

Abstract
A biocatalytic platform that employs the final two monomodular type I polyketide synthases (PKS) of the pikromycin pathway in vitro followed by direct appendage of D-desosamine and final C-H oxidation(s) in vivo was developed and applied toward the synthesis of a suite of 12-and 14-membered ring macrolide natural products. This methodology delivered both compound classes in thirteen steps (longest linear sequence) from commercially available (R)-Roche ester in >10% overall yields.
Keywords: Chemoenzymatic Synthesis, Biocatalysis, Pikromycin, Macrolides, Polyketide Synthase
Introduction
Biocatalysis offers tremendous potential to complement conventional methods in synthetic chemistry.1 Traditionally used for discrete chemical reactions, recent chemoenzymatic advances have enabled cascades that streamline synthesis by reducing step counts, increasing yields, and minimizing waste generation.2–3 While such processes are becoming increasingly prevalent, they are typically employed in the production of relatively simple synthons for starting material or later incorporation into mature molecules. Conversely, the biosynthetic machinery involved in the generation of complex secondary metabolites could theoretically be leveraged at late stages of a synthesis to achieve similar improvement in step counts and overall yields. Specifically, modular type I polyketide synthases (PKSs) are of interest as a single polypeptide chain is capable of performing multiple regio-and stereospecific reactions while stabilizing otherwise labile oligoketide substrates.
Pikromycin (1) is a PKS derived macrolide antibiotic belonging to the ketolide subclass, originally isolated in 1950.4–5 The subsequent discovery of erythromycin A (2) and its success as a broad spectrum antibiotic initiated drug discovery efforts through isolation and semi-synthetic modification,6 with 3rd generation macrolides treating front line infections almost sixty years later. Through on-going strain improvement programs, fermentation-derived erythromycin provides the starting material for a number of clinically relevant antibiotics including 2nd generation clarithromycin, azithromycin and the 3rd generation ketolides telithromycin (4), cethromycin, and solithromycin. Despite the availability of erythromycin as a commodity finechemical, rigorous structure activity relationship (SAR) studies of the ketolide class are constrained by the inherent limitations of semi-synthesis using this complex natural product. For example, methods to directly remove, add, or modify alkyl groups in the macrolactone ring remain elusive with current synthetic technology. In contrast, de novo production of ketolides though total synthesis offers the opportunity to explore previously inaccessible chemical space for discovery of new anti-infective agents.7–10 At present, this remains a formidable goal as conventional synthetic approaches understandably suffer from practical limitations, even in the case of natural macrolides or their simplified macrolactone11–12 skeletons. Total syntheses have been previously reported for erythromycin13–15 and pikromycin,16 however, the innate complexity of these compounds requires high step counts, resulting in low overall yields. An underexplored route to macrolactone scaffolds employs PKS enzymes in a controlled in vitro environment. Development of biocatalytic approaches using modular PKSs has the potential to provide ready access to natural macrolactones, and increase our functional understanding of these complex proteins toward creating libraries of novel macrolides in an efficient manner.
Bacterial PKS pathways mediate the extension and processing of polyketide chains in a stepwise stereospecific manner, typically adding two (acetate) to three (propionate) carbon atoms per module.17 In the case of propionate extension, methylmalonyl-coenzyme A (MM-CoA, 5) serves as a masked enolate of the extender unit. The acyl transferase (AT) domain selects for methylmalonate and transfers this subunit to the phosphopantetheine (Ppant) arm of the acyl carrier protein (ACP), where the ketosynthase (KS) domain accepts a growing chain elongation intermediate from the previous module and catalyzes a decarboxylative Claisen condensation placing the extended chain onto the ACP for reductive tailoring. The ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) domains modify the chain at the β position, (depending on the module and the presence of processing domains), while the terminal thioesterase domain (TE) catalyzes cyclization of the macrolactone product.18–21
Streptomyces venezuelae ATCC 15439 produces a suite of 12- and 14-membered ring macrolide antibiotics with late-stage PKS modules (e.g. PikAIII and PikAIV) in the biosynthetic pathway demonstrating substrate flexibility.22–23 The unnatural PikAIII-TE fusion protein produces the 12-membered ring macrolactone 10-deoxymethyonolide (10-dml, 7) from the N-acetylcysteamine thioester (NAC) (21) of the natural Pik pentaketide (6). Incubation with both PikAIII and PikAIV monomodules results in two propionate extensions followed by macrocyclization to yield the 14-membered ring aglycone narbonolide (nbl, 8).24 Although considerable progress has been made in micro-scale analysis of modular PKS systems using advanced polyketide chain elongation intermediates,19,23–24 we were motivated to explore the possibility of a more efficient and scalable biocatalytic system.
