

As a consequence of their unique chemical, physical and biological properties, silyl-substituted arenes have attracted wide interest from the synthetic,[1] material science,[2] and pharmaceutical sectors.[3] Traditionally, silyl substituents have been introduced via addition of aryl lithium or magnesium reagents to silicon electrophiles, although this often necessitates the use of protecting groups.[4] Consequently, transition metal catalyzed cross couplings of aryl halides with disilanes or hydrosilanes have grown in popularity, especially if base-or nucleophile-sensitive functionality are present.[5] More recently, direct C-H functionalization[6] has emerged as a conceptually and economically attractive alternative for the direct silylation of arenes. It is evident, however, that challenges remain and the general utility of this strategy is restricted in many instances by, inter alia, (1) poor regioselectivity, (2) stringent structural requirements, (3) noncommercial/expensive reagents, (4) harsh reaction conditions (typically 120°–200°C), and/or (5) impractical ratios of arene to silicon reagent (10:1 to 60:1).[7–9] Herein, we report an efficient, regioselective protocol for the bipyridine-ligated, iridium-catalyzed[9] C-H functionalization/silyation of a wide variety of heteroarenes under comparatively mild conditions. Importantly, the reaction does not require protection of N-H groups and uses only a small excess (3 equiv) of inexpensive triethylsilane (eq 1).
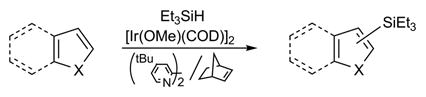 |
(1) |
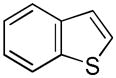
Indole 1 was selected as the model heteroarene for initial studies due to its prominence in many natural products and pharmaceuticals.[10] While several laboratories have cogently demonstrated arylation[11] and borylation[12] of indoles via transition metal catalyzed C-H functionalization, corresponding silylations are much less efficient (Figure 1). In an instructive example, Ishiyama et al. reported[13] unprotected 1 gave only complex product mixtures using the catalytic system [Ir(OMe)(COD)]2/4,4-di-tert-butyl-2, 2-bipyridine (dtbpy) in octane; silylated adducts could only be obtained using N-substituted indoles at elevated temperatures (120°C) and if a large excess of substrate was utilized. It was, thus, not surprising that 1 was unreactive to triethylsilane under comparable reaction conditions and could be recovered unchanged despite prolonged heating (Table 1, Entry 1). However, switching to THF as solvent and lowering the temperature to 80°C rewarded us with 2-(triethylsilyl)-1H-indole 2,[14] albeit in just 4% yield (Entry 2). This was then boosted to 15% upon addition of cyclopentene as a co-reactant (Entry 3). Optimization ultimately led to the more hindered olefin 2-norbornene[15] and a dramatically improved yield of 2 (Entry 4).[16,17] Notably, no 3-(triethylsilyl)-1H-indole was detected in the crude reaction mixture by 1H/13C NMR. Other solvents including octane (Entry 5), DME (Entry 6), and dioxane (Entry 7) were less satisfactory. Reducing the equivalents of Et3SiH and 2-norbornene from 5 equiv to 3 equiv had no effect on the yield or reaction rate (Entry 8), but further reductions in either reagent were detrimental to the yield (Entries 9–11).
Figure 1.
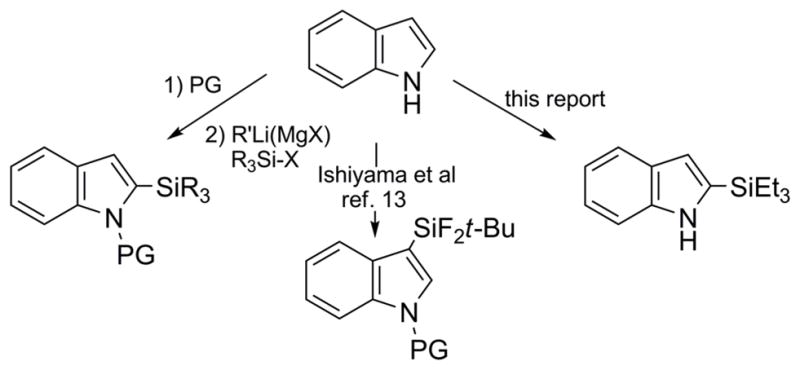
Comparison of silyl indole syntheses.
