

Abstract
ATP-binding proteins play significant roles in numerous cellular processes. Here, we introduced a novel isotope-coded ATP-affinity probe (ICAP) as acylating agent to simultaneously enrich and incorporate isotope label to ATP-binding proteins. By taking advantage of the quantitative capability of this isotope-coded probe, we devised an affinity profiling strategy to comprehensively characterize ATP-protein interactions at the entire proteome scale. False-positive identification of ATP-binding sites derived from non-specific labeling was effectively minimized through the comparison of the labeling behaviors of lysine residues with the use of low and high concentrations of the ICAP reagents. A total of 258 previously known ATP-binding proteins from lysates of Hela-S3 and Jurkat-T cells were validated with this affinity profiling assay. Additionally, we demonstrated that this novel quantitative ATP-affinity profiling strategy is particularly useful for unveiling previously unrecognized nucleotide-binding sites in ATP-binding proteins. For example, our profiling results revealed K356 as a new ATP-binding site in HSP90. Furthermore, 293 proteins without documented ATP-binding GO were predicted to be ATP-binding proteins on the basis of our quantitative affinity profiling results. We also uncovered, for the first time, the ATP-binding capability of human proliferating cell nuclear antigen (PCNA), identified the lysine residue involved in ATP binding, and validated the protein’s capacity in ATP binding with an independent assay. The ICAP approach described in the present paper should be generally applicable for the quantitative assessment of ATP-binding proteins in proteomic samples from cells and tissues.
Introduction
Adenosine triphosphate (ATP)-binding proteins, including ATP-binding cassette transporters (ABC transporters), chaperones, and kinases, are one of the most important superfamily of proteins that are involved in a variety of pivotal cellular processes, such as cell signaling, proliferation, differentiation, and apoptosis. For example, numerous ABC transporters catalyze the hydrolysis of ATP to provide energy required for translocation of various substrates across cell membranes.1 Additionally, kinases, as one of the largest superfamilies of enzymes in higher eukaryotes, mediate cellular protein and lipid phosphorylation to regulate downstream signaling cascade.2
Despite the importance of ATP-binding proteins in cellular functions, the proteome-wide picture of ATP-binding proteins is far from complete. The abundance of individual ATP-binding proteins may be assessed by using immunoblot;3 however, global study of the expression levels of ATP-binding proteins can hardly be achieved with the use of conventional methods owing to the enormous diversity of the ATP-binding protein family. The development of mass spectrometry (MS) instrumentation and bioinformatic tools provides the opportunity to identify and quantify up to several thousand proteins in complex samples. However, proteomic studies of ATP-binding proteins, including different ATPases and kinases, by MS are still a challenge, which is mainly attributed to the under-representation of some ATP-binding proteins in the proteome. For instance, protein kinases, despite being the largest family of enzymes in mammals, constitute only 1.7% of the human genome2. Therefore, selective enrichment of ATP-binding proteins from cellular extracts is essential for the comprehensive identification and quantification of ATP-binding proteins.
Much remains to be learnt about protein-ATP interactions in human proteome. Although a large number of known ATP-binding proteins are documented in gene ontology (GO) annotation database, numerous proteins with available sequence information remain unannotated with respect to their nucleotide-binding affinity.4 Furthermore, experimental characterization of ATP-protein interaction often relies on radioactivity-based ultrafiltration assay5 or fluorescence-based binding assay.6 These traditional methods are usually costly and time-consuming because they require the use of purified proteins, which prevents high-throughput studies at the whole proteome level. Most importantly, none of these approaches permit the robust discovery of ATP-binding site, or ‘ATP-interacting residues’, which is another aspect of significant interest in nucleotide-protein interaction studies. The ATP binding sites, where ATP is captured and hydrolyzed to ADP, are particularly important because they are responsible for changing the conformation and/or modulating the catalytic activity of proteins.1 Additionally, many anti-cancer drugs target ATP-binding sites in ATPases and kinases.1 Therefore, new methods for robust and systematic characterizations of protein-ATP interactions as well as for the identification of ATP-interacting residues at a global proteome scale are invaluable for understanding better the regulatory mechanisms of ATP-related protein functions.