From a practical perspective, preparative scale (>1 mmol) biocatalysis by type I modular PKSs has been impeded by a number of technical challenges including (i) slow loading of substrate onto the KS (ii) stoichiometric and cost-prohibitive cofactors (iii) the large, fragile nature of PKS proteins. Accordingly, we sought to address these challenges and develop multifunctional PKS enzymes as options in the toolbox of synthetic chemistry. Finally, tailoring of the macrolactones to macrolides was envisioned through whole cell biotransformation to append D-desosamine and perform C-H oxidation(s) by the PikC monooxygenase (Scheme 2).
Scheme 2.
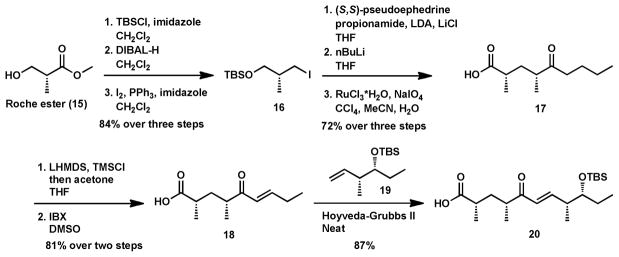
Second generation24 approach to Pik pentaketide.
Results
Synthesis of Pik Pentaketide
Retrosynthetically, we envisioned disconnection at the enone double bond, simplifying the necessary building blocks to α,β unsaturated ketone 18, a type II olefin,25 matched to undergo cross metathesis with type I fragment 19. Synthesis of enone 18 began with TBS protection of (R)-Roche ester (15),26–27 which was subsequently reduced and iodinated to yield 16. Alkylation with (S,S)-pseudoephedrine propionamide,28 displacement of the auxiliary with n-BuLi and RuO4 oxidation29 furnished saturated acid 17. Conversion of 17 to enone 18 was envisioned through oxidation of the corresponding silyl enol ether.30 Trapping the kinetic enolate of 17 with LDA/TMSCl at −78 °C proved problematic as epimerization was observed through a presumed silyl ketene intermediate. Employment of LHMDS/TMSCl at −78 °C facilitated smooth trapping of the enolate31 and subsequent IBX oxidation of the crude silyl enol ether yielded compound 18.30 This two-step sequence initially demonstrated poor reproducibility but was improved through quenching excess LHMDS/TMSCl with a drop wise addition of acetone prior to an aqueous quench to provide 18 in good yield.32 Common metathesis conditions were evaluated with 1.5 equivalents of silyl ether 19 and 3 mol% of HG-II, 33 where the highest yields were achieved when the reaction was run neat at 50 °C for 12 hours. Cleavage of the silyl ether with hydrogen fluoride in acetonitrile afforded the unactivated pentaketide 5. With the desired substrate 20 in hand, we considered alternatives to NAC thioesters in an effort to increase efficiency of loading target KS domains to initiate PKS chain extension.
Synthesis and Evaluation of Pentaketide Thioesters
We envisioned four Pik pentaketide substrates of varying reactivity and aqueous solubility for direct PKS loading. NAC 21 was synthesized as described previously,24 while 22–24 were generated from corresponding disulfides.34 Activated pentaketides 21–24 were incubated with purified PikAIII-TE, extender unit, and hydride donor under non-optimized conditions (Figure 2). QTOF-LC/MS analysis of 50 μL reaction mixtures after one hour revealed significant differences in conversion between aryl thioesters and traditionally employed NAC thioesters. Thiophenol activated 22 showed a 4-fold increase in conversion to 10-dml while 23 and 24 demonstrated 2-fold increases. Furthermore, 22 showed no detectable hydrolysis in the absence of enzyme or in the presence of denatured enzyme, while both 23 and 24 underwent detectable hydrolysis. Compounds 21 and 22 were further analyzed via HPLC calibrated against a 10-dml standard curve providing absolute quantification along the reaction time course. Having demonstrated the superiority of thiophenol-activated pentaketide 22 to the previously employed 21, attention was turned toward exploring alternatives to superstoichiometric quantities of MM-CoA (5) and NADPH as essential co-factors.
