. Boron-containing inhibitors of synthetases. Chem Soc Rev. 2011;40:4279–4285. doi: 10.1039/c0cs00131g. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs RT, Plattner JJ, Nare B, Wring SA, Chen D, Freund Y, et al. Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Future Med Chem. 2011;3:1259–1278. doi: 10.4155/fmc.11.80. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soriano-Ursúa MA, Mancilla-Percino T, Correa-Basurto J, Querejeta E, Trujillo-Ferrara JG. Give boron a chance: Boron containing compounds reach ionotropic and metabotropic transmembrane receptors. Mini Rev Med Chem. 2011;11:1031–1038. doi: 10.2174/138955711797247743. [DOI] [PubMed] [Google Scholar]
- 24.Sekhon BS. Boron neutron capture therapy: An overview. Pharmainfo.net. 2008;6(2) Available from: http://www.pharmainfo.net/reviews/boron-neutron-capture-therapy-overview . [Google Scholar]
- 25.Sekhon BS, Gandhi L. Medicinal uses of inorganic compounds-I. Resonance. 2006;11(4):75–89. [Google Scholar]
- 26.Sekhon BS, Gandhi L. Medicinal uses of inorganic compounds-II. Resonance. 2006;11(8):62–68. [Google Scholar]
- 27.Wittig A, Collette L, Appelman K, et al. EORTC trial 11001: Distribution of two 10B-compounds in patients with squamous cell carcinoma of head and neck, a translational research/phase 1 trial. J Cell Mol Med. 2009;13:1653–1665. doi: 10.1111/j.1582-4934.2009.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kankaanranta L, Seppälä T, Koivunoro H, Saarilahti K, Atula T, Collan J, et al. Boron neutron capture therapy in the treatment of locally recurred head-and-neck cancer: Final analysis of a phase I/II trial. Int J Radiat Oncol Biol Phys. 2012;82:e67–e75. doi: 10.1016/j.ijrobp.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 29.McCallum RI. Antimony in Medical History. Edinburgh, UK: The Pentland Press; 1999. [Google Scholar]
- 30.Frézard F, Martins PS, Barbosa MCM, Pimenta AMC, Ferreira WA, de Melo JE, et al. New insights into the chemical structure and composition of the pentavalent antimonial drugs, meglumine antimonate and sodium stibogluconate. J Inorg Biochem. 2008;102:656–665. doi: 10.1016/j.jinorgbio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Baiocco P, Colotti G, Franceschini S, Ilari A. Molecular basis of antimony treatment in leishmaniasis. Med Chem. 2009 Apr 23;52(8):2603–2612. doi: 10.1021/jm900185q. [DOI] [PubMed] [Google Scholar]
- 32.Sharma P, Perez D, Cabrera A, Rosas N, Arias JL. Perspectives of antimony compounds in oncology. Acta Pharmacol Sin. 2008;29:881–890. doi: 10.1111/j.1745-7254.2008.00818.x. [DOI] [PubMed] [Google Scholar]
- 33.Ge R, Sun H. Bioinorganic chemistry of bismuth and antimony: target sites of metallodrugs. Acc Chem Res. 2007;40:267–274. doi: 10.1021/ar600001b. [DOI] [PubMed] [Google Scholar]
- 34.Sundar S, Chakravarty J. Antimony toxicity. Int J Environ Res Public Health. 2010;7:4267–4277. doi: 10.3390/ijerph7124267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haldar AK, Sen P, Roy S. Use of antimony in the treatment of Leishmaniasis: Current status and future directions. Mol Biol Int. 2011;2011 doi: 10.4061/2011/571242. Article ID 571242, doi: 10.4061/2011/571242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musa A, Khalil E, Hailu A, Olobo J, Balasegaram M, Omollo R, et al. Sodium stibogluconate (SSG) & paromomycin combination compared to SSG for visceral leishmaniasis in East Africa: A randomised controlled trial. PLoS Negl Trop Dis. 2012 Jun;6(6):e1674. doi: 10.1371/journal.pntd.0001674. doi:10.1371/journal.pntd.0001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freitas-Junior LH, Chatelain E, Kim HA, Siqueira-Neto JL. Visceral leishmaniasis treatment: What do we have, what do we need and how to deliver it? Int J Parasitol.: Drugs and Drug Resistance. 2012;2:11–19. doi: 10.1016/j.ijpddr.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bienert GP, Schüssler MD, Jahn TP. Metalloids: essential, beneficial or toxic. Major intrinsic proteins sort it out? Trends Biochem. 2008;33:20–26. doi: 10.1016/j.tibs.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Biyani N, Mandal S, Seth C, Saint M, Natarajan K, Ghosh I, et al. Characterization of Leishmania donovani aquaporins shows presence of subcellular aquaporins similar to tonoplast intrinsic proteins of plants. PLoS One. 2011;6:e24820. doi: 10.1371/journal.pone.0024820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thayer JS. Teaching bio-organometal chemistry. I. The metalloids. 1977;54:604. doi: 10.1021/ed054p662. [DOI] [PubMed] [Google Scholar]