Table 1.
Reaction parameters for C-H functionalization of indole 1 to 2.[a]
| ||||
|---|---|---|---|---|
| Entry | Et3SiH (equiv) | Co-Reactant (equiv) | Solvent | Yield [%] |
| 1 | 5 | none | Octane | 0[b] |
| 2 | 5 | none | THF | 4 |
| 3 | 5 | Cyclopentene (5) | THF | 15 |
| 4 | 5 | 2-norbornene (5) | THF | 85 |
| 5 | 5 | 2-norbornene (5) | Octane | 22 |
| 6 | 5 | 2-norbornene (5) | DME | 49 |
| 7 | 5 | 2-norbornene (5) | dioxane | 59 |
| 8 | 3 | 2-norbornene (3) | THF | 87 |
| 9 | 1.5 | 2-norbornene (1.5) | THF | 15 |
| 10 | 3 | 2-norbornene (0.5) | THF | 7 |
| 11 | 1.5 | 2-norbornene (3) | THF | 43 |
Reaction conditions: 5 mol% [Ir(OMe)(COD)]2 and 10 mol% 4,4-di-tert- butyl-2,2-bipyridine (dtbpy) at 80°C for 24 h.
At 120°C for 24 h.
Having established suitable reaction conditions, it was of interest to explore the scope and generality of the methodology starting with substituted indoles (Table 2). Generally, silylations of indoles bearing electron-donating substituents proceeded well, e.g., 5-methoxy (3→4, Entry 1), 4-tert-butyldimethylsilyloxy (5→6, Entry 2), and 6-methyl (7→8, Entry 3). 3-Methylindole (9→10, Entry 4), on the other hand, reacted sluggishly, which we attribute to steric congestion, but continued the strict C(2)-regioselectivity found with the others of the series. Electron-withdrawing substituents were tolerated including 5-fluoro (11→12, Entry 5) and 7-chloro (13→14, Entry 6). More modest yields of silylated adducts were obtained with 5-bromo (15→16, Entry 7) and 4-cyano (17→18, Entry 8) which were accompanied by lesser amounts of unidentified byproducts; ca. 20% of the starting material was recovered.
Table 2.
C-H functionalization/triethylsilylation of substituted indoles.[a]
| Entry | Indole | Adduct | Yield [%] |
|---|---|---|---|
| 1 |
 3 |
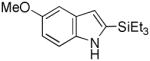 4 |
90 |
| 2 |
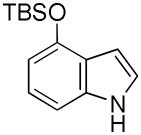 5 |
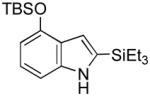 6 |
82 |
| 3 |
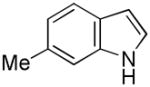 7 |
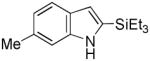 8 |
86 |
| 4 |
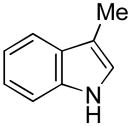 9 |
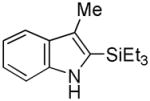 10 |
55[b] |
| 5 |
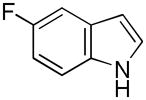 11 |
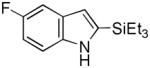 12 |
76 |
| 6 |
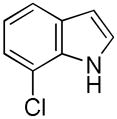 13 |
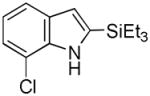 14 |
64 |
| 7 |
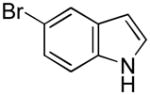 15 |
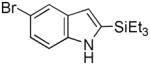 16 |
52[b] |
| 8 |
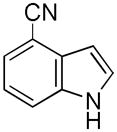 17 |
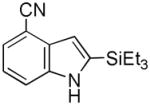 18 |
41[b] |
| 9 |
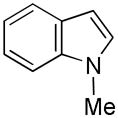 19 |
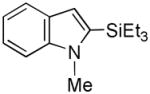 20 |
49 |
| 10 |
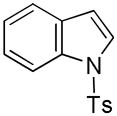 21 |
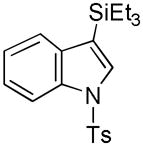 22 |
70 |
Reaction conditions: 5 mol% [Ir(OMe)(COD)]2, 10 mol% 4,4-di-tert- butyl-2,2-bipyridine (dtbpy), Et3SiH (3 equiv), and norbornene (3 equiv) in THF at 80°C for 24 h.