Figure 2.

Synthesis and evaluation of activated Pik pentaketides. Enzymatic reaction conditions: 1mM Pik pentaketide, 10mM MM-NAC (vide infra), 0.5mM NADP+, 2.5mM glucose-6-phosphate, 0.5 unit/mL glucose-6-phosphate dehydrogenase, 8mM 2-vinylpyridine, and 1μM purified PikAIII-TE, 1–24 hours. Conversion of 21 and 22 to 10-deoxymethyonolide (7) was monitored (HPLC) with data represented as mean ± standard deviation. n=3
Evaluation of Superstoichiometric Co-Factors
Replacement of MM-CoA is requisite to attain a practical chemoenzymatic platform due to its cost (>$1000/25 mg)35 and poor atom economy. Khosla and coworkers reported methylmalonyl N-acetylcysteamine (MM-NAC, 25) as a surrogate for MM-CoA in the erythromycin pathway (DEBS) albeit at higher concentrations.36 In our hands, gram scale synthesis of MM-NAC was hindered by difficulties in separation from side-products due to the extreme hydrophilicity of the compound. An alternative two-step route proceeded through compound 27, a crystalline solid available in a single step from methyl Meldrum’s acid (26).37 Transthioesterification38 in aqueous sodium bicarbonate followed by acidification with cation exchange resin, washing of the aqueous phase with CH2Cl2 and subsequent lyophilization provided rapid access to decagrams of 25. The stoichiometric dependence of MM-NAC was examined in a stop point assay after 4 hours with both PikAIII-TE and PikAIII/PikAIV (Figure 3). While enzymatic NADPH recycling was potentially problematic in the presence of electrophilic thioesters, the glucose-6-phosphate/glucose-6-phosphate dehydrogenase system39 proved sufficiently robust to be employed without modification. Ultimately, NADP+ could be recycled effectively at 10 mol% relative to pentaketide substrate.
Figure 3.

Synthesis and evaluation of methylmalonyl N-acetylcysteamine (MM-NAC, 25) Enzymatic reaction conditions: 1mM Pik pentaketide 22, 1–20mM MM-NAC, 0.5mM NADP+, 2.5mM glucose-6-phosphate, 0.5 unit/mL glucose-6-phosphate dehydrogenase, 8mM 2- vinylpyridine, and 1μM purified PikAIII-TE or PikAIII/PikAIV, 4 hours. Conversion of pentaketides 21 and 22 to 10-deoxymethyonolide (6) or narbonolide (7) was monitored (HPLC) with data represented as mean ± standard deviation. n=3
Semi-Preparative and Preparative PKS catalysis
Following co-factor optimization, we examined the scalability of the in vitro macrolactone-forming PKS reactions. Scale-up of analytical 50 μL reactions to semi-preparative 200 mL reactions (0.2 mmol 22) while maintaining identical concentrations and stoichiometry provided 10-dml (7) in 53% yield and acetyl-narbonolide (28) in 47% yield, respectively after silica gel chromatography (Figure 4). The observed precipitation of modular PKS proteins during the reaction spurred a final evaluation of conditions, and transitioning from purified protein to crude cell lysate while maintaining identical concentrations and stoichiometry demonstrated modest improvement generating 10-dml in 62% yield or acetyl-narbonolide in 55% yield. Increased enzymatic stability enabled subsequent reactions to be conducted at a four-fold greater concentration while decreasing the stoichiometry of MM-NAC by a factor of two, affording 10-dml in 66% yield or acetyl-narbonolide in 55% yield. With final reaction conditions in hand, preparative-scale catalysis with 0.5 g of Pik pentaketide 22 (1.43 mmol) generated 10-dml in 60% yield, and acetyl-narbonolide in 49% yield.
Figure 4.