- 41.Milan S, Oscar B, Joan D. Spirogermanium: A new investigational drug of novel structure and lack of bone marrow toxicity. Invest New Drugs. 1983;1:225–234. doi: 10.1007/BF00208894. [DOI] [PubMed] [Google Scholar]
- 42.Lin MH, Hsu TS, Yang PM, Tsai MY, Perng TP, Lin LY. Comparison of organic and inorganic germanium compounds in cellular radiosensitivity and preparation of germanium nanoparticles as a radiosensitizer. Int J Radiat Biol. 2009;85:214–226. doi: 10.1080/09553000902748583. [DOI] [PubMed] [Google Scholar]
- 43.Sabbioni E, Fortaner S, Bosisio S, Farina M, Del Torchio R, Edel J, et al. Metabolic fate of ultratrace levels of GeCl 4 in the rat and in vitro studies on its basal cytotoxicity and carcinogenic potential in Balb/3T3 and HaCaT cell lines. J Appl Toxicol. 2010;30:34–41. doi: 10.1002/jat.1469. [DOI] [PubMed] [Google Scholar]
- 44.Lloyd NC, Morgan HW, Nicholson BK, Ronimux RS. The composition of Ehrlich’s salvarsan: resolution of a century-old debate. Angew Chem Int Ed Engl. 2005;44:941–944. doi: 10.1002/anie.200461471. [DOI] [PubMed] [Google Scholar]
- 45.Roy P, Saha A. Metabolism and toxicity of arsenic: A human carcinogen. Curr Sci. 2002;82:38–45. [Google Scholar]
- 46.Jahn TP. MIPS and their role in the exchange of metalloids. Springer: 2010. [PubMed] [Google Scholar]
- 47.Dilda PJ, Hogg PJ. Arsenic based drugs. Cancer Treatment Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 48.Chen SJ, Zhou GB, Zhang XW, Mao JH, de The H, Chen Z. From an old remedy to a magic bullet: Molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. 2011;117:6425–6437. doi: 10.1182/blood-2010-11-283598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yedjou C, Tchounwou P, Jenkins J, McMurray R. Basic mechanisms of arsenic trioxide (ATO)-induced apoptosis in human Leukemia (HL-60) cells. J Hematol Oncol. 2010;3:28. doi: 10.1186/1756-8722-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ralph SJ. Arsenic-based antineoplastic drugs and their mechanisms of action. MetBased Drugs. 2008;2008:260146. doi: 10.1155/2008/260146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodgers J, Jones A, Gibaud S, Bradley B, McCabe C, Barrett MP, et al. Melarsoprol cyclodextrin inclusion complexes as promising oral candidates for the treatment of human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1308. doi: 10.1371/journal.pntd.0001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hikita E, Arai M, Tanaka S, Onda K, Utsumi H, Yuan B, et al. Effects of inorganic and organic arsenic compounds on growth and apoptosis of human T-Lymphoblastoid Leukemia cells. Anticancer Res. 2011;31:4169–4178. [PubMed] [Google Scholar]
- 53.Prajapati V, Kale RK, Singh RP. Arsenic and its combinations in cancer therapeutics. Ther Del. 2011;2:793–806. doi: 10.4155/tde.11.51. [DOI] [PubMed] [Google Scholar]
- 54.Subbarayan PR, Lee K, Ardalan B. Arsenic trioxide suppresses thymidylate synthase in 5-FU-resistant colorectal cancer cell line HT29 in vitro re-sensitizing cells to 5-FU. Anticancer Res. 2010;30:1157–1162. [PubMed] [Google Scholar]
- 55.Lima CBC, Arrais-Silva WW, Oliveira RL, Cunha R, Giorgio S. A novel organotellurium compound (RT-01) as a new antileishmanial agent. Korean J Parasitol. 2009;47:213–218. doi: 10.3347/kjp.2009.47.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cunha RLOR, Gouvea IE, Juliano L. A glimpse on biological activities of tellurium compounds. Annals Braz Acad Sci. 2009;81:393–407. doi: 10.1590/s0001-37652009000300006. [DOI] [PubMed] [Google Scholar]
- 57.Sredni B. Immunomodulating tellurium compounds as anti-cancer agents. Semin Cancer Biol. 2012;22:60–69. doi: 10.1016/j.semcancer.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Sredni B, Geffen R, Duan W, Albeck M, Shalit F, Lander H, et al. Multifunctional tellurium molecule protects and restores dopaminergic neurons in Parkinson’s disease models. The FASEB J. 2007;21:1870–1883. doi: 10.1096/fj.06-7500com. [DOI] [PubMed] [Google Scholar]