Same as [a], except 10 mol% [Ir(OMe)(COD)]2 and 20 mol% dtbpy for 36–48 h.
While the mechanistic details have yet to be elucidated, the observed regioselectivity is most consistent with a chelation-assisted reaction, i.e., coordination between the iridium catalyst and indole nitrogen followed by insertion into the adjacent C-H.[12a] As anticipated, N-methylindole (19→20, Entry 9) behaved similarly, although the increased steric hinderance around the nitrogen suppressed the yield somewhat (cf., Entry 4). Notably, tosylation of the ring nitrogen redirected the regioselectivity towards C(3)-silylation (21→22, Entry 10), suggesting a change in mechanism or metal migration from C(2) to C(3) as proposed for palladium-mediated indole C-H functionalizations.[6b]
It was gratifying to find S- and O-heteroarenes were also suitable substrates (Table 3). Thiophene (23) was smoothly functionalized to the 2,5-disilyl derivative (24) in excellent yield (Entry 1) when 4 equivalents each of triethylsilane and norbornene were used.[18] Not surprisingly, a 2-substituted thiophene underwent C(5)-mono-silylation in very good yield (25→26, Entry 2) whereas a 3-substituted thiophene gave rise to a 2:1 mixture of 5-triethylsilyl (67%) and 2,5-bis-silylated (33%) adducts (27→28, Entry 3). Benzofuran (31) and furan (33) gave analogous results (Entries 5 and 6, respectively).
Table 3.
C-H functionalization/triethylsilylation of S- and Oheteroarenes.[a]
| Entry | Heteroarene | Adduct | Yield [%] |
|---|---|---|---|
| 1 |
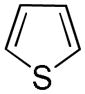 23 |
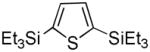 24 |
98[b] |
| 2 |
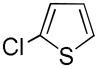 25 |
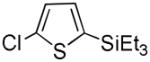 26 |
91 |
| 3 |
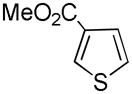 27 |
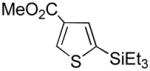 28 |
67[c] |
| 4 |
 29 |
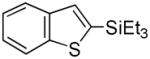 30 |
99 |
| 5 |
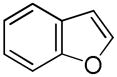 31 |
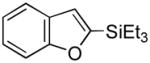 32 |
83 |
| 6 |
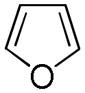 33 |
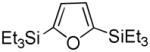 34 |
93 |
Reaction conditions described in Table 2, footnote [a].
Used triethylsilane (4 equiv) and norbornene (4 equiv).
2,5-bis-TES adduct obtained in 33% yield.
In summary, this report describes an efficient Ir-catalyzed C-H functionalization/silylation of N-/S-/O-heteroarenes including N-unsubstituted indoles under mild conditions and only a modest excess (3 equiv) of Et3SiH. The silylation is strongly promoted by 2- norbornene and features a high level of regioselectivity. Initial results indicate the general procedure described herein is also applicable to other triorganosilyl groups including those suitable for further transformations, e.g., 29 →35 (eq 2).[19]
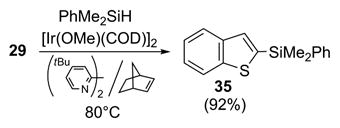 |
(2) |
Experimental section
General procedure
A flamed-dried Schlenk tube was charged with heteroarene (0.2 mmol), [Ir(OMe)(COD)]2 (6.6 mg, 0.01 mmol) and dtbpy (5.4 mg, 0.02 mmol), then evacuated and flushed with argon three times. Under a positive flow of argon, 2-norbornene (56 mg, 0.6 mmol) and dry THF (1 mL) were added. After stirring for 5 min, triethylsilane (98 μL, 0.6 mmol) was added dropwise and the reaction mixture was heated at 80°C for 24 h or indicated time. The solvent was concentrated under reduced pressure. The residue was purified by flash chromatography using silica gel to give 2-(triethylsilyl)heteroarene.
For 1-tosyl-indole, the 3-silylation product was obtained using the above general procedure.
For furan (0.2 mmol) and thiophene (0.2 mmol), 2-norbornene (75 mg, 0.8 mmol) and triethylsilane (130 μL, 0.8 mmol) were employed.
Footnotes
We thank the NIH (GM31278, DK38226, AI077853) and the Robert A. Welch Foundation for financial support.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Recent examples: Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. 2. De Meijere A, Diederich F, editors. Vol. 1. Wiley-VCH Verlag GmbH & Co; KGaA, Weinheim (Germany): 2004. pp. 163–216.Nakao Y, Sahoo AK, Imanaka H, Yada A, Hiyama T. Pure Appl Chem. 2006;78:435.Suzawa K, Ueno M, Wheatley AEH, Kondo Y. Chem Commun. 2006:4850. doi: 10.1039/b611090h.Pierrat P, Gros P, Fort Y. Org Lett. 2005;7:697. doi: 10.1021/ol047482u.Abele E, Lukevics E. Heterocycles. 2002;57:361.
- 2.a) Bai D, Han S, Lu ZH, Wang S. Can J Chem. 2008;86:230. [Google Scholar]; b) Kumagai T, Itsuno S. Macromolecules. 2002;35:5323. [Google Scholar]
- 3.Franz AK. Curr Opin Drug Dis Develop. 2007;10:654. [PubMed] [Google Scholar]
- 4.a) Nguyen TH, Castanet AS, Mortier J. Org Lett. 2006;8:765. doi: 10.1021/ol0530427. [DOI] [PubMed] [Google Scholar]; b) Hartung CG, Fecher A, Chapell B, Snieckus V. Org Lett. 2003;5:1899. doi: 10.1021/ol0344772. [DOI] [PubMed] [Google Scholar]
- 5.a) Denmark S, Kallemeyn JM. Org Lett. 2003;5:3483. doi: 10.1021/ol035288m. [DOI] [PubMed] [Google Scholar]; b) McNeill E, Barder TE, Buchwald SL. Org Lett. 2007;9:3785. doi: 10.1021/ol701518f. [DOI] [PubMed] [Google Scholar]; c) Hamze A, Provot O, Alami M, Brion JD. Org Lett. 2006;8:931. doi: 10.1021/ol052996u. [DOI] [PubMed] [Google Scholar]
- 6.a) Bergman RG. Nature. 2007;446:39. doi: 10.1038/446391a. [DOI] [PubMed] [Google Scholar]; b) Sergin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; d) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. [Google Scholar]; d) Kakiuchi F. In: Handbook of C-H Transformations. Dyker G, editor. Vol. 1. Wiley-VCH Verlag GmbH & Co; KGaA, Weinheim (Germany): 2005. pp. 131–137.pp. 265–266. [Google Scholar]
- 7.Ru catalysis: Kakiuchi F, Tsuchiya K, Matsumoto M, Mizushima E, Chatani N. J Am Chem Soc. 2004;126:12792. doi: 10.1021/ja047040d.Kakiuchi F, Matsumoto M, Tsuchiya K, Igi K, Hayamizu T, Chatani N, Murai S. J Organomet Chem. 2003;686:134.Kakiuchi F, Igi K, Matsumoto M, Hayamizu T, Chatani N, Murai S. Chem Lett. 2002:396.
- 8.Pt catalysis: Murata M, Fukuyama N, Wada JI, Watanabe S, Masyda Y. Chem Lett. 2007;36:910.Tsukada N, Hartwig JF. J Am Chem Soc. 2005;127:5022. doi: 10.1021/ja050612p.Uchimaru Y, El Sayed AMM, Tanaka M. Organometallics. 1993;12:2065.
