

Abstract
The maximum per capita rate of population growth, r, is a central measure of population biology. However, researchers can only directly calculate r when adequate time series, life tables and similar datasets are available. We instead view r as an evolvable, synthetic life-history trait and use comparative phylogenetic approaches to predict r for poorly known species. Combining molecular phylogenies, life-history trait data and stochastic macroevolutionary models, we predicted r for mammals of the Caniformia and Cervidae. Cross-validation analyses demonstrated that, even with sparse life-history data, comparative methods estimated r well and outperformed models based on body mass. Values of r predicted via comparative methods were in strong rank agreement with observed values and reduced mean prediction errors by approximately 68 per cent compared with two null models. We demonstrate the utility of our method by estimating r for 102 extant species in these mammal groups with unknown life-history traits.
Keywords: Caniformia, intrinsic rate of natural increase, macroevolutionary model, mammal life history, population doubling time, species conservation
1. Introduction
The maximum per capita rate of population growth, r, also called the intrinsic rate of increase, is a central measure of population biology. It helps to determine the stability and dynamics of populations and to differentiate species with regard to extinction risks, conservation needs and invasion potential [1–3]. Despite the broad relevance of r to theoreticians and empirical biologists alike, the time series, life tables and other datasets from which r can be calculated directly are available for only a tiny subset of wild species [4]. As a result, researchers have long relied upon approximations of r based on life-history traits, allometric regressions and correlative analyses [5–10]. Despite widespread use, some of these methods are badly biased [11,12] and others are often imprecise [13]. None of these methods take into consideration species' shared evolutionary histories.
Historically, ecologists have turned to the Euler equation from demography [14] to calculate r. The Euler equation is
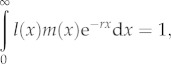 |
1.1 |
where l(x) is the survivorship to age x (i.e. the proportion of individuals that survive to age x), and m(x) is the per capita fecundity of female offspring at age x [15]. To obtain r from equation (1.1), one must have empirical survivorship and fecundity schedules, or must make assumptions about the shape and scale of those schedules.
In broad comparative analyses, one cannot generally apply equation (1.1) directly because survivorship and fecundity schedules are so rarely available. For example, Lynch & Fagan [11] found lifetables for only 58 species across all approximately 5400 mammals, and the comADRe database, which is a global compilation of demographic projection matrices, includes matrices for only 139 mammal species [16]. In addition, because mammals differ so much in generation times, analyses based on equation (1.1) can be mathematically unwieldy in multispecies comparisons [17].
Given these difficulties, comparative analyses exploring the relationship between life-history and population growth rate are generally based on an approximation of equation (1.1). Many authors have adopted a step-function approximation to l(x), first proposed by Cole [5], which assumes an extreme form of type I survivorship [18,19]. Although this approximation is convenient and commonly used, its approach to survivorship, and its assumption that reproduction occurs on a strictly annual basis, leads to extreme overestimates of r [11].
An alternative approximation to equation (1.1) assumes type II (exponential) survivorship and allows for episodic, pulsed reproduction rather than continuous reproduction [17]. Both of these biologically realistic modifications are especially appropriate for mammalian life history. With these changes, equation (1.1) becomes
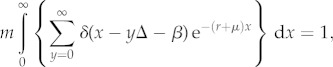 |
1.2 |
where r is the maximum population growth rate, m is the maximum number of female offspring per reproductive episode (litter), Δ is the average interval between litters, β is the minimum age of first reproduction and μ is the average mortality rate. δ(z) is an interval delta function that equals 1/T for 0 < z < T and is zero otherwise, where T is the duration of the mammalian ‘birth pulse’, which is taken to be one day [17]. This model does not constrain reproduction to occur on an annual basis but does assume constant fecundity per birth event. The integral and sum in equation (1.2) can be evaluated, yielding
| 1.3 |
In a survivorship analysis spanning 58 mammal species, values of r from equation (1.2) provided excellent matches to values obtained via the full empirical schedules, whereas those from the Cole approximation to equation (1.1) were badly biased [11,12]. Because equation (1.2) requires only life-history trait data to estimate r but yields estimates that agree closely with those obtained from full lifetable data, this method of calculating r balances reasonable outputs with limited data requirements [11,12,17]. (Note that the variant of r given in equation (1.2) was denoted 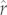 in previous work to differentiate it from other population growth measures [11,17]. However, to avoid confusion with the statistical estimates of the trait that appear below, we have opted for the simpler notation, r, here)
in previous work to differentiate it from other population growth measures [11,17]. However, to avoid confusion with the statistical estimates of the trait that appear below, we have opted for the simpler notation, r, here)
The parameters m, Δ, β and μ are all fundamental life-history traits shaped by evolution [20]. Collectively, these traits contribute to functional and performance-related variation among taxa under conceptual frameworks such as the fast–slow life-history continuum [21,22]. As calculated in equations (1.1)–(1.2), r represents a synthetic measure of a species maximum per capita rate of population growth in the absence of density dependence. Consequently, r is both a central measure of population biology and a conservation-relevant metric that, among other things, characterizes how quickly a population could increase in size, such as when recovering after a major disturbance or population collapse. Note that r is not the realized population growth rate, which will reflect year-to-year variation in density, demographic and environmental stochasticity and other factors. Even though estimating r via equation (1.3) is far less data-intensive than other widely used methods, this approach still requires data on life-history traits that are lacking for many species. In those cases, a method is needed to predict species' capacities for population growth in the absence of species-specific life-history data.