Semipreparative and preparative PKS cascade to macrolactones. Enzymatic reaction conditions: 1mM-4mM Pik pentaketide 22, 10–20 equiv. MM-NAC, 0.1mM NADP+, 2.5–10mM glucose-6-phosphate, 0.5–2 unit/mL glucose-6-phosphate dehydrogenase, 1–4μM purifieda or cell freeb PikAIII-TE or PikAIII/PikAIV, 4 hours. Narbonolide was acetylated to 28 prior to chromatography to prevent degradation. Yields are calculated from isolated macrolactone after chromatography.
Biotransformation of Macrolactones to Macrolides
As the final step toward targeted macrolides, we evaluated whole cell biocatalysis for introduction of D-desosamine and the final p450-mediated C-H oxidation(s). Engineered variants of Streptomyces venezuelae ATCC 15439 designated strains DHS200140 and YJ11241 were assayed for their ability to transform macrolactones to macrolides (Figure 5). Observed variation in levels of bioconversion from culture to culture (normalized by time and based on initial culture inoculation to addition of macrolactone) motivated studies to isolate and evaluate growth phase dependence. Addition of the macrolactone substrate to high OD600 DHS2001 cultures resulted in fast initial rates, but incomplete conversions. By contrast, addition of macrolactone to pre-log phase cultures of resulted in highest overall conversion to macrolide products. While conversion of 10-dml to methymycins (11–14) was undetectable with either strain, this was overcome by addition of acetyl-narbonolide (28)42 to afford >99% conversion to target macrolides over 48 hours. Addition of acetyl-narbonolide to narbonolide biotransformations increased rates and overall conversion (>99%) in a similar manner. Bioconversion of 10-dml with DHS2001 predominately afforded neomethymycin and methymycin, with low levels of novamethymycin isolated as well. S. venezuelae strain YJ112 mediated conversion of 10-dml with superior access to higher oxidation states including increased levels of novamethymycin, and enabled discovery of the new macrolide ketomethymycin arising from a presumed dihydroxylation at the C-12 position. Addition of narbonolide to S. venezuelae strain YJ112 provided pikromycin in 72% yield.
Figure 5.
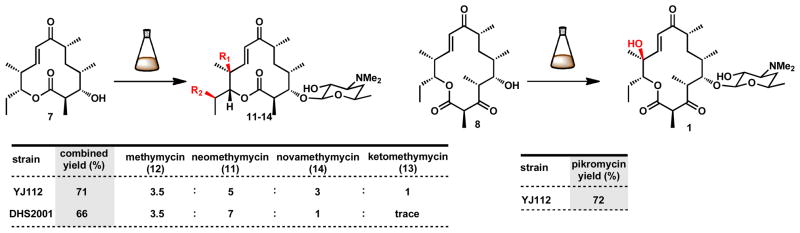
Biotansformation of macrolactones to macrolides. Biotransformation conditions: macrolactone added to 100 mg/L with 2.5 mg/L acetyl-narbonolide (28) when culture reached OD600 0.1, incubated for 48 hours. methymycin (12): R1=OH, R2=H, neomethymycin (11): R1=H, R2=OH, novamethymycin (14): R1=OH, R2=OH, ketomethymycin (13): R1=H, R2=O. Yields are calculated as the sum of differentially oxidized macrolides after HPLC purification.
Discussion
Modern synthesis of polyketide natural products is routinely enabled by chiral auxiliary methodology that predictably forms C-C bonds in a stereoselective, biomimetic manner. Prominent examples, including Myers alkylation28,43 and Evans aldol44 protocols and variants thereof, are commonly employed in constructing these complex secondary metabolites. Indeed, a recent synthesis of narbonolide [>19 steps (longest linear sequence) and <3% overall yield]12 and related macrolactone 6-dEB [22 steps (longest linear sequence) and 7.8% overall yield]45 relied heavily on these “practical laboratory emulations of the series of acylation/reduction reactions performed by polyketide synthases.”46 For example, an aldol reaction with N-propionyl-2-oxazolidone and propionaldehyde mimics one round of KS catalyzed propionate extension and KR reduction. In this approach, each new C-C bond formed requires further steps to protect installed functionality, displace the auxiliary, and oxidize or otherwise activate the chain for the next C-C bond construction. The thiophenol activated Pik pentaketide 22 was synthesized in this manner [11 linear steps and 38% yield], demonstrating the typical strengths (>90% yield per operation) and shortcomings (numerous redox and protecting group manipulations) of this approach. PKS catalysis offers an attractive alternative to synthetic emulators, as post C-C bond redox steps are performed on the protein with effective stabilization of labile polyketide chains. Covalent attachment of the chain elongation intermediate to the ACP eliminates the need for protective groups prior to subsequent elongation or cyclization.
