

Abstract
Purpose of review
Preclinical research in epileptology has been very successful in producing effective drugs. Unfortunately, however, seizures are still not adequately controlled in a third of the affected individuals, and comorbidities still impose a major burden on quality of life. New preclinical and clinical drug development strategies are needed to identify drugs that target these unmet medical needs.
Recent findings
Even in recent years, the antiseizure approach based on screenings has contributed to the identification of new drugs. Thus, it should not be abandoned. However, we propose that a radically new approach, specifically designed to tackle the existing gaps in care, should be developed to complement the traditional screening. This new approach will require integrated strategies for preclinical screening and experimental trial design. In this review, we will attempt to address some of the issues that must be resolved to engage this effort. Are there suitable models to tackle the unmet therapeutic needs in epilepsy? Are there ways de-risk the transition from pre-clinical to clinical studies? Are there ways to improve the efficiency of clinical trials and to design ad hoc trials for the unmet therapeutic needs?
Summary
Development and validation of a new, integrated strategy for anti-epilepsy drug development is needed to identify truly innovative drugs.
Keywords: Anti-seizure drug, Anti-epileptogenesis, Disease modification, Co-morbidity, Biomarker
INTRODUCTION
Preclinical research in epileptology has been very successful in producing effective drugs. Since the identification of phenytoin using maximal electroshock (MES) and, subsequently, of trimethadione using subcutaneous pentylenterazole (PTZ), these predictive seizure models have been used for decades, allowing the development of many effective drugs that prevent seizures in approximately 70% of the patients. However, while all epileptic syndromes share the common symptom of seizures, they dramatically differ in terms of etiology, other symptomatology, natural history, sensitivity to available drug treatments, and all available AED are symptomatic agents that may control the seizures but do not modify the disease [1, 2▪ ▪]. Thus, we will avoid here the commonly used term anti-epileptic drugs (AED) and, to describe drug effects, we will use instead the words “antiseizure” for symptomatic effects, “antiepileptogenic” for prevention and cure, and “antiepilepsy” as a general term [2▪ ▪].
Screening programs based on the classic acute seizure models (MES and PTZ), more recently integrated with other models like kindling and 6 Hz electrical stimulation, continue to identify new antiseizure drugs. For example, the anticonvulsant screening program (ASP) of the American National Institutes for Neurological Disorders and Stroke (NINDS) have tested more than 30,000 potential drugs, identifying or contributing to the identification of at least nine new drugs [3]. Availability of these new drugs have expanded the therapeutic armamentarium and offered alternatives in patients suffering side effects from previous drugs, but have not significantly decreased the proportion of patients with drug-resistant epilepsy [1]. This suggests that, while current screening approaches remain valuable and may continue to identify drugs in the future, the newly found drugs are likely to have an incremental value, with improved tolerability and pharmacokinetics profiles, but little additional benefit for drug-resistant patients. Moreover, they will not tackle other crucial therapeutic gaps in care for epilepsy, namely: disease modification (none of the existing AED can alter the natural history of epilepsy or prevent development of epilepsy in at-risk patients); comorbidities (many patients with epilepsy also suffer other diseases like depression, cognitive impairment, anxiety, that are not attenuated by AEDs); targeted and optimized intervention on specific epileptic subtypes [2–4]. Another important consideration is the frequency of failures in the clinical development. The success rate of phase III clinical studies for AED has gradually declined to about 10%. As a result there is increasing skepticism regarding the possibility of identifying truly transformational new drugs for epilepsy using conventional screening and concern of the high risk associated with the clinical phase, with the interest of pharmaceutical industries in embarking in development of new AED reduced dramatically in the past few years.