Recently, we and others reported the application of biotin-conjugated acyl nucleotide probes for the enrichment and identification of ATP-binding proteins from complex protein mixtures.7, 8 In this context, the entire proteome is treated with an acyl phosphate-linked ATP affinity probe, which allows for the incorporation of a biotin tag to lysine residue on a subset of proteins with ATP-binding affinity. It was demonstrated that this ATP-affinity probe can be harnessed as an enrichment tool to enable the subsequent LC-MS/MS identification and quantification of ATP-binding proteins at the global proteome scale. Moreover, this approach could be employed to characterize, at the entire proteome level, site-specific interaction between ATP and its binding proteins. We demonstrated previously that the lysine residue labeled by the biotin-ATP probe may represent the specific lysine residue that directly contacts the γ-phosphate group of the bound ATP. However, both applications rely on stable isotope labeling by amino acids in cell culture (SILAC) because proteins or peptides in the different experimental groups need to be differentiated to achieve quantitative comparison. However, the SILAC labeling approach may not be compatible with some applications of the ATP affinity probe. For instance, clinical samples (e.g. biological fluids, tissue samples) are not amenable to SILAC.9 In addition, chemical isotopic labeling methods like dimethyl labeling10 are prone to additional experimental errors because the isotopic tags are usually incorporated in the late stage of the sample preparation.
To overcome this limitation, we introduced the desthiobiotin-based Isotope-Coded ATP-affinity Probe (ICAP) as an acylating agent to selectively label and enrich ATP-binding proteins from the human proteome. With the use of this novel probe, ATP-binding proteins from whole cell lysate can be simultaneously enriched and isotopically labeled. In addition, we developed a novel quantitative profiling strategy with the use of ICAP to characterize ATP-protein interactions and determine specific ATP-binding sites in ATP-binding proteins at the entire proteome scale.
Experimental Procedures
Cell lysate preparation and labeling with the nucleotide affinity probe
The biotinylated nucleotide affinity probes were prepared following previously published procedures with minor modifications (see Supporting Information).11 HeLa-S3 cells were purchased from the National Cell Culture Center (Minneapolis, MN). Jurkat-T cells (ATCC; Manassas, VA) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and penicillin (100 IU/mL). Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. Approximately 2×107 cells were harvested, washed with cold PBS for three times, and lysed in 1-mL lysis buffer, which contained 0.7% CHAPS, 50 mM HEPES (pH 7.4), 0.5 mM EDTA, 100 mM NaCl, and 10 OL (1:100) protease inhibitor cocktail on ice for 30 min. The cell lysates were centrifuged at 16000g at 4°C for 30 min, and the resulting supernatants were collected and subjected to gel filtration separation using NAP-25 columns (Amersham Biosciences) to remove free endogenous nucleotides. Cell lysates were eluted into a 2 mL buffer, containing 50 mM HEPES (pH 7.4), 75 mM NaCl, and 5% glycerol. The resulting proteins in cell lysates were quantified using Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA) and stored at −80°C. Immediately prior to the labeling reaction, MgCl2, MnCl2, and CaCl2 were added to the concentrated cell lysate until their final concentrations reached 50, 5, and 5 mM, respectively. Approximately 1 mg cell lysate was treated separately with light and heavy labeled desthiobiotin-ATP affinity probe at concentrations of 10 and 100 OM, respectively. Labeling reactions were carried out with gentle shaking at room temperature for 1.5 h. After the reaction, the remaining probes in the cell lysates were removed by buffer exchange with 25 mM NH4HCO3 (pH 8.5) using Amicon Ultra-4 filter (10,000 NMWL, Millipore).
In-solution enzymatic digestion and affinity purification
After addition of 8 M urea for protein denaturation, and dithiothreitol and iodoacetamide for cysteine reduction and alkylation, the labeled proteins were digested with modified sequencing-grade trypsin (Roche Applied Science) at an enzyme/substrate ratio of 1:100 in 25 mM NH4HCO3 (pH 8.5) at 37°C for overnight. The peptide mixture was subsequently dried in a Speed-vac and redissolved in 1 mL of 100 mM potassium phosphate and 0.15 M NaCl (pH 7.5, PBS buffer), to which solution was subsequently added 200 OL avidin-agarose resin (Sigma-Aldrich). The mixture was then incubated at 25°C for 1 hr with gentle shaking. The agarose resin was then washed with 3 mL PBS buffer and 3 mL H2O to remove unbound peptides, and the labeled peptides were subsequently eluted with 1% TFA in CH3CN/H2O (7:3, v/v) at 65°C.
Fractionation of biotinylated peptides with high-pH reversed-phase chromatography (High-pH-RP-LC)
The above enriched desthiobiotinylated peptides were redissolved in 20 μL H2O and subjected to high-pH-RP-LC separation on an Agilent 1100 system using a 0.5 mm × 150 mm Zorbax SB-C18 column (5 μm in particle size, Agilent Technologies). Mobile phase A was 15 mM aqueous ammonium formate (pH 7.5), and mobile phase B contained 15 mM ammonium formate in water/acetonitrile (1:9). Peptides were eluted with a 60-min linear gradient of 5–40% B and six fractions were collected. Each fraction was dried and reconstituted in 0.1% formic acid prior to LC-MS/MS analysis, the details of which are shown in on-line Supporting Information.