Despite the clear potential for synthetic utility and more than two decades of investigation,47–48 the development of modular PKS based biocatalytic systems has been hindered by the inherent complexity of PKS enzymes. Initial development was complicated by codependent factors that precluded optimizing individual reaction parameters in a stepwise manner. For example, while sensitive radio-assays have been necessary to compensate for the inefficiency of most in vitro PKS reactions and the micro-scale at which they are conducted, their qualitative readout and low-throughput nature make them unsuitable for screening reaction conditions. Fortunately, increased accessibility to high resolution LC/MS enabled us to avoid use of radioactive substrates and establish basic parameters (Figure 2) from which to proceed.
While evaluation of thioester leaving groups by QTOF-LC/MS illuminated striking improvement in substrate conversion and ultimately enabled the development of simple HPLC assays, initial experiments appeared less than promising. The NAC activated Pik pentaketide 21 is sparingly water soluble despite the polarity imparted by NAC, and substitution to more hydrophobic thiophenol 22 led to a visible dispersion at 1mM concentrations. Fortunately, loss of turbidity could be used to visually track the progress of the reaction, with consumption of starting material rendering a clear solution. The superior conversion of aryl thioesters compared to conventional NAC thioesters suggests that increased reactivity trumps possible KS-Ppant interactions. Of particular note is the apparent preference for the Pik pentaketide substrate to load the KS active site cysteine over the ACP Ppant arm, as concomitant ACP acylation would effectively remove the module from participating in catalysis. In retrospect, observed fidelity in loading the PikAIII KS domain active site cysteine residue is surprising given the demonstrated efficacy of thiophenol thioesters in loading Ppant arms of PCP domains.49–52 Ultimately, PKS mediated conversion of Pik pentaketide proceeded with modest yield to 10-dml [60% yield, 23% overall from 15] or acetyl-narbonolide [49% yield, 18% overall from 15] at preparative scale, generating ~250 mg of either macrolactone.
Chemical glycosylation of both macrolactones with D-desosamine53–54 and subsequent C-H oxidation through PikC monooxygenase have been described,55 though complications obtaining and attaching D-desosamine significantly impede this approach. This amino sugar can be chemically synthesized in nine steps,54,56 though it is more commonly harvested from degradation of erythromycin A.57–58 Utilizing D-desosamine acquired from degradation requires a multi-step sequence of protection, activation, glycosylation, and deprotection. Further, glycosylation of hindered secondary alcohols in either macrolactone has been challenging and proceeds with moderate yields.16,54
Alternatively, the direct biotransformation of macrolactones to macrolides presents an attractive option in converting 10-dml to methymycins (11–14) and narbonolide to pikromycin (1).59 In this approach, the 10-dml and narbonolide macrolactones are natural substrates for the D-desosamine biosynthetic pathway and subsequent PikC oxidation within engineered variants of S. venezuelae ATCC 15439. Indeed, strain YJ122 efficiently converted crude narbonolide to pikromycin [72% yield, 13% overall from 15]. Bioconversion of 10-dml to methymycins proved to be tunable depending on the strain employed, with DHS2001 biotransformation affording primarily monohydroxylated products [combined 66% yield, 15% overall from 15]. S. venezuelae strain YJ112 mediated conversion of 10-dml with increased accessibility to dihydroxylated novamethymycin, and provided access to previously unknown ketomethymycin [combined 71% yield, 16% overall from 15]. Careful analysis of wild type S. venezuelae ATCC 15439 culture revealed production of a compound having an identical mass and retention time to synthetic ketomethymycin, suggesting that this compound is indeed a secondary metabolite made in trace quantities under normal laboratory culture conditions.
Conclusion
Advances in polyketide chain elongation intermediate activation and optimization of in vitro reaction parameters yielded practical type I PKS biocatalysis for scalable de novo macrolide production. The flexibility of the Pik pathway enabled divergent generation of either 10-dml or narbonolide and optimized whole cell biotransformations effectively tailored macrolactones to macrolides, with S. venezuelae strain YJ112 enabling access to ketomethymycin bearing a previously unobserved oxidation state at the C-12 position. This platform offers practical entry to natural 12-and 14 membered ring macrolides from the Pik pathway, and the potential of this system to produce unnatural macrolides will be reported in due course.
Supplementary Material
Figure 1.

Macrolide antibiotics including ketolide 3,60 an intermediate en route to telithromycin. from erythromycin A.
Scheme 1.

Retrosynthetic analysis. PikC: P450 catalyzing mono-and dihydroxylation of YC-17 (9) and narbomycin (10) macrolides. DesI-VIII: D-desosamine biosynthetic pathway catalyzing glycosylation of 10-deoxymethynolide (7) and narbonolide (8) macrolactones. PikAIII-TE or PikAIII/AIV: PKS modules capable of converting Pik pentaketide (6) to macrolactones 10- deoxymethynolide (7) or narbonolide (8).
Acknowledgments
This research was supported by Rackham Merit and American Foundation for Pharmaceutical Education pre-doctoral fellowships (D.A.H.), the Life Sciences Research Foundation (A.R.H.N), and NIH grant GM076477 and the Hans W. Vahlteich Professorship (to D.H.S.).
Footnotes
Supporting Information Available: Full experimental details and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 2.Li T, Liang J, Ambrogelly A, Brennan T, Gloor G, Huisman G, Lalonde J, Lekhal A, Mijts B, Muley S, Newman L, Tobin M, Wong G, Zaks A, Zhang X. J Am Chem Soc. 2012;134:6467. doi: 10.1021/ja3010495. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg WA, Varvak A, Hanson SR, Wong K, Huang H, Chen P, Burk MJ. Proc Natl Acad Sci USA. 2004;101:5788. doi: 10.1073/pnas.0307563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockmann H, Henkel W. Naturwissenschaften. 1950;37:138. [Google Scholar]
- 5.Brockmann H, Henkel W. Chem Ber. 1951;84:284. [Google Scholar]
- 6.Katz L, Ashley GW. Chem Rev. 2005;105:499. doi: 10.1021/cr030107f. [DOI] [PubMed] [Google Scholar]
- 7.Velvadapu V, Paul T, Wagh B, Klepacki D, Guvench O, MacKerell A, Jr, Andrade RB. ACS Med Chem Lett. 2011;2:68. doi: 10.1021/ml1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagh B, Paul T, Glassford I, DeBrosse C, Klepacki D, Small MC, MacKerell AD, Andrade RB. ACS Med Chem Lett. 2012;3:1013. doi: 10.1021/ml300230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velvadapu V, Glassford I, Lee M, Paul T, DeBrosse C, Klepacki D, Small MC, MacKerell AD, Jr, Andrade RB. ACS Med Chem Lett. 2012;3:211. doi: 10.1021/ml200254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velvadapu V, Paul T, Wagh B, Glassford I, DeBrosse C, Andrade RB. J Org Chem. 2011;76:7516. doi: 10.1021/jo201319b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao X, Woo SK, Krische MJ. J Am Chem Soc. 2013;135:4223. doi: 10.1021/ja4008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xuan R, Oh HS, Lee Y, Kang HY. J Org Chem. 2008;73:1456. doi: 10.1021/jo702384d. [DOI] [PubMed] [Google Scholar]