Altogether, these considerations suggest that, while the traditional antiseizure approach based on screenings should not be abandoned, a radically new approach should be developed to complement it. To pursue this goal, however, a series of critical questions must be addressed. Are there suitable models to tackle the unmet therapeutic needs (disease modification, pharmaco-resistance, comorbidities, targeted intervention)? Are these models sufficiently validated and suitable for drug identification? Can we de-risk the transition from pre-clinical to clinical studies, preventively identifying putative drugs that would fail in phase III? Are there ways to improve the efficiency of clinical trials and to design ad hoc trials for the unmet therapeutic needs?
In this article, we will briefly address these questions and propose specific solutions that we believe may improve the situation. This review is based on the work of a joint ILAE-AES task force for the optimization of preclinical epilepsy therapy discovery and, in particular, on the results of the discussion in a two-day workshop held in London in September 2012.
ANIMAL MODELS TO DEVELOP DRUGS FOR RELEVANT THERAPEUTIC NEEDS AND ISSUES RELATED TO THEIR USE
Preclinical epilepsy research has the privilege of hosting the largest number of animal models among neurological disciplines. Yet, this also reflects the multivariate types of seizures, epilepsies and pathogenetic mechanisms. With the possible exception of certain genetic or inbred models, most of the available animal models of seizures are induced. Induction methods include (a) focal or systemic exposure to drugs that activate excitatory or inhibit inhibitory signaling systems and have been used to model status epilepticus (SE) and post-SE epilepsy (kainic acid, pilocarpine), clonic seizures (flurothyl, PTZ) or absence seizures (low dose PTZ); (b) electrical stimulation models (electroshock or kindling); (c) physical models that reproduce specific epilepsy-related insults (traumatic brain injury, hypoxia-ischemia). Furthermore, models of conditions predisposing to epilepsy (febrile seizures, methylazoxymethanol acetate-induced dysplasias) exist. Rodents are the most popular species although lower (i.e. drosophila) or higher species (dogs, rabbits, primates) have been utilized. Despite the significant preclinical advances, at a time when the goals of epilepsy care are shifting from stopping seizures to curing epilepsy and its comorbidities, it may be worth discussing ways to improve translation into clinically relevant applications.
Preclinical models generate relatively homogeneous populations of animals in regards to etiology, pathology and seizure types. This approach minimizes the sample size and costs of the study, but cannot reproduce the variable presentations of human epilepsy syndromes. Also, the induction methods are more commonly experimental than naturally encountered in human patients. Validation of a drug’s efficacy across models and etiologies, where possible, might perhaps help better determine whether the treatment effect is etiology (model) specific or might target a common pathogenetic pathway of the studied seizure or epilepsy type.
In most acute models of seizures, drugs are given prior to seizure induction. Preemptive antiseizure treatment is not usually, however, possible in the clinical practice. This creates a gap in translation, since the efficacy of a drug is often tightly connected with the timing of its administration. Benzodiazepines are more effective if given early in the course than if given during established SE, because GABAA receptors are internalized [5]. Testing therefore a drug’s efficacy by delivering it after seizure onset and providing the therapeutic time window of administration may better guide subsequent clinical trials.
Animal models of acute seizures are typically done on otherwise naïve animals, representing therefore models of “first seizure”. This may be fine for projects aiming to develop better therapies for SE, which need better therapies even at first occurrence, but not sufficient to identify better antiseizure drugs for patients with recurrent or drug-resistant seizures. Prior seizures or exposure to antiseizure drugs may alter the expression of neurotransmitter systems, the connectivity patterns in the affected areas, but also the accessibility of drugs to the epileptogenic focus due to the underlying pathology or the expression of multidrug transporters [6–9]. These issues can to some extent be overcome by testing the antiseizure effect on spontaneous seizures using models of chronic epilepsy or of drug resistant seizures.
Chronic models of epilepsies are an excellent setting to test antiepileptogenesis and disease-modifying treatments, albeit these are not available for all epilepsy syndromes. Such studies require robust sample sizes and long periods of observation, including after treatment washout, and extensive testing with continuous video-EEG and/or additional behavioral endpoints. As in humans, spontaneous seizures in animals are known to cluster or show progressively increased frequencies with time or have variable inter-seizure intervals which may make data analysis cumbersome [10]. Treatment effects may vary according to the timing of administration, dose, model, species, and measured outcome, as shown for rapamycin [11–13]. Due to the significant time and cost requirements for preclinical and clinical studies of antiepileptogenesis and disease modification, it is imperative to obtain early on guidance for therapeutic time window, target mechanism relevance in the target population, objective measures of target engagement, and clinically relevant measures of success, so as to minimize the chance of failure to translate in the clinical trials.
Early life epilepsies hold a special place among epilepsies in need for better therapies as they leave lifelong sequelae, both because of poor epilepsy therapies and of the associated comorbidities. Treatments developed in adult models may not have the same efficacy of tolerability in early life seizures or epilepsies [14–16]. A number of animal models of early life epilepsies are emerging, such as models of infantile spasms [17, 18], yet more needs to be done.
Animal models have been and will be invaluable in drug discovery, even though species and model-related differences may hinder translation into clinically successful interventions. Identification of clinically relevant biomarkers of treatment relevance, implementation, and success might significantly help filter out the possible winner drugs, and de-risk the transition to clinical trials.
DE-RISKING THE TRANSITION FROM PRE-CLINICAL TO CLINICAL DRUG DEVELOPMENT
There is increasing concern from industry and other funders of drug development that results of pre-clinical drug testing studies are often not replicable on independent testing, including in-house testing by industry [19, 20]. Furthermore there is concern that the results of preclinical studies often fail to translate into positive results in clinical trials [19–21]. The failure of clinical trials is very expensive and provides a major disincentive for further development in the field. There are likely multiple reasons for the failure of replication and translation, but methodological issues such as inadequately powered studies, publication and other biases, and lack of study design rigor likely play a role [19, 20].
The Workshop proposed a potential solution to address this problem by implementing a “Phase II” multi-center pre-clinical trial paradigm. This paradigm would be modeled on the methodology used for double-blinded multicenter clinical trials. Phase II trials would be adequately powered with a larger number of animals studied than single lab studies can undertake. This process would reduce biases related to individual laboratory practices and conditions, implement rigorous blinding and statistical design, and incorporate independent monitoring of data collection and analysis.
Current pre-clinical drug testing usually involves a relatively small number of animals from one or few laboratories using the particular animal models in which they have experience and expertise. While we believe that this should remain as the mainstay of drug discovery, Phase II trials would represent a second optional state of testing that aims to improve the evidence base for efficacy prior to proceeding to expensive clinical trials. A candidate therapy moving into Stage II multicenter preclinical studies could either be a “pure” proof of concept compound or therapy or a “true” preclinical drug candidate compound (i.e. one that has potential to ultimately be a therapeutic drug in clinical practice).
It is envisaged that 5–20 laboratories would participate in a particular Phase II pre-clinical trial, with 2–5 different models used, with more than one model for each participating group. The choice of the models to be used would be selected on the basis that they have readily quantifiable efficacy endpoints that relate to specific syndromes and gaps in clinical care (i.e. drug resistant seizures, anti-epileptogenesis, disease modification or co-morbidities). The numbers of animals per study would be based on power calculation from Stage I studies performed by biomedical statisticians with experience in clinical trial design. This is estimated to likely be between 40–200, with the number of subjects required to be studied likely to be less than in clinical studies because of the less variability (i.e. noise) in the experimental models, study conditions and endpoint assessments than with clinical studies (e.g. measuring seizures with continuous EEG recordings rather than patient reported seizures). Phase II studies would be pre-registered as is currently done for clinical trials, to reduce the potential for publication bias [21].