Results
1. Working principle of the isotope-coded ATP-affinity probe (ICAP)
Bioinformatic studies showed that most ATP-binding proteins carry at least one lysine residue in a consensus amino acid sequence motif known as the phosphate-binding loop (P-loop), which constitutes a nucleotide-binding site and is important for the ATPase activity.12 Therefore, nucleotide affinity probes bearing an acyl phosphate moiety were developed to target the lysine residue in the nucleotide binding site.7 Here, we devised a novel ICAP-based strategy to simultaneously enrich and isotopically label ATP-binding proteins from cell lysates. The ICAP reagent contains three components (Figure 1), namely, a binding moiety (ATP), an enrichment moiety (i.e. desthiobiotin) facilitating downstream purification, and an isotope-coded linker present in light (contains six hydrogens) or heavy form (contains six deuterons). The isotope-coded linker and ATP are conjugated through an acyl phosphate. Upon binding to ATP-binding proteins, the acyl-phosphate component of the ICAP reacts with the ε-amino group of the P-loop lysine residue to yield a stable amide bond. As a result, desthiobiotin along with light or heavy isotope-coded linker is covalently attached to the lysine residue on ATP-binding proteins (Supplementary Figure S1A).
Figure 1.
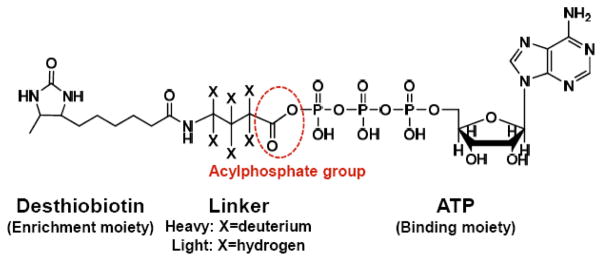
The structure of isotope-coded ATP affinity probe (ICAP).
A general experimental procedure with the use of ICAP includes the following steps (Supplementary Figure 2): (1) ATP-binding proteins from a protein sample representing one experimental state are derivatized with the isotopically light form of the ATP affinity probe, whereas the ATP-binding proteins from protein sample representing another experiment state are modified with the heavy isotope-coded ATP probe. (2) The two protein samples are combined and digested with trypsin and the resulting light/heavy desthiobiotin-labeled peptide pairs can be enriched with avidin agarose. (3) The affinity-purified peptides with the desthiobiotin tag are analyzed by LC-MS/MS. The MS peak intensity ratios of the light and heavy forms of desthiobiotin-modified peptide pairs can be utilized to obtain accurate quantification results of the corresponding ATP-binding proteins originating from the two experiment states.
2. Strategy for proteome-wide characterization of ATP-protein interactions
Building upon the quantitative capability of the ICAP-based method, we first applied these probes for the characterization of ATP-protein interactions at the global proteome scale. As reported previously,7 owing to its relatively high reactivity, apart from the lysine residue(s) located at the nucleotide binding site, other lysine residues may also be modified by the nucleotide affinity probe through non-specific electrostatic interactions. To distinguish specific from non-specific labelings, we developed a novel strategy to unambiguously characterize ATP-binding affinity of proteins at the entire proteome scale.
Intrigued by a previous study in reactivity profiling of functional cysteines by comparing the extent of cysteine alkylation under different probe concentrations,13 we reasoned that the binding of the ATP component of the probe to a protein renders the acyl phosphate moiety conducive for coupling with the lysine residue at the ATP binding site of the protein, thereby greatly promoting the rate for amide bond formation.14 Therefore, lysine residues involved in ATP binding and those that are not would exhibit distinct labeling behaviors with low and high concentrations of ATP affinity probe. At low probe concentration, the lysine residue at the ATP-binding site possesses hyper-reactivity and will be completely labeled. However, lysines not involved with ATP binding will only be partially labeled because the limited amount of labeling reagent reacts preferentially with the lysine at the ATP-binding site. At high probe concentration, the ATP-binding lysine will still be labeled to completion, whereas non-ATP binding lysine will be labeled to a much greater extent than that at low probe concentration.
Based on the above hypothesis, we devised an affinity profiling strategy by allowing a low (10 μM) concentration of the light ATP probe and a high (100 μM) concentration of the heavy ATP probe to react separately with the same amount of cell lysates (forward experiment). To minimize the bias introduced by the labeling, reverse labeling experiment was also performed, where the same amount of cell lysates were treated individually with a high (100 μM) concentration of the light ATP probe and a low (10 μM) concentration of the heavy ATP probe. As a result, peak intensity ratios of light and heavy desthiobiotin-labeled peptides can be used to derive ATP-binding affinity ratio, RATP10/1, which reflects the relative binding affinities of ATP towards specific lysine residues in individual proteins (Figure 2). With this assumption, specific ATP-binding lysine will generate an RATP10/1 close to 1 since similar amount of ATP-binding lysine will be labeled regardless of the probe concentration. By contrast, non-specifically labeled lysine will display concentration-dependent increase in ATP probe labeling, which results in a RATP10/1 ≫1.