- 13.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Auyeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, Chenevert RB, Fliri A, Frobel K, Gais HJ, Garratt DG, Hayakawa K, Heggie W, Hesson DP, Hoppe D, Hoppe I, Hyatt JA, Ikeda D, Jacobi PA, Kim KS, Kobuke Y, Kojima K, Krowicki K, Lee VJ, Leutert T, Malchenko S, Martens J, Matthews RS, Ong BS, Press JB, Rajanbabu TV, Rousseau G, Sauter HM, Suzuki M, Tatsuta K, Tolbert LM, Truesdale EA, Uchida I, Ueda Y, Uyehara T, Vasella AT, Vladuchick WC, Wade PA, Williams RM, Wong HNC. J Am Chem Soc. 1981;103:3210. [Google Scholar]
- 14.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Auyeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, Chenevert RB, Fliri A, Frobel K, Gais HJ, Garratt DG, Hayakawa K, Heggie W, Hesson DP, Hoppe D, Hoppe I, Hyatt JA, Ikeda D, Jacobi PA, Kim KS, Kobuke Y, Kojima K, Krowicki K, Lee VJ, Leutert T, Malchenko S, Martens J, Matthews RS, Ong BS, Press JB, Rajanbabu TV, Rousseau G, Sauter HM, Suzuki M, Tatsuta K, Tolbert LM, Truesdale EA, Uchida I, Ueda Y, Uyehara T, Vasella AT, Vladuchick WC, Wade PA, Williams RM, Wong HNC. J Am Chem Soc. 1981;103:3213. [Google Scholar]
- 15.Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Auyeung BW, Balaram P, Browne LJ, Card PJ, Chen CH, Chenevert RB, Fliri A, Frobel K, Gais HJ, Garratt DG, Hayakawa K, Heggie W, Hesson DP, Hoppe D, Hoppe I, Hyatt JA, Ikeda D, Jacobi PA, Kim KS, Kobuke Y, Kojima K, Krowicki K, Lee VJ, Leutert T, Malchenko S, Martens J, Matthews RS, Ong BS, Press JB, Rajanbabu TV, Rousseau G, Sauter HM, Suzuki M, Tatsuta K, Tolbert LM, Truesdale EA, Uchida I, Ueda Y, Uyehara T, Vasella AT, Vladuchick WC, Wade PA, Williams RM, Wong HNC. J Am Chem Soc. 1981;103:3215. [Google Scholar]
- 16.Oh HS, Kang HY. J Org Chem. 2012;77:1125. doi: 10.1021/jo201158q. [DOI] [PubMed] [Google Scholar]
- 17.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 18.Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL. Nat Chem Biol. 2006;2:537. doi: 10.1038/nchembio824. [DOI] [PubMed] [Google Scholar]
- 19.Aldrich CC, Venkatraman L, Sherman DH, Fecik RA. J Am Chem Soc. 2005;127:8910. doi: 10.1021/ja0504340. [DOI] [PubMed] [Google Scholar]
- 20.Giraldes JW, Akey DL, Kittendorf JD, Sherman DH, Smith JL, Fecik RA. Nat Chem Biol. 2006;2:531. doi: 10.1038/nchembio822. [DOI] [PubMed] [Google Scholar]
- 21.Mortison JD, Sherman DH. J Org Chem. 2010;75:7041. doi: 10.1021/jo101124n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kittendorf JD, Sherman DH. Bioorg Med Chem. 2009;17:2137. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortison JD, Kittendorf JD, Sherman DH. J Am Chem Soc. 2009;131:15784. doi: 10.1021/ja9060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aldrich CC, Beck BJ, Fecik RA, Sherman DH. J Am Chem Soc. 2005;127:8441. doi: 10.1021/ja042592h. [DOI] [PubMed] [Google Scholar]
- 25.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 26.Goodhue CT, Schaeffer JR. Biotechnol Bioeng. 1971;13:203. doi: 10.1002/bit.260130204. [DOI] [PubMed] [Google Scholar]
- 27.Cohen N, Eichel WF, Lopresti RJ, Neukom C, Saucy G. J Org Chem. 1976;41:3505. doi: 10.1021/jo00884a002. [DOI] [PubMed] [Google Scholar]
- 28.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496. [Google Scholar]
- 29.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936. [Google Scholar]
- 30.Nicolaou KC, Gray DLF, Montagnon T, Harrison ST. Angew Chem, Int Ed. 2002;41:996. doi: 10.1002/1521-3773(20020315)41:6<996::aid-anie996>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 31.Heathcock CH, Buse CT, Kleschick WA, Pirrung MC, Sohn JE, Lampe J. J Org Chem. 1980;45:1066. [Google Scholar]