- 59.Makarovsky D, Kalechman Y, Sonino T, Freidkin I, Teitz S, Albeck M, et al. Tellurium compound AS101 induces PC12 differentiation and rescues the neurons from apoptotic death. Ann N Y Acad Sci. 2003;1010:659–666. doi: 10.1196/annals.1299.120. [DOI] [PubMed] [Google Scholar]
- 60.Brodsky M, Yosef S, Galit R, Albeck M, Longo DL, Albeck A, et al. The synthetic tellurium compound, AS101, is a novel inhibitor of IL-1beta converting enzyme. J Interferon Cytokine Res. 2007;27:453–462. doi: 10.1089/jir.2007.0168. [DOI] [PubMed] [Google Scholar]
- 61.Brodsky M, Halpert G, Albeck M, Sredni B. The anti-inflammatory effects of the tellurium redox modulating compound, AS101, are associated with regulation of NFêB signaling pathway and nitric oxide induction in macrophages. J Inflammation. 2010;7:3. doi: 10.1186/1476-9255-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brodsky M, Yosef S, Galit R, Albeck M, Longo DL, Albeck A, et al. The synthetic tellurium compound, AS101, is a novel inhibitor of IL-1beta converting enzyme. J Interferon Cytokine Res. 2007;27:453–462. doi: 10.1089/jir.2007.0168. [DOI] [PubMed] [Google Scholar]
- 63.Alfthan O, Andersson L, Esposti PL, Fosså SD, Gammelgaard PA, Gjöres JE, et al. Cisobitan in treatment of prostatic cancer. A prospective controlled multicentre study. Scand J Urol Nephrol. 1983;17:37–43. doi: 10.3109/00365598309179778. [DOI] [PubMed] [Google Scholar]
- 64.Molnar J, Mucsi I, Nacsa J, Hevér A, Gyémánt N, Ugocsai K, et al. New silicon compounds as resistance modifiers against multidrug-resistant cancer cells. Anticancer Res. 2004;24:865–871. [PubMed] [Google Scholar]
- 65.Fusi F, Ferrara A, Zalatnai A, Molnar J, Sgaragli G, Saponara S. Vascular activity of two silicon compounds, ALIS 409 and ALIS 421, novel multidrug-resistance reverting agents in cancer cells. Cancer Chemother Pharmacol. 2008;61:443–451. doi: 10.1007/s00280-007-0488-6. [DOI] [PubMed] [Google Scholar]
- 66.Zalatnai A, Molnár J. Effect of SILA-409, a new organosilicon multidrug resistance modifier, on human pancreatic cancer xenografts. In Vivo. 2006;20:137–140. [PubMed] [Google Scholar]
- 67.Pooni PK, Showell GA. Silicon switches of marketed drugs. Mini Rev Med Chem. 2006;6:1169–1177. doi: 10.2174/138955706778560120. [DOI] [PubMed] [Google Scholar]
- 68.Vallet-Regí M, Colilla M, González B. Medical applications of organic-inorganic hybrid materials within the field of silica-based bioceramics. Chem Soc Rev. 2011;40:596–607. doi: 10.1039/c0cs00025f. [DOI] [PubMed] [Google Scholar]
- 69.Lukevics E, Ignatovich L. In: 14Si biological activity of organosilicon compounds, Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The use of metals in medicine. Gielen M, Tiekink E R, editors. John Wiley & Sons, Ltd; 2005. [Google Scholar]
- 70.Bo Y, Singh S, Duong HQ, Cao C, Sieburth SM. Efficient, enantioselective assembly of silanediol protease inhibitors. Org Lett. 2011;13:1787–1789. doi: 10.1021/ol2002978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simovic S, Ghouchi-Eskandar N, Sinn AM, Losic D, Prestidge CA. Silica materials in drug delivery applications. Curr Drug Discov Technol. 2011;8:269–726. doi: 10.2174/157016311796799026. [DOI] [PubMed] [Google Scholar]
- 72.Vivero-Escoto JL, Slowing II, Trewyn BG, Lin VS. Mesoporous silica nanoparticles for intracellular controlled drug delivery. Small. 2010;6:1952–1967. doi: 10.1002/smll.200901789. [DOI] [PubMed] [Google Scholar]
- 73.Quintanar-Guerrero D, Ganem-Quintanar A, Nava-Arzaluz MG, Piñón-Segundo E. Silica xerogels as pharmaceutical drug carriers. Expert Opin Drug Deliv. 2009;6:485–498. doi: 10.1517/17425240902902307. [DOI] [PubMed] [Google Scholar]
- 74.Bernik DL. Silicon based materials for drug delivery devices and implants. Recent Pat Nanotechnol. 2007;1:186–192. doi: 10.2174/187221007782360402. [DOI] [PubMed] [Google Scholar]
- 75.Kumar DS, Banji D, Madhavi B, Bodanapu V, Dondapati S, Sri AP. Nanostructured porous silicon - a novel biomaterial for drug delivery. Int J Pharm Pharmaceu Sci. 2009;1:8–16. [Google Scholar]
- 76.Gately S, West R. Novel therapeutics with enhanced biological activity generated by the strategic introduction of silicon isosteres into known drug scaffolds. Drug Development Res. 2007;68:156–163. [Google Scholar]