Figure 2.

A general strategy for quantitative ATP-affinity profiling using ICAP.
3. Proteome-wide profiling of ATP-binding proteins
To test this proposed strategy, we performed quantitative ATP-affinity profiling of whole cell lysate from Hela-S3 cells using paired light and heavy ICAP reagents at 10 μM and 100 μM concentrations. We were able to quantify a total of 1575 proteins, including more than 4600 light or heavy desthiobiotin tag-modified lysine residues. As depicted in Figure 3A, a large number of peptides with desthiobiotin modification exhibited significantly different probe labeling efficiency under low and high probe concentrations, generating RATP10/1≫1. However, a small portion of the desthiobiotin-modified peptides bear RATP10/1 close to 1, indicating that they share similar labeling efficiency at low and high probe concentrations.
Figure 3.

(A) Measured RATP10/1 ratio from Hela-S3 cell lysates with low (10 μM) and high (100 μM) concentrations of ICAP; (B) GO analysis of proteins with different RATP10/1 ratios using both Hela-S3 (top) and Jurkat-T (bottom) cell lysates.
We next sought to assess if these peptides with low RATP10/1 ratios are indeed derived from ATP-binding proteins. Similar as the previous quantitative reactivity profiling of functional cysteines in proteins,13 we arbitrarily consider lysine residue in peptides with RATP10/1<2 as ATP-binding lysine. In addition, only those modified lysine residues being successfully quantified at least twice are retained on the list. With the use of these criteria, we obtained 496 unique desthiobiotin-modified peptides, representing approximately 11% of total identified peptides, from 333 unique proteins. We considered these 333 proteins as possible ATP-binding proteins (Supplementary Table S2). GO analysis using DAVID15 indicated that ATP binding and kinase activity are significantly over-represented in this protein group. As indicated in Figure 3B and Supplementary Table S1, more than 50% of the 333 proteins are with known ATP-binding GO, suggesting a 5-fold increase in enrichment relative to the entire human proteome with a p-value of 1.9E-77. In contrast, the percentage of known ATP-binding proteins in the protein group containing peptides with RATP10/1>2 is only 11%. More significantly, among the 496 peptides from the possible ATP-binding protein group, peptides of known ATP-binding proteins were on average identified and quantified three times more frequently than those of proteins without ATP-binding GO, suggesting even more pronounced ATP-binding enrichment efficiency (77% of all quantification events, Supplementary Table S2). Accordingly, the percentages of proteins with kinase activity GO and protein kinase activity GO in the possible ATP-binding protein group are 5- and 6-fold higher than those in the rest protein group. By contrast, 54 out of 65 identified known GTP-binding proteins were excluded from possible ATP-binding protein groups, indicating that a large proportion of non-specific labeling arising from electrostatic interaction can be discerned based on the large RATP10/1 ratio. For example, K134 from GTP-binding nuclear protein Ran (Uniprot: P62826) was found to be modified with desthiobiotin with a large RATP10/1 ratio of 6.64, indicating that labeling of this lysine arises likely from non-specific binding. These findings demonstrated that the above-described quantitative affinity profiling could serve as an effective strategy to validate and discover ATP-binding proteins by effectively eliminating false-positive targets, which often compromise the application of this type of affinity reactive probes in protein-ligand binding studies.
Similar results were obtained when the same procedure was applied for quantitative profiling of ATP-protein interaction in the whole cell lysate of Jurkat-T cells. We were able to obtain a total of 504 unique desthiobiotin-labeled peptides from 323 unique proteins satisfying the aforementioned criteria (Supplementary Table S3). According to GO annotation (Figure 3B and Supplementary Table S1), 50% and 29% of the 323 proteins are known ATP-binding proteins and kinases, respectively. This result reveals that our quantitative ATP-affinity profiling strategy is generally applicable for protein mixtures derived from any biological samples and not restricted to a particular cell line.
By combining the quantitative ATP-affinity profiling results from Hela-S3 and Jurkat-T cells (Supplementary Figure S3), a total of 258 proteins with known ATP-binding affinity were validated with this method, among which 71 were commonly found for both cell lines. In addition, 138 proteins in our predicted ATP-binding protein list are known to possess kinase activity. Furthermore, 166 proteins from Hela-S3 cells and 161 proteins from Jurkat-T cells are predicted to be ATP-binding proteins by our quantitative affinity profiling strategy, though they do not possess documented ATP-binding GO. These results provide a wealth of information to further validate ATP-protein interaction and discover novel ATP-binding sites.