- 32.Trace (isopropenyloxy)trimethysilane appears to have little effect on the subsequent IBX oxidation
- 33.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
- 34.Corey EJ, Nicolaou KC. J Am Chem Soc. 1974;96:5614. [Google Scholar]
- 35.Sigma Aldrich #M1762
- 36.Pohl NL, Gokhale RS, Cane DE, Khosla C. J Am Chem Soc. 1998;120:11206. [Google Scholar]
- 37.Magdziak D, Lalic G, Lee HM, Fortner KC, Aloise AD, Shair MD. J Am Chem Soc. 2005;127:7284. doi: 10.1021/ja051759j. [DOI] [PubMed] [Google Scholar]
- 38.Padmakumar R, Gantla S, Banerjee R. Anal Biochem. 1993;214:318. doi: 10.1006/abio.1993.1494. [DOI] [PubMed] [Google Scholar]
- 39.Wong CH, Whitesides GM. J Am Chem Soc. 1981;103:4890. [Google Scholar]
- 40.Jung WS, Lee SK, Hong JSJ, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH, Yoon YJ. Appl Microbiol Biotechnol. 2006;72:763. doi: 10.1007/s00253-006-0318-5. [DOI] [PubMed] [Google Scholar]
- 41.Jung WS, Jeong SJ, Park SR, Choi CY, Park BC, Park JW, Yoon YJ. Appl Environ Microbiol. 2008;74:1972. doi: 10.1128/AEM.02296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.acetyl-narbolide rapidly accelerates biotransformations (manuscript in preparation)
- 43.Morales MR, Mellem KT, Myers AG. Angew Chem, Int Ed. 2012;51:4568. doi: 10.1002/anie.201200370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127. [Google Scholar]
- 45.Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans DA, Kim AS, Metternich R, Novack VJ. J Am Chem Soc. 1998;120:5921. [Google Scholar]
- 47.Caffrey P, Bevitt DJ, Staunton J, Leadlay PF. FEBS Lett. 1992;304:225. doi: 10.1016/0014-5793(92)80624-p. [DOI] [PubMed] [Google Scholar]
- 48.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 49.Grunewald J, Marahiel MA. Microbiol Mol Biol Rev. 2006;70:121. doi: 10.1128/MMBR.70.1.121-146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sieber SA, Tao JH, Walsh CT, Marahiel MA. Angew Chem, Int Ed. 2004;43:493. doi: 10.1002/anie.200352787. [DOI] [PubMed] [Google Scholar]
- 51.Grunewald J, Sieber SA, Marahiel MA. Biochemistry. 2004;43:2915. doi: 10.1021/bi036140d. [DOI] [PubMed] [Google Scholar]
- 52.Kopp F, Mahlert C, Gruenewald J, Marahiel MA. J Am Chem Soc. 2006;128:16478. doi: 10.1021/ja0667458. [DOI] [PubMed] [Google Scholar]
- 53.Oh HS, Kang HY. J Org Chem. 2012;77:1125. doi: 10.1021/jo201158q. [DOI] [PubMed] [Google Scholar]
- 54.Oh HS, Xuan R, Kang HY. Org Biomol Chem. 2009;7:4458. doi: 10.1039/b911200f. [DOI] [PubMed] [Google Scholar]
- 55.Li S, Ouellet H, Sherman DH, Podust LM. J Biol Chem. 2009;284:5723. doi: 10.1074/jbc.M807592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee DM, Lee H, Kang HY. Bull Korean Chem Soc. 2008;29:535. [Google Scholar]
- 57.Li S, Chaulagain MR, Knauff AR, Podust LM, Montgomery J, Sherman DH. Proc Natl Acad Sci USA. 2009;106:18463. doi: 10.1073/pnas.0907203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen H, Yamase H, Murakami K, Chang C-w, Zhao L, Zhao Z, Liu H-w. Biochemistry. 2002;41:9165. doi: 10.1021/bi020245j. [DOI] [PubMed] [Google Scholar]
- 59.Venkatraman L, Salomon CE, Sherman DH, Fecik RA. J Org Chem. 2006;71:9853. doi: 10.1021/jo062047u. [DOI] [PubMed] [Google Scholar]
- 60.Wei X, You Q. Org Process Res Dev. 2006;10:446. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.