There would be a central coordinating site for each study, which should be independent from the data collections sites. Data collected at individual sites could be analyzed (blinded to treatment arm) in at least part by the local sites for relevant endpoints (e.g. seizure quantification, neuro-behavioral testing). The raw and/or analyzed data would be transmitted to the central coordinating site for any higher level analysis and pooling with data from other sites for the final data analysis. There would also be independent monitoring of data collection and analysis, as currently occurs with multicenter clinical trials. Standards for reporting the outcomes of these trials would be adopted [19].
While not the primary endpoints of the Phase II studies, ideally some toxicity and PK data could be collected during these studies. This would involve obtaining sufficient PK measurements to ensure that the parent compound reaches the target and drive the pharmacological read outs – in particular in models or paradigms necessitating chronic administration. Conventional chronic toxicity testing would not be necessary but the efficacy experiments should be associated with a behavioral assessment of adverse effects.
It is recognized that these Phase II multicenter pre-clinical studies will be expensive, and resource and time intensive, compared with traditional pre-clinical studies. However the cost is likely to be significantly less that of failed Phase II/III clinical studies, and therefore overall should prove to be cost-effective. The funding model will likely require a combination of government funding and industry funding. The government funding will establish the basic structures, protocols, laboratory credentialing, databases etc. Industry or venture capital would fund the primary costs of undertaking the study, potentially supplemented by grants from government and philanthropy. Participating investigators would receive “investigator payments” to cover the costs of undertaking the studies in the laboratories, and appropriate infrastructure costs. The intellectual property for the compounds would remain owned by the “sponsor” of the compound.
IMPROVED DESIGN OF CLINICAL TRIALS AND DESIGN OF SPECIFIC CLINICAL TRIALS FOR THE THERAPEUTIC NEEDS
Once a new chemical entity is identified and validated, it will go through preclinical assessment to ensure that it is likely to be safe, and that the pharmacological properties (eg half-life, route of elimination, drug interactions) are acceptable. This will be followed by testing in human volunteers. Subsequently, clinical trials must be performed to determine whether the compound will be efficacious in the condition for which it will be used. At present, anti-seizure drugs typically undergo efficacy trials that assess their impact over a 3 month evaluation period when added on to the patient’s existing regimen, in comparison to placebo (Figure 1). These trials are usually done in treatment resistant patients who are having very frequent partial-onset seizures [22].
Figure 1.
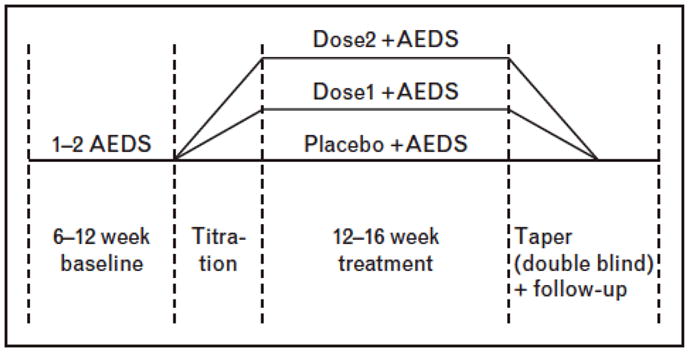
Typical placebo-controlled add-on trial of new anti-seizure drugs in treatment-resistant focal epilepsy.
This standard trial design has been effective in supporting the approval of a score of AEDs over the last 2 decades, and in that sense has been very successful. Yet, it is quite likely that there will be changes needed for future trials. These are necessitated on a number of grounds. The first is that the feasibility of the present design is in question. The design requires patients to possibly receive placebo for up to three months, despite the fact that they maybe experiencing a large number of seizures. This was acceptable when there were a few antiepileptic drugs available to patients. In contrast, when there are up to 17 drugs available, patients do not want to wait three months before they are to be tried on the next potentially successful therapy. This has made recruitment to these trials challenging. Moreover, recent data suggests that patients remaining in an add on placebo arm of a randomized controlled trial may have an increased risk of sudden unexplained death [23]. This raises the possibility that this trial design may pose potential danger to patients. For these reasons, new trial designs that reduce exposure to placebo are optimal, and are being considered. These might take the form, for example, of “time to nth seizure”, a design which allows the patient to exit from the study after they have experienced sufficient seizures to determine that the study intervention (whether active drug or placebo) has failed [24].
Current epilepsy trials are not set up to determine whether one drug is superior to another, as all drugs are compared to placebo. Differences in effect size from trial to trial could be a result of minor differences in trial design, outcome measures, or patient population enrolled. In the future, it is to be hoped that new therapies will actually be substantial improvements over available agents. If this is the case, current treatment options could be used as a comparison instead of placebo, with the expectation that the investigational therapy would demonstrate superiority. There are issues inherent to such “active control comparisons”, which are currently being addressed through modified trial designs [25].
In the future, it is to be hoped that in addition to anti-seizure agents that are currently in use, novel therapeutics will also be developed that have either disease modifying or anti-epileptogenic properties. These properties would not be uncovered through standard trial designs. At present, epilepsy prevention trials are typically done in patients after traumatic brain injury. However, other trial populations (for example post refractory status epilepticus, encephalitis or stroke) need to be explored [26]. There is also an urgent need for new trial designs to assess disease modification. No such designs currently exist.
CONCLUSIONS
The traditional screening approach and the current clinical trial design have been successful in identifying effective AED and should not be abandoned. Nonetheless, these approaches are not capable to tackle crucial therapeutic gaps in care for epilepsy like drug-resistance, disease modification, co-morbidities. We believe that the development of new, truly innovative anti-epileptic drugs will require an intense effort in multiple directions: employment of new epilepsy models; rigorous experimental design; new kinds of clinical trials. To ensure optimal translation of preclinical findings and to de-risk costly development efforts, these efforts should be orchestrated in an integrated manner, involving both preclinical and clinical scientists. One essential tool will be the identification of biomarkers or surrogate endpoints that reliably predict the efficacy of a potential AED without the need to wait for another seizure (antiseizure effects in diagnosed patients) or of spontaneous seizures (anti-epileptogenic effect) [27]. Predictive, noninvasive biomarkers would facilitate development of anti-seizure and anti-epileptogenic compounds as well as clinical trials, reducing their costs.
To help pursuing these efforts, a Cochrane-like collaboration that publishes systematic reviews of preclinical data would be very useful. Such reviews should address appropriateness and best utilization of animal models, tools for behavioral or outcome assessment, methodological design, and critical, comparative evaluation of the preclinical efficacy data for specific seizure types or syndromes. For these systematic reviews, it will be essential to access negative as well as positive data, and therefore it will also be essential to provide a forum to publish negative studies.
Acknowledgments
The support of the International League Against Epilepsy (ILAE), American Epilepsy Society (AES), Citizens United for Research in Epilepsy (CURE), Autism Speaks, and Epilepsy Therapy Project for the London workshop on optimization of preclinical epilepsy therapy discovery is gratefully acknowledged by all authors. We would also like to acknowledge the participants of the London workshop for their valuable input.
A.S.G. has received research grant support by NINDS NS078333, NS020253 and Autism Speaks, the Heffer Family Foundation, and the Siegel family Foundation.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪ ▪ of outstanding interest
- 1.Loscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–678. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 2▪ ▪.Galanopoulou AS, Buckmaster PS, Staley KJ, et al. Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia. 2012;53:571–582. doi: 10.1111/j.1528-1167.2011.03391.x. This is a review that provides a comprehensive overview of the major issues related to the development of new anti-epileptic therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The NIH/NINDS Anticonvulsant Screening Program (ASP) Recommendations from the working group’s 2012 review of the Program. Epilepsia. 2012;53:1837–1839. doi: 10.1111/j.1528-1167.2012.03641.x. [DOI] [PubMed] [Google Scholar]
- 4.Simonato M, Löscher W, Cole AJ, et al. Finding a better drug for epilepsy: Preclinical screening strategies and experimental trial design. Epilepsia. 2012 doi: 10.1111/j.1528-1167.2012.03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodkin HP, Sun C, Yeh JL, et al. GABA(A) receptor internalization during seizures. Epilepsia. 2007;48 (Suppl 5):109–113. doi: 10.1111/j.1528-1167.2007.01297.x. [DOI] [PubMed] [Google Scholar]
- 6.Brooks-Kayal AR. Rearranging receptors. Epilepsia. 2005;46 (Suppl 7):29–38. doi: 10.1111/j.1528-1167.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 7.Isaeva E, Isaev D, Khazipov R, et al. Long-term suppression of GABAergic activity by neonatal seizures in rat somatosensory cortex. Epilepsy Res. 2009;87:286–289. doi: 10.1016/j.eplepsyres.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pekcec A, Unkrüer B, Stein V, et al. Over-expression of P-glycoprotein in the canine brain following spontaneous status epilepticus. Epilepsy Res. 2009;83:144–151. doi: 10.1016/j.eplepsyres.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Eyal S, Lamb JG, Smith-Yockman M, et al. The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat liver. Brit J Pharmacol. 2006;149:250–260. doi: 10.1038/sj.bjp.0706830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek FE, Staley KJ. The time course of acquired epilepsy: implications for therapeutic intervention to suppress epileptogenesis. Neuroscience Lett. 2011;497:240–246. doi: 10.1016/j.neulet.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 11.Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raffo E, Coppola A, Ono T, et al. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–329. doi: 10.1016/j.nbd.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bittigau P, Sifringer M, Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann NY Acad Sci. 2003;993:103–114. doi: 10.1111/j.1749-6632.2003.tb07517.x. [DOI] [PubMed] [Google Scholar]
- 15.Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–2189. doi: 10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 16.Mackay MT, Weiss SK, Adams-Webber T, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62:1668–1681. doi: 10.1212/01.wnl.0000127773.72699.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chudomelova L, Scantlebury MH, Raffo E, et al. Modeling new therapies for infantile spasms. Epilepsia. 2010;51 (Suppl 3):27–33. doi: 10.1111/j.1528-1167.2010.02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coppola A, Moshe SL. Animal models. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. Amsterdam: Elsevier; 2012. pp. 63–98. [DOI] [PubMed] [Google Scholar]
- 19.Landis SC, Amara SG, Asadullah K, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 21.Kimmelman J, Anderson JA. Should preclinical studies be registered? Nat Biotechnol. 2012;30:488–489. doi: 10.1038/nbt.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter RJ, Baulac M, Nohria V. Clinical development of drugs for epilepsy: a review of approaches in the United States and Europe. Epilepsy Res. 2010;89:163–175. doi: 10.1016/j.eplepsyres.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol. 2011;10:961–968. doi: 10.1016/S1474-4422(11)70193-4. [DOI] [PubMed] [Google Scholar]
- 24.Friedman D, French JA. Clinical trials for therapeutic assessment of antiepileptic drugs in the 21st century: obstacles and solutions. Lancet Neurol. 2012;11:827–834. doi: 10.1016/S1474-4422(12)70177-1. [DOI] [PubMed] [Google Scholar]
- 25.French JA, Kryscio RJ. Active control trials for epilepsy: avoiding bias in head-to-head trials. Neurology. 2006;66:1294–1295. doi: 10.1212/01.wnl.0000218836.23697.f9. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt D. Is antiepileptogenesis a realistic goal in clinical trials? Concerns and new horizons. Epileptic Disord. 2012;14:105–113. doi: 10.1684/epd.2012.0512. [DOI] [PubMed] [Google Scholar]
- 27.Engel J., Jr Biomarkers in epilepsy: foreword. Biomarkers Med. 2011;5:529–530. doi: 10.2217/bmm.11.63. [DOI] [PubMed] [Google Scholar]