4. Validation and discovery of ATP-binding sites in known ATP-binding proteins
As mentioned above, aside from the study of ATP affinity at the protein level, the ATP-binding site, or ‘ATP-interacting residues’, is also important in nucleotide-protein interaction studies because these specific residues generally play crucial roles in maintaining the ATP-binding affinity. Therefore, mutations of these identified ATP-interacting residues serve as an effective means to diminish the ATPase and/or kinase activities of the ATP-binding proteins.16 However, identification of residues that interact with ATP often relies on X-ray crystal structure17 or site-directed mutagenesis,18 which is often costly and time-consuming.
Due to the specificity and precise quantification capability, our proposed ATP-affinity profiling strategy could serve as a quantitative and direct method to accurately assign ATP-binding lysine residues, which directly interact with γ-phosphate group of the bound ATP,7 without the availability of 3-dimensional structure information of the protein. For example, we found one desthiobiotin-labeled lysine site from two peptides (AVLLGPPGAGK#GTQAPR and AVLLGPPGAGK#) in ATP-binding protein adenylate kinase 2 (AK2), where K28 displays a low RATP10/1 of 1.2 (Figure 4). The small RATP10/1 suggests that K28 is a possible ATP-binding lysine in AK2, which is consistent with the observation that K28 is responsible for ATP binding and phosphohydrolase activity (PDB entry: 3TLX). Moreover, although only a single modified lysine was detected in the majority of identified possible ATP-binding proteins, multiple lysines were labeled for some proteins. However, these lysine residues often exhibited markedly different RATP10/1 ratios, which can be utilized to differentiate ATP-binding lysine from those that are non-specifically labeled. For instance, two lysine residues, K130 and K201, from cyclin-dependent kinase 1 were labeled in our quantitative profiling experiment; however, K201 shows a much larger RATP10/1 than K130, indicating that K130 is the true ATP-binding lysine and the labeling of K201 emanates from non-specific binding (Figure 4). Crystal structure of Cdk1 homolog, Cdk2 (PDB entry: 1HCK), reveals that K130 is in close proximity to the γ-phosphate group of ATP. Therefore, our strategy allows for site-specific determination of relative ATP-binding affinities of different binding sites in proteins by monitoring RATP10/1, thereby minimizing false-positive identification of ATP-binding sites.
Figure 4.

Light- and heavy-labeled peptides from forward- and reverse-affinity profiling experiments using ICAP. (A, B) Peptide AVLLGPPGAGK28#GTQAPR with a low RATP10/1 ratio from adenylate kinase 2; (C, D) Peptide DLK130#PQNLLIDDK with a low RATP10/1 ratio from cyclin-dependent kinase 1; (E, F) Peptide K201#PLFHGDSEIDQLFR with a high RATP10/1 ratio from cyclin-dependent kinase 1. “#” indicates the desthiobiotin-labeling site.
Benefited from this high-throughput proteome-wide quantitative analysis, we sought to analyze local sequence context surrounding the desthiobiotin-modified lysine, which is considered as the ATP-binding site in proteins. First, we examined all the desthiobiotin-labeled peptides with RATP10/1 ratios being smaller than 2 from profiling experiments of Hela-S3 and Jurkat-T cells. Similar as our previous results,7 three unique motifs were successfully identified using motif-X (Supplementary Figure S4)19. The well-known P-loop sequence motif of GxxxxGK12 displays a 39-fold enrichment with respect to the occurrence frequency in our input sequence compared to the entire proteome. In addition, another two unique motifs of HRDxKxxN and xAVK, which were identified with significant enrichment factors (185 and 1.8, respectively), are derived exclusively from kinases. These identified motifs provide invaluable information to study the functions of ATPase and kinases by generating catalytically inactive mutants, especially in large scale.16
More importantly, this novel quantitative ATP-affinity profiling strategy is particularly useful for unveiling the lysine residues that are involved in ATP-binding, but not present in any of the known ATP-binding motif. For instance, K176 in ribose-phosphate pyrophosphokinase 3 (PRPS1L1), which is not situated in any consensus motif known to be important in ATP-binding, was labeled by the desthiobiotin-tag with a low RATP10/1 of 1.3. This result strongly suggests that K176 is involved with ATP binding, which is supported by the crystal structure of the PRPS1L1-AMP complex (PDB entry: 2HCR).
We also noted that some ATP-binding proteins possess multiple lysine residues with low RATP10/1 ratios, raising the possibility that more than one lysine may contribute to ATP-protein interaction. For instance, for double-stranded RNA-activated protein kinase (PKR), apart from K416 residing in the HRDxKxxN motif (RATP10/1 ratio of 1.19), another peptide containing K296 (K.TYVIK#R.V) without any clear motif feature also displays ATP-affinity with an RATP10/1 ratio of 1.32 (Figure 5A). Indeed site-directed mutagenesis studies showed that substitution of K296 with an arginine gave rise to a catalytically dead mutant that completely abolishes the kinase activity of PKR.20 In another example, we found two lysine residues (K112 and K356) from heat shock protein 90 (HSP90), an ATP-binding chaperone, exhibit low RATP10/1 ratios of 0.92 and 1.08, respectively (Figure 5B). These two lysine residues are remote from each other in the crystal structure of HSP90 (Supplementary Figure S5, PDB entry: 2CG9), suggesting that HSP90 may contain two distinct ATP-binding sites. Interestingly, a previous study demonstrated that, in addition to the N-terminal ATP-binding site, a second C-terminal ATP-binding site in HSP90 becomes accessible after the first N-terminal site is occupied with ATP.21 Consistent with the low RATP10/1 ratio observed for K112, crystal structure (Supplementary Figure S5) revealed the direct contact between ATP and K112. However, due to the lack of conclusive experimental evidence, the exact position of C-terminal ATP-binding site on HSP90 remains elusive. On the basis of our quantitative proteomic results, we deduce that K356 may represent the other ATP-binding site located in C-terminal domain of HSP90. This notion is further supported by a previous study, where ATP binding induces the generation of two C-terminal fragments of 70 kDa (C70) and 39 kDa (C39) of HSP90 upon iron-catalyzed chemical cleavage.22 Mapping these two fragments to the sequence of HSP90 revealed that C70 fragment matches the cleavage site around N-terminal ATP-binding site of K112, whereas the C39 fragment suggests that C-terminal ATP-binding site may localize in the region harboring amino acid residues 350-450. This is consistent with our finding that K356 may constitute the second ATP-binding lysine (Figure 5B). Therefore, our results provided the first line of direct evidence to support that the second ATP-binding site resides on the C-terminal domain of HSP90 and K356 is involved with this ATP binding.
Figure 5.

Chromatograms represent multiple lysine sites with low RATP10/1 identified from PKR (A) and HSP90 (B) in quantitative ATP-affinity profiling assay. Blue and red profiles depict chromatograms of peptides from cell lysates labeled with high and low concentrations of the probe, respectively. Two sequence fragments containing intact C terminal domain generated from ATP binding-induced, iron-catalyzed chemical cleavage of HSP90 are also shown in (B).
These examples clearly illustrated the capability of our approach in characterizing the nucleotide-binding affinity of both known binding motifs and, more importantly, previously unrecognized nucleotide-binding site of ATP-binding proteins. This facilitates the identification of the lysine residues that are the most relevant for ATP-related function of proteins, which may provide invaluable information to generate activity-deficient mutants for functional studies of ATPase and kinases.
5. Exploration of novel ATP-binding proteins and ATP-binding sites
As mentioned above, more than 160 proteins without documented ATP-binding GO from Hela-S3 cells and Jurkat-T cells, respectively, are predicted to be ATP-binding proteins based on our quantitative affinity profiling results. To explore novel ATP-binding proteins and ATP-binding sites, we first examined the 22 proteins that were identified with the same peptides from the two cell lines (Supplementary Table S4).
GO analysis reveals that most of the 22 proteins have reported nucleotide-binding affinity. Four of them belong to NAD-binding proteins, such as NAD-dependent malic enzyme and glyceraldehyde-3-phosphate dehydrogenase, which is consistent with our previous findings that this ATP-affinity probe can also capture NAD-binding proteins.11 Only 8 proteins do not possess any nucleotide-related GO annotations. However, literature search reveals that some of them are also closely associated with ATP-binding. For example, K212 in phosphopantothenate-cysteine ligase (PPCS) was identified with low RATP10/1 ratios in affinity profiling experiments with both Hela-S3 and Jurkat-T lysates (1.24 and 0.87, respectively). This is in accordance with the previous finding that PPCS catalyzes the conversion of pantothenate to coenzyme A by utilizing ATP as a substrate.23 In another example, K375 in protein disulfide-isomerase (PDI) exhibits low RATP10/1 ratios in ATP profiling experiments for both cell lines, strongly suggesting that this protein possesses ATP-binding property. PDI is a multifunction protein and it is involved in protein folding.24 Although the sequence of PDI does not bear any motifs or homologies of kinases, ATPases or chaperones, Guthapfel et al.25 demonstrated that PDI was directly associated with ATP-dependent autophosphorylation with clear ATP binding and hydrolysis properties. This notion is supported by our results. Moreover, our study also provides the information about the ATP-binding site in PDI.
We also made an interesting observation with proliferating cell nuclear antigen (PCNA). In eukaryotes, including humans, PCNA forms a homotrimer, which complexes with the five-subunit replication factor C (RFC) as clamp loader to encircle duplex DNA, forming mobile tethers for polymerases and thereby enabling processive DNA synthesis.26 Previous studies suggest that ATP binding and hydrolysis in ATPase cycle is closely associated with every step of the DNA-clamp loading reaction. RFC, with conserved ATP binding/hydrolysis motifs, is therefore believed to serve as the primary ATP-binding sites to enable utilization of ATP for driving the complex clamp loading reaction. Intriguingly, our ATP affinity profiling with both Hela-S3 and Jurkat-T cell lysates strongly suggest that PCNA itself may also be involved in ATP-binding. We note that K80 in PCNA was quantified multiple times with an average RATP10/1 ratio of 1.16 (Figure 6). Careful examination of crystal structure of RFC-ATP-PCNA complex reveals five ATP-binding sites on RFC (Figure 6C, PDB entry: 1SXJ). In our ATP-affinity profiling assay, we successfully quantified lysine residues located in GxxxxGK motifs from RFC2 (RATP10/1 = 1.4) and RFC4 (RATP10/1 = 1.1) with low RATP10/1 ratios (Supplementary Tables S2 and S3), which are consistent with the ATP-binding sites of RFC as revealed by the crystal structure. Furthermore, as indicated in the crystal structure, these five binding sites on RFC are far from K80 in PCNA, indicating that desthiobiotin labeling on PCNA is unlikely derived from crossover labeling from the bound ATP probe on RFC protein.
Figure 6.

(A, B) Light- and heavy-labeled peptide ILK80#CAGNEDIITLR with low RATP10/1 ratio from PCNA in quantitative ATP-affinity profiling assay; “#” indicates the desthiobiotin labeling site. (C) Crystal structure of RFC-PCNA complex bound with ATP. Displayed is the relationship between K80 in PCNA and ATP-binding sites on RFC complex. (D) Fluorescence emission spectra of 50 mM PBS (red), PCNA alone (orange), TNP-ATP alone (yellow), or TNP-ATP together with PCNA (green) and a mixture of TNP-ATP, PCNA and ATP (blue).
To examine PCNA’s ATP-binding ability, we performed a binding assay using a widely used fluorescent ATP analog, 2′,3′-O-(2,4,6-trinitrophenyl)-ATP (TNP-ATP) (Supporting Information).27, 28 TNP-ATP is only moderately fluorescent in aqueous solution, while a substantial fluorescence enhancement and blue shift are observed upon binding to ATP-binding proteins. As revealed in Figure 6D, 25 μM of TNP-ATP displays minimal fluorescence in solution with a λmax of 540 nm. However, when 6 μM PCNA was added to TNP-ATP, the relative fluorescence increases by approximately 4.5-fold, indicating the direct binding of PCNA with the ATP analog. These effects were diminished when 2 mM free ATP was added. To estimate the binding affinity of TNP-ATP to PCNA, we titrated a constant concentration of PCNA solution with increasing amounts of TNP-ATP. Fluorescence intensity versus TNP-ATP concentration was plotted and fit into single binding model of a hyperbolic function29, 30 to obtain a dissociation constant (Kd) of 13 μM for interaction between PCNA and TNP-ATP. For comparison, similar experiments were performed with known ATP-binding proteins, YADH and creatine phosphokinase, and similar fluorescence enhancement response and Kd values of 13 and 27 μM were determined for the two proteins (Supplementary Figure S6). In this context, Fukuda et al.31 observed that the mutation of K80 in human PCNA to an alanine results in a greater than 2-fold reduction of its capability in stimulating polymerase δ-mediated DNA synthesis. Thus, our results suggest that the direct binding of ATP to PCNA may play a significant role in PCNA’s ability to promote processive DNA replication. Moreover, our results also unveiled the lysine residue in PCNA that is involved in ATP-binding.
Conclusions
Here, we introduced ICAP to simultaneously enrich and isotopically label ATP-binding proteins in the whole proteome. It has several advantages for studying nucleotide-binding proteins. First, unlike other chemical labeling methods including dimethyl labeling, the label is introduced onto the protein in the early stage of experiment, which minimizes inaccurate protein quantification emanating from variations in subsequent steps of sample preparation such as protein reduction, alkylation and proteolytic digestion. Furthermore, the ICAP-based strategy should be applicable for any biological samples, including clinical samples that are not amenable to metabolic labeling. Therefore, ICAP exhibits great potential in quantitative studies of nucleotide-binding proteins in the whole proteome scale.
This multifunctional affinity probe may be applied for two aspects of nucleotide-binding protein studies. In this vein, we have devised a quantitative affinity profiling strategy, encompassing the use of ICAP, to comprehensively characterize ATP-protein interactions at the entire proteome scale. A key advantage in charactering ATP-protein interaction with our affinity probe over previous binding assay lies in that our strategy allows for unambiguous determination of the interaction between ATP ligand and lysine residues in proteins of interest. False-positive identification of ATP-binding sites and the corresponding proteins due to non-specific labeling can be effectively minimized due to the specificity and quantitative nature of our strategy. Thus, ATP-protein interaction studies can be, for the first time, extended to quantitative surveys of specific binding regions of proteins at the entire proteome scale. It can be envisaged that similar nucleotide-affinity probes can be generally applied for the capture and characterization of proteins that can bind to other nucleotides. Moreover, the isotope-coded ATP probe with both enrichment and labeling capability facilitates future quantitative comparison of expression levels of ATPase and kinases originating from any biological samples.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 ES019873 to Y.W.) and Y.X. was supported by a Dissertation Research Award from the California Tobacco-Related Disease Research Program (20DT-0040). The authors want to thank Prof. Zhihao Zhuang for providing recombinant human PCNA and Prof. Huiwang Ai for help with the fluorescence microplate reader.
Footnotes
Supporting Information Available. Experimental procedures, GO analysis results, and detailed lists of possible ATP-binding proteins identified from Hela-S3 and Jurkat-T cells. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chene P. Nat Rev Drug Discov. 2002;1:665–673. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Stemmann O, Mann M. Mol Cell. 2008;31:438–448. doi: 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YN, Yu DJ, Li SS, Fan YX, Huang Y, Shen HB. BMC Bioinformatics. 2012;13:118. doi: 10.1186/1471-2105-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ormö M, Sjöberg BM. Anal Biochem. 1990;189:138–141. doi: 10.1016/0003-2697(90)90059-i. [DOI] [PubMed] [Google Scholar]
- 6.Guarnieri MT, Blagg BS, Zhao R. Assay Drug Dev Technol. 2011;9:174–183. doi: 10.1089/adt.2010.0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao Y, Guo L, Jiang X, Wang Y. Anal Chem. 2013;85:3198–3206. doi: 10.1021/ac303383c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Biochemistry. 2006;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 9.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 10.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJR. Nat Protocols. 2009;4:484–494.1. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 11.Qiu H, Wang Y. Anal Chem. 2007;79:5547–5556. doi: 10.1021/ac0622375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saraste M, Sibbald PR, Wittinghofer A. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 13.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Liu DR. Angew Chem Int Ed. 2004;43:4848–4870. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Nat Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 16.Varjosalo M, Björklund M, Cheng F, Syvänen H, Kivioja T, Kilpinen S, Sun Z, Kallioniemi O, Stunnenberg HG, He WW, Ojala P, Taipale J. Cell. 2008;133:537–548. doi: 10.1016/j.cell.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 17.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. J Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz D, Gygi SP. Nat Biotech. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 20.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Soti C, Vermes A, Haystead TAJ, Csermely P. Eur J Biochem. 2003;270:2421–2428. doi: 10.1046/j.1432-1033.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- 22.Soti C, Racz A, Csermely P. J Biol Chem. 2002;277:7066–7075. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- 23.Manoj N, Strauss E, Begley TP, Ealick SE. Structure. 2003;11:927–936. doi: 10.1016/s0969-2126(03)00146-1. [DOI] [PubMed] [Google Scholar]
- 24.Noiva R, Lennarz WJ. J Biol Chem. 1992;267:3553–3556. [PubMed] [Google Scholar]
- 25.Guthapfel R, Guéguen P, Quéméneur E. J Biol Chem. 1996;271:2663–2666. doi: 10.1074/jbc.271.5.2663. [DOI] [PubMed] [Google Scholar]
- 26.Chen S, Levin MK, Sakato M, Zhou Y, Hingorani MM. J Mol Biol. 2009;388:431–442. doi: 10.1016/j.jmb.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubala M, Plasek J, Amler E. Physiol Res. 2004;53:109–113. [PubMed] [Google Scholar]
- 28.Stewart RC, VanBruggen R, Ellefson DD, Wolfe AJ. Biochemistry. 1998;37:12269–12279. doi: 10.1021/bi980970n. [DOI] [PubMed] [Google Scholar]
- 29.Ni Q, Shaffer J, Adams JA. Protein Sci. 2000;9:1818–1827. doi: 10.1110/ps.9.9.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marek M, Milles S, Schreiber G, Daleke DL, Dittmar G, Herrmann A, Müller P, Pomorski TG. J Biol Chem. 2011;286:21835–21843. doi: 10.1074/jbc.M111.244525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T. J Biol Chem. 1995;270:22527–22534. doi: 10.1074/jbc.270.38.22527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.