- 9.Ir catalysis: Saiki T, Nishio Y, Ishiyama T, Miyaura N. Organometallics. 2006;25:6068.Ishiyama T, Sato K, Nishio Y, Miyaura N. Angew Chem Int Ed. 2003;42:5346. doi: 10.1002/anie.200352399.
- 10.Borschberg H-J. Curr Org Chem. 2005;9:1465. [Google Scholar]
- 11.a) Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; b) Deprez NR, Kalyani D, Krause A, Sanford MS. J Am Chem Soc. 2006;128:4972. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]; c) Grimster NP, Gauntlett C, Godfrey CRA, Gaunt MJ. Angew Chem Int Ed. 2005;44:3125. doi: 10.1002/anie.200500468. [DOI] [PubMed] [Google Scholar]; d) Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578. doi: 10.1021/ja035054y. [DOI] [PubMed] [Google Scholar]
- 12.a) Paul S, Chotana GA, Holmes D, Reichle R, Maleczka PE, Smith MR. J Am Chem Soc. 2006;128:15552. doi: 10.1021/ja0631652. [DOI] [PubMed] [Google Scholar]; b) Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]; c) Ishiyama T, Nobuta Y, Hartwig JF, Miyaura N. Chem Comm. 2003:2924. doi: 10.1039/b311103b. [DOI] [PubMed] [Google Scholar]; d) Ishiyama T, Miyaura N. Pure Appl Chem. 2006;78:1369. [Google Scholar]
- 13.Ishiyama T, Sata K, Nishio Y, Saiki T, Miyaura N. Chem Comm. 2005:5065. doi: 10.1039/b511171d. [DOI] [PubMed] [Google Scholar]
- 14.Alternative synthesis of 2-triethylsilylindole: Kamikawa K, Kinoshita S, Furusyo M, Takemoto S, Matsuzaka H, Uemura M. J Org Chem. 2007;72:3394. doi: 10.1021/jo0700427.
- 15.Other unsaturated molecules, e. g., tetramethylethylene, 1-hexene, 1-heptyne, and diisopropyl azodicarboxylate, had no effect on the yield of 2.
-
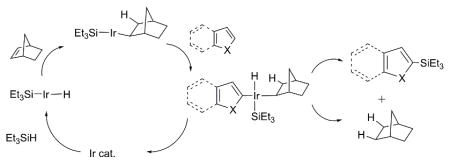
16.For prior use of 2-norbornene in catalytic C-H activations, see references 7a,c. The exact role of 2-norbornene as a promotor of the Ir-catalyzed reactions reported herein is unclear. The identification of norbornane by GC-MS in the reaction mixture suggests it functions as a sink for the hydrogen generated during the reaction. Additionally, it may play a more intimate role as a participant in the catalytic cycle. We speculate norbornene’s bicyclic skeleton affords greater steric shielding and/or stability towards unproductive β-eliminations than comparable Ir-complexes with cyclic or acyclic olefins. Unfortunately, attempts to detect Ir-norbornene intermediates by NMR were disappointing.
- 17.Equivalent reactions of 1 in THF at 80°C for 24 h using Et3SiH (3 equiv), 2-norbornene (3 equiv), and Ru3(CO)12, [CpRhCl2]2, or [RhCl(COD)]2, instead of [Ir(OMe)(COD)]2/4,4-di-tert-butyl-2,2-bipyridine, produced little, if any, 2.
- 18.1.5 equivalents of Et3SiH/1.5 equivalents of 2-norbornene provided 24 (8%) and 2-triethylsilylthiophene (23%). Both the yield and ratio of 24/2-triethylsilylthiophene increased progressively as the number of equivalents of Et3SiH increased from 1.5 to 4.0.
- 19.Tamao-Kumada-Fleming oxidation review: Mullins RJ, Jolley SL, Knapp AR. In: Name Reactions for Functional Group Transformations. Li JJ, Corey EJ, editors. John Wiley & Sons, Inc; Hoboken (USA): 2007. pp. 237–247.