Here, we illustrate how phylogenetic comparative methods can greatly expand the suite of species for which it is possible to estimate r. Because individual life-history and ecological strategies are often phylogenetically structured [23], we view r as a synthetic life-history trait that varies among species within a clade. This approach is warranted because inheritance from a common ancestor coupled with phylogenetic inertia routinely yields situations in which similar trait values cluster across related species [24]. Furthermore, it is exactly these types of relationships that, along with shared environmental factors, underpin the phylogenetic structuring of extinction risk and endangerment status across species [25–27]. Using established phylogenies, we examine how successfully macroevolutionary models recover r-values for well-studied species. We then leverage phylogenetic relationships and r-values obtained for well-studied species to predict r for more poorly studied species.
2. Material and methods
(a). Tree-based models
We use phylogenetic independent contrasts (PIC) to predict values of r for extant species in the context of their shared evolutionary history [28–30]. This technique assumes a Brownian motion model for trait evolution, where the variance parameter σ2 describes the scale of fluctuations in the unbiased random walk. Because r is fundamental to a species' survival, we expect it to evolve gradually rather than wildly, reflecting critical life-history trade-offs such as longevity versus fecundity [20].
We consider two PIC models. In the first, r for an extant species is predicted only from the values of r known for other extant species and the phylogeny of the clade (‘PIC-r’ model; [29]). In the second, female body mass is additionally included as a covariate (‘PIC-r-mass’ model; [30]). Body mass estimates were available for all species included in the phylogenies we used, except for four species from the Caniformia. We log-transformed body mass for all comparative analyses. Comparisons among these two PIC models and the two alternative null models described below illuminate the forces underlying interspecific variation in r.
Under either model, the phylogenetic position is known for each species whose value of r was to be predicted. For each of these ‘unknown’ species in turn, the expected value of r and its uncertainty was obtained, as detailed by Garland et al. [29, eqn A10] and Garland & Ives [30, eqn A15] and summarized here. First, the tree was pruned to contain only the unknown species and all species with known r-values. Second, the tree was temporarily rooted at the node immediately parental to the unknown tip species. Third, PIC was used to estimate r at this root. Finally, this root estimate was extended along the branch to the unknown tip. Under PIC-r, the expected value of r at the unknown tip was the same as at the temporary root. Under PIC-r-mass, it depended also on the difference between tip and root body mass, where the latter was similarly estimated by PIC. The variance of the estimate for r was increased by σ2 (also estimated by PIC) multiplied by the unknown tip branch length.
We examine the performance of these approaches via cross-validation analyses for two mammalian clades, each of which includes species of conservation interest: the Caniformia (where r is calculable via equation (1.3) for many species) and the Cervidae (where data density are lower). We then apply them to predict r-values for species currently lacking estimates.
(b). Life-history data
(i). Empirical estimates of r
Values of r were calculated by inserting life-history trait data obtained from published compilations [31–33] into equation (1.3) [17] (see the electronic supplementary material, appendix C). We refer to these calculated values as our observations, against which we contrast our predictions from the PIC-r and PIC-r-mass models and two alternative null models (detailed below). When multiple values for particular life-history traits led to multiple values of r for a single species, we used the geometric mean of those values in our analyses. Future efforts will incorporate intraspecific variation in estimates of r and/or estimates of other measures of population growth rate [12] that are independently available.
(ii). Body mass
Body mass values were obtained from published compilations [17,31,32,34].
(c). Phylogenetic trees
Because both of the evolutionary models discussed above work best in clades with good phylogenetic resolution [35], we examined the evolution of r in two mammalian groups with well-studied phylogenies (see the electronic supplementary material, appendix C). We investigated the carnivore suborder Caniformia because of the relative wealth of life-history data from which we could calculate r for caniform species worldwide (65 of 140 species, see below). We obtained the molecular (cytB), species-level phylogeny of the order Carnivora (parent to the Caniformia) from Agnarsson et al. [36], which includes 82 per cent of taxa in the order Carnivora, and smoothed it to an ultrametric tree using the R function chronopl [37].
We also examined the family Cervidae in which estimates of r were more sparsely available. We used a lower-resolution phylogeny for Cervidae, extracted from the Bininda-Emonds et al. [38] mammalian supertree in an already ultrametric form. This phylogeny includes 76 per cent of cervid taxa with 45 per cent bifurcation completeness [38].
Additionally, we considered separately three caniform clades (the Pinnipedia, Musteloidea and Canidae) and one cervid clade (the Plesiometacarpalia), each of which had 10 or more species with estimates of r available. These additional analyses allow us to consider the possibility that r may evolve differently on different parts of the tree.
To assess the appropriateness of applying Brownian motion-based phylogenetic comparative methods to our data, we first tested for significant phylogenetic signal [39] using a randomization test implemented in the R package picante. We also calculated Pagel's λ [40,41] and used a likelihood ratio test [42] to assess whether a branch length transform was necessary for the amount of trait evolution to be proportional to branch length.
(d). Model assessment
(i). Cross-validation
We used leave-one-out cross-validation to test model performance. For each species with a known r-value, taken one at a time, this value was ignored and the species was treated as ‘unknown’ in the prediction procedure. This was carried out for each model, the PIC models described above and the null models outlined below.
Model performance was assessed in three ways. First, we assessed general agreement of the predicted and observed values by examining the relationship between the predicted 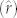 and observed values for each group of species. Second, we assessed accuracy by comparing proportional prediction errors, computed as
and observed values for each group of species. Second, we assessed accuracy by comparing proportional prediction errors, computed as 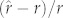 for each species. (Absolute prediction errors
for each species. (Absolute prediction errors  are considered in the electronic supplementary material, appendix B.) Third, we assessed accuracy by scoring the proportion of species for which the 95% prediction intervals from the model included the observed r-values.
are considered in the electronic supplementary material, appendix B.) Third, we assessed accuracy by scoring the proportion of species for which the 95% prediction intervals from the model included the observed r-values.
(ii). Alternative null models
As a benchmark against which to judge the predictive improvement provided by tree-based PIC models for r, we considered an allometric model in which we used phylogenetically corrected least-squares regression to account for correlated errors owing to phylogenetic relatedness [43]. Further justification for the use of this model, details on its implementation and variants, and discussion of its implications for metabolic theory appear in the electronic supplementary material, appendix A. The allometric analysis incorporates non-independence owing to shared evolutionary history among species when determining the relationship between body size and r, but it does not consider the phylogenetic position of an unknown species when predicting its value of r. In other words, the allometric null regression models represent static mappings between female body size and r for the suite of species under consideration, whereas the PIC models customize predictions for the target species based additionally on their shared evolutionary history with the rest of the clade [30].
As a second alternative, we considered a null model that incorporates neither body mass nor phylogenetic information. In this model, observed r-values are treated as independent samples from a normal distribution, for which the mean and variance are estimated from the known species in the clade. This model, which we term the Brownian motion null model, is equivalent to the Brownian motion model of r evolution used by the PIC-r model, but on a star-shaped rather than bifurcating tree. The allometric null and Brownian motion r-only models provide contrasts, respectively, with the PIC-r-mass and PIC-r tree-based models.
(e). Prediction
Using the PIC-r and PIC-r-mass approaches described above, we estimated values for those members of the Caniformia and Cervidae (75 and 27 species, respectively) for which r was not calculable from available life-history data. This procedure considered each unknown tip in turn, analogous to the cross-validation technique.
3. Results
(a). Quantifying phylogenetic conservatism of r
The Caniformia data exhibited a statistically significant level of phylogenetic signal for population growth rate r (p < 0.001, with Blomberg's K = 0.68) and did not require a branch length transform to improve the fit of the Brownian motion trait evolution model that underlies PIC (λ = 0.96, p = 0.46). The same was true for Cervidae, with significant phylogenetic signal (p = 0.001, with K = 1.42) and no need for a branch length transform (λ = 0.95, p = 0.81). We therefore proceeded with the PIC analyses on these data.
(b). General agreement of tree-based estimates and observed values of r
In cross-validation tests, predictions from the tree-based PIC-r and PIC-r-mass models showed good general agreement with observed values calculated from life-history traits using equation (1.3) for both the Caniformia and the Cervidae (figure 1). In the Caniformia, prediction errors from PIC-r were distributed roughly equally around a 1 : 1 line of correspondence with two exceptions, a diverse group of species with small–medium-observed values of r (which were overestimated) and two species of weasels with large-observed values of r (which were underestimated; figure 1). Overall, for both taxa, both the PIC-r and PIC-r-mass models tended to overestimate r for species with small values of r and to underestimate r for species with large values of r.
Figure 1.

Relationship between observed r as calculated from equation (1.3), and predicted r as estimated by the (a,b) PIC-r model and (c,d) PIC-r-mass model for (a,c) Caniformia and (b,d) Cervidae. Dashed lines are 1 : 1, representing perfect prediction by the model. Solid lines represent an ordinary least-squares fit (Caniformia: y = 0.52x + 0.35, R2 = 0.57; Cervidae: y = 0.22x + 0.25, R2 = 0.22). In (a,c), individual families are: Ailuridae (closed circles), Canidae (open circles), Mephitidae (closed triangles), Mustelidae (open triangles), Otariidae (closed diamonds), Phocidae (open diamonds), Procyonidae (closed squares) and Ursidae (open squares).
For both groups of species, at least one of the tree-based models yielded a significant rank correlation between the predicted and observed values. For the Caniformia, rank correlations were quite strong for both the PIC-r model (Spearman's ρ = 0.82; p < 0.0001) and PIC-r-mass model (Spearman's ρ = 0.79; p < 0.0001). Thus, the tