

Abstract
Janus Kinases (JAKs) are essential mediators of almost all biological signalling events initiated by haemopoietic and immune cytokines. However, aberrant and/or prolonged JAK- induced signalling is detrimental and can give rise to a number of inflammatory and proliferative pathologies. For this reason the tyrosine kinase activity of the JAKs is carefully regulated at a number of different levels. Primarily this is achieved by: (1) Ensuring the catalytic domain is “switched off” under basal conditions and (2) Inhibiting the activity of JAK after it has been switched on. Whilst the first mode of inhibition is mediated by JAK’s own pseudokinase domain (JH2 domain) as well as the action of phosphatases, the second is achieved by the action of the SOCS (Suppressor of Cytokine Signalling) proteins, negative feedback inhibitors of JAK-mediated signalling. This review focuses on the mode of action of SOCS1 and SOCS3, the two most potent JAK inhibitors.
Keywords: Janus Kinases, SOCS, cytokine signalling, JAK/STAT
Introduction
Cytokines signal by binding to specific cell-surface receptors on their target cell. The cytoplasmic domains of these cytokine-receptors are bound by one or more members of the JAK (Janus Kinase) family[1, 2]. Prior to cytokine stimulation these JAKs are found in an “inactive” state. Cytokine exposure leads to a change in the conformation and/or oligomeric state of the receptor which allows trans-activation of JAKs [3, 4]. Once activated, JAKs phosphorylate the receptor and subsequently the Signal Transducers and Activators of Transcription (STAT) family of transcription factors. STATs are normally sequestered in the cytoplasm but once activated, they dimerise and translocate into the nucleus, where they upregulate transcription of the appropriate genes and thereby effect the appropriate biological response. [2, 5]
Negative feedback inhibition of JAK-mediated signalling: The SOCS family
As part of the response to cytokine stimulation, STATs directly upregulate the transcription of SOCS proteins [6–9], which inhibit the signalling cascade, creating a negative feedback loop (See Figure 1). This prevents prolonged cytokine signalling, which could result in chronic inflammation and promote aberrant proliferation and tumourigenesis.
Figure 1. Regulation of JAK/STAT signalling by SOCS3.
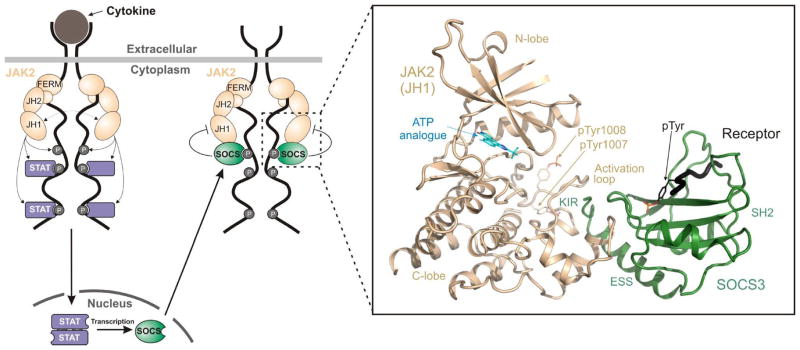
Schematic diagram of cytokine-induced JAK/STAT signalling. SOCS proteins are targets for STAT-induced upregulation whereupon they inhibit signalling, forming a negative feedback loop. The two most potent members of the SOCS family, SOCS1 and SOCS3, act by directly inhibiting the catalytic domain (JH1 domain) of JAK. The boxed area indicates the ternary complex structure solved in [17] and shown in the inset. Inset, the crystal structure of SOCS3 bound to JAK2 and a fragment of the IL-6 receptor (gp130 chain). PDB: 4GL9. Figure is reproduced with kind permission from [17].
There are eight classical SOCS proteins encoded in the human genome, SOCS1-7 and CIS [6]. All contain a central SH2 domain and a short, C-terminal domain termed the “SOCS box”. The SOCS box recruits the E3 ubiquitin ligase scaffold Cullin5 [10, 11] which then catalyses the ubiquitination of phosphorylated signalling intermediates bound to the SOCS SH2 domains [12–14]. These substrates are presumed to include both the JAKs themselves as well as the cytokine receptors to which they are bound although, to date, there are few validated targets for SOCS-mediated ubiquitination [12].
The two most potent suppressors of cytokine signalling are SOCS1 and SOCS3. In addition to ubiquitin ligase activity, these two proteins also suppress signalling via a second mechanism, making them distinct from the rest of SOCS family. This second mechanism involves a short motif immediately upstream of the SH2 domain, known as the KIR (kinase inhibitory region) [15, 16]. This enables SOCS1 and SOCS3 to directly inhibit the kinase activity of JAK and is their dominant mode-of-action in vivo. The molecular mechanism of this mode of inhibition was recently determined upon solving the crystal structure of SOCS3 bound to JAK2 [17] and is presented below.
The mechanism of JAK inhibition by SOCS3
In the late 1990s, Yoshimura and colleagues first demonstrated that overexpression of either SOCS3 or SOCS1 leads to reduced JAK autophosphorylation as well as reduced levels of phosphorylated STAT downstream [15, 16]. Using mutagenesis they identified a 12 aminoacid region upstream of the SH2 domain in both SOCS1 and SOCS3 that is required for this activity and hypothesized that these two SOCS proteins may bind directly to JAKs and inhibit their catalytic activity. Our recent biochemical [18] and structural [17] data support Yoshimura’s hypothesis and have uncovered the molecular mechanism of SOCS3- (and by analogy SOCS1-) mediated inhibition of JAK.
Using an in vitro kinase assay system consisting of purified recombinant enzyme (JAK2 kinase domain), substrate (gp130 cytoplasmic domain) and inhibitor (SOCS3) we demonstrated that SOCS3 inhibits the catalytic (JH1) domain of JAK2 with an IC50 of approximately 1 μM [18]. The IC50 was identical using a number of different substrates including proteins, peptides and synthetic polypeptides (poly-Glu4Tyr), indicating SOCS3 was interacting directly with JAK and not with substrate. Point mutations in either the KIR (F25A) or the SH2 domain (R71A) completely abrogated this inhibition. These data fit the previously proposed model in which SOCS3 tethers itself to JAK by binding phosphotyrosines in the JAK activation loop via its SH2 domain and then blocking the JAK active site using its KIR. Surprisingly however, the addition of large molar excess of a competitor phosphopeptide did not interfere with JAK inhibition, seemingly contradicting the R71A mutation data, and implying that SOCS3 does not bind JAK2 using the canonical phosphotyrosine-binding site of the SH2 domain. The explanation for this came when Kershaw et al., solved the structure of SOCS3 bound to JAK2 using X-ray crystallography [17].
The SOCS3:JAK2 crystal structure shows that SOCS3 does not interact with phosphotyrosines on JAK2 (Figure 1 inset). Instead, whilst SOCS3 binds JAK via its SH2 domain, it does so using a surface that is on the opposite side of this domain when compared to the canonical phosphotyrosine-binding groove. This surface consists of the ESS (the extended SH2 subdomain, an α-helical N-terminal extension to the SH2 domain found in all SOCS proteins), the loop between β-sheets B and C in the SH2 domain, the KIR, and regions in between. This leaves the phosphotyrosine binding groove open and able to bind a different ligand. Indeed, the structure solved by Kershaw et al., contained a phosphopeptide from the gp130 shared co-receptor (the Interleukin-6 receptor β chain) that was found to be located in the phosphotyrosine-binding groove of the SOCS3 SH2 domain and did not interfere with JAK binding.
SOCS3 uses a largely hydrophobic surface to anchor to the JAK2 catalytic domain and then inhibits the enzyme by placing the KIR (residues 22–29, which is unstructured in the absence of JAK [19, 20]) into the substrate-binding groove. The first residue of the KIR (Leu22) occupies the “P+1” binding pocket of JAK (Figure 2B), where the residue immediately C-terminal to the substrate tyrosine is predicted to bind. SOCS3 thereby inhibits JAK by blocking substrate from docking. This proposed mode-of-action has been supported biochemically in two ways: (A) Mutant forms of SOCS3 that incorporate a tyrosine residue 1–3 residues N-terminal to Leu22 of the KIR (i.e. residues 19–21 of the protein) are extremely efficient substrates for JAK-catalysed phosphorylation (Figure 2C) [17]. This is consistent with a model in which JAK2 remains catalytically active with SOCS3 bound but cannot phosphorylate substrates because SOCS3 blocks them from binding, however when SOCS3 is the substrate then catalysis can proceed efficiently; (B) truncating the KIR by 1–2 residues results in a SOCS3 construct that can bind but no longer completely inhibit JAK. For example, when JAK2 is saturated with a fragment of SOCS3 lacking only the first residue of the KIR it retains 25% of its activity. This data is consistent with a model in which truncated forms of SOCS3 cannot completely block substrate binding because of a reduced overlap between the KIR and the substrate (Figure 2D). In support of this, we found that when this overlap was reduced even further by using a C-terminally truncated form of the substrate, which only contained a single residue downstream of the tyrosine, inhibition was even less complete.
Figure 2. SOCS3 inhibits JAK1, JAK2 and TYK2 by blocking substrate binding.

(A)Schematic diagram of the domain structure of JAK2 and SOCS3. (B) SOCS3 (green, cartoon representation) docks onto the GQM motif of JAK (electrostatic surface representation) and places its KIR in the substrate binding groove.. The numbering indicates the exact fragments present in the crystal structure of PDB: 4GL9. (C) Close-up of the JAK:KIR interaction with a substrate peptide (white) modeled. *indicates that ATP and substrate are modeled based on the IRK:substrate:ATP structure (PDB 1IR3). The KIR of SOCS3 (green) blocks substrate binding, the first residue of the KIR, Leu22, is located where the P+1 residue would reside, this is indicated schematically in (D). Arg21 may act as a true pseudosubstrate residue however was not part of the crystallized construct and is therefore shown semi-transparent.
We would like to highlight the fact that the only fragment of SOCS3 we have been able to crystallise in complex with JAK lacks the first 21 residues of the native protein. Our structure hints that residue 21 may bind in the tyrosine binding pocket, thereby acting as a pseudosubstrate residue (See Figure 2D). This is supported by the fact that a tyrosine placed at residue 21 is an extremely efficient substrate [17]. Residue 21 in the native SOCS3 sequence is an arginine and in SOCS1 is a histidine and these are absolutely conserved throughout evolution. Both histidine and arginine have sidechains that are long enough to access the tyrosine binding pocket on JAK and also contain planar elements at their distal end that may stack onto Pro1017 of JAK2 in the same way that a substrate tyrosine would. Despite this Arg21 of SOCS3 did not provide any extra affinity for JAK2 and was not required for complete inhibition. Therefore any interactions it makes seem to be dispensable to SOCS3 function.
Overall our structural and biochemical data strongly imply that SOCS3 inhibits JAK by blocking substrate binding although one interesting conundrum remains: a steady-state analysis of SOCS3 inhibition of JAK2 revealed that SOCS3 displays non-competitive inhibitory kinetics both as regards ATP and substrate [18]. Whilst the SOCS3:JAK2 structure shows that SOCS3 does not compete with ATP binding, it does appear to compete with substrate binding and should therefore give rise to competitive kinetics when analysed under steady-state conditions. The reason for this disparity is currently unclear but the explanation may lie in the fact that whilst the KIR competes with substrate for binding, SOCS3 is tethered to JAK by a much larger surface that provides the majority of the affinity and does not compete with substrate.
Comparison to other kinase inhibitory systems
The SOCS3:JAK2 structure revealed parallels between SOCS3 inhibition of JAK and other pseudosubstrate-based kinase inhibitory systems, in particular Grb14 inhibition of the insulin receptor tyrosine kinase (IRK). Grb14 also acts by blocking substrate binding using a KIR-like region (termed the BPS “Between PH and SH2” region). This region is unstructured in the absence of IRK in the same way that the SOCS3 KIR is unstructured in the absence of JAK [21]. However, Grb14 anchors itself to IRK by using its SH2 domain to bind phosphotyrosines on IRK. In contrast SOCS3 anchors itself to a different surface on JAK2 via a phospho-independent mechanism. The surface targeted by SOCS3 is centered upon the N-terminal section of the αG-helix in JAK2 (and the important GQM motif immediately upstream, see next section). This helix is conserved amongst kinases and is close to the substrate binding site, therefore it is targeted by a number of pseudosubstrate-based inhibitors of other kinases including the auto-inhibitory regions of type I and type II PAKs (p21 associated kinases)[22, 23] and the viral protein K3L from vaccinia that inhibits Protein Kinase R[24].
SOCS3 targets specific JAK/Receptor pairs
There are four mammalian JAKs (JAK1-3 and TYK2) and these are unique amongst kinases in containing an approximately 20-residue insertion in their kinase domain structures between the F-helix and the aforementioned G-helix, termed the JAK insertion loop [25]. The last three residues of this insertion are Gly-Gln-Met (GQM) in JAK1, JAK2 and TYK2 but these are not conserved in JAK3 (Figure 3A). Using in vitro inhibition assays we showed that SOCS3 directly inhibits JAK1, JAK2 and TYK2, with similar affinity, but does not inhibit JAK3[18] and that this inhibition requires the presence of this GQM motif. Mutating either the Gly or the Met within the JAK2 GQM motif to the corresponding residues in JAK3 (Asp and Pro respectively), generated a mutant JAK2 protein that was fully active but no longer responsive to SOCS3-mediated inhibition. Our crystal structure confirms that this region encoding the GQM motif is important for the SOCS/JAK interaction.
Figure 3. SOCS3 inhibits JAK1, JAK2 and TYK2 but not JAK3 due to a three residue motif (GQM) in the JAK insertion loop.

(A) Sequence alignment of JAKs. Highly conserved residues are shown boxed in grey, the JAK insertion loop is indicted in red above the sequence and the GQM motif shown boxed in yellow. *Zebrafish JAK2b is grouped with TYK2 in this figure. (B) Sequence alignment of SOCS1/SOCS3 conserved residues in the KIR are shaded black. (C) The structure of JAK2, PDB ID 2B7A. The GQM motif is solvent exposed and shown in yellow and the JAK insertion loop shown in red. Figure is reproduced with kind permission from [18] (copyright Elsevier Inc.).
An examination of JAK evolution is telling in this regard. Only vertebrates have evolved an expanded JAK system, consisting of four JAKs whereas lower organisms, such as insects, contain only a single JAK homologue (Hopscotch in Drosophila). Likewise, only vertebrates have evolved SOCS1 and SOCS3 homologues with a kinase inhibitory region (Figure 3B). The GQM motif is conserved in JAK1, JAK2 and TYK2 in all vertebrates and is always absent in JAK3 (Figure 3A). Therefore vertebrates have evolved a more complex JAK repertoire alongside the ability to inhibit only a subset of them. The advantage of having JAK3 insensitive to SOCS-mediated inhibition remains to be determined.
In addition to specificity towards particular JAKs, SOCS3 also targets particular cytokine receptors by binding specific phosphotyrosine motifs (Table 1). These include the receptors for G-CSF[26] and Leptin[27] and most importantly the gp130 shared co-receptor[28] which is used by a number of different cytokines including IL-6, IL-11, Leukemia inhibitory factor (LIF), Granulocyte colony-stimulating factor (G-CSF) and Oncostatin M (OSM). The crystal structure of the SOCS3:JAK2 complex shows that SOCS3 can bind JAK2 and gp130 at the same time, using two distinct surfaces. In vivo, JAK is also bound to gp130 (via its FERM domain), setting up a three-way interaction, whereby each component of the system can interact with the other two simultaneously. This implies that whilst SOCS3 binds gp130 or JAK2 alone with moderate affinity (0.1–1 μM) it will bind a JAK2/gp130 heterodimer with much higher avidity. The dissociation constant could be as strong as 10−12 M given that, theoretically, a ligand that binds two individual and non-overlapping sites on a receptor simultaneously does so with an affinity that approaches the product of the individual Kd values rather than the sum. Further work is required to determine if this is truly the case with SOCS3.
Table 1.
Cytokines inhibited by SOCS3*.
| Cytokine | Receptor Chain 1† | Receptor chain 2 | SOCS3 pTyr site | Associated JAKs |
|---|---|---|---|---|
| IL-6 LIF CNTF |
IL-6Ra LIFR LIFR |
gp130 | pY757 | JAK1, JAK2, TYK2 |
| Leptin | LepR | LepR | pY985 | JAK2 |
| G-CSF | GCSFR | GCSFR | pY729 | JAK1, JAK2, TYK2 |
As demonstrated by genetic deletion. See [29] for review.
Receptors and JAKs shown in bold contain SOCS3 binding sites
Taken together, the affinity of SOCS3 for particular JAKs and particular receptors allows it to target certain signalling pathways with high specificity. These pathways are those that signal through JAK1, JAK2 or TYK2 via a receptor that contains a SOCS3 binding motif. Currently there are a number of cytokines that fit these criteria, namely: IL-6, IL-11, G-CSF, LIF, OSM, Leptin and potentially EPO. The importance of SOCS3 for IL-6, LIF, Leptin and G-CSF has been demonstrated by genetic deletion (see Table 1).
It appears likely that SOCS1 acts via the same mechanism as SOCS3. Not only does the KIR of SOCS1 share a high degree of sequence similarity to that of SOCS3 but the amino acid residues that form the 3-dimensional surface of SOCS3 that contacts JAK are almost all conserved in SOCS1 [17]. Therefore, we would predict that SOCS1 will target the same JAKs as SOCS3 (JAK1, JAK2 and TYK2) but will target different receptors due to sequence divergence within the pTyr-binding grooves of their SH2 domains. To date, little is known about which sites on which receptors are SOCS1 targets, with the exception of pTyr441 in the interferon-γ receptor. It is of great importance to determine which JAKs and receptors are targeted by SOCS1 in order to determine the full repertoire of cytokines regulated by these two potent and important signalling inhibitors. Our current efforts lie in this direction.
Acknowledgments
The original research described in this review was supported by the National Health and Medical Research Council of Australia (program grant nos. 461219 and 487922, 1011804), the U.S. National Institutes of Health (grant no. CA22556), the Victorian State Government Operational Infrastructure Support Grant, and the NHMRC Independent Research Institutes Infrastructure Support Scheme (361646). N.A.N. acknowledges fellowship support from the National Health and Medical Research Council, J.M.M and J.J.B. from the Australian Research Council.
Abbreviations
- JAKs
Janus Kinases
- SOCS
Suppressor of Cytokine Signalling)
- STAT
Signal Transducers and Activators of Transcription
- IRK
insulin receptor tyrosine kinase
References
- 1.Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86:1603–1607. doi: 10.1073/pnas.86.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilks AF, Oates AC. The JAK/STAT pathway. Cancer Surv. 1996;27:139–163. [PubMed] [Google Scholar]
- 3.Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JE. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993;366:580–583. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 5.Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Thierfelder WE, Kreider B, Silvennoinen O. Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem Sci. 1994;19:222–227. doi: 10.1016/0968-0004(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 6.Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 8.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 9.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 10.Babon JJ, Sabo JK, Soetopo A, Yao S, Bailey MF, Zhang JG, Nicola NA, Norton RS. The SOCS box domain of SOCS3: structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J Mol Biol. 2008;381:928–940. doi: 10.1016/j.jmb.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babon JJ, Sabo JK, Zhang JG, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol. 2009;387:162–174. doi: 10.1016/j.jmb.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, Hattori K, Hatakeyama S, Yada M, Morita S, Kitamura T, Kato H, Nakayama K, Yoshimura A. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276:12530–12538. doi: 10.1074/jbc.M010074200. [DOI] [PubMed] [Google Scholar]
- 13.Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang JG, Metcalf D, Rakar S, Asimakis M, Greenhalgh CJ, Willson TA, Starr R, Nicholson SE, Carter W, Alexander WS, Hilton DJ, Nicola NA. The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc Natl Acad Sci U S A. 2001;98:13261–13265. doi: 10.1073/pnas.231486498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, Sasaki M, Johnston JA, Yoshimura A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes to Cells. 1999;4:339–351. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- 16.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. Embo J. 1999;18:1309–1320. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–476. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36:239–250. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babon JJ, Yao S, DeSouza DP, Harrison CF, Fabri LJ, Liepinsh E, Scrofani SD, Baca M, Norton RS. Secondary structure assignment of mouse SOCS3 by NMR defines the domain boundaries and identifies an unstructured insertion in the SH2 domain. Febs J. 2005;272:6120–6130. doi: 10.1111/j.1742-4658.2005.05010.x. [DOI] [PubMed] [Google Scholar]
- 20.Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, Willson TA, Hilton DJ, Nicola NA, Baca M, Nicholson SE, Norton RS. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. 2006;22:205–216. doi: 10.1016/j.molcel.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 21.Moncoq K, Broutin I, Larue V, Perdereau D, Cailliau K, Browaeys-Poly E, Burnol AF, Ducruix A. The PIR domain of Grb14 is an intrinsically unstructured protein: implication in insulin signaling. FEBS Lett. 2003;554:240–246. doi: 10.1016/s0014-5793(03)01095-0. [DOI] [PubMed] [Google Scholar]
- 22.Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ, Harrison SC. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- 23.Ha BH, Davis MJ, Chen C, Lou HJ, Gao J, Zhang R, Krauthammer M, Halaban R, Schlessinger J, Turk BE, Boggon TJ. Type II p21-activated kinases (PAKs) are regulated by an autoinhibitory pseudosubstrate. Proc Natl Acad Sci U S A. 2012;109:16107–16112. doi: 10.1073/pnas.1214447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elde NC, Child SJ, Geballe AP, Malik HS. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 2009;457:485–489. doi: 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–183. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 26.Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, Zhang JG, Hilton DJ, Nicola NA, Alexander WS, Roberts AW. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20:153–165. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- 27.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 28.Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, Metcalf D, Hilton DJ, Nicola NA, Baca M. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A. 2000;97:6493–6498. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babon JJ, Nicola NA. The biology and mechanism of action of suppressor of cytokine signaling 3. Growth Factors. 2012;30:207–219. doi: 10.3109/08977194.2012.687375. [DOI] [PMC free article] [PubMed] [Google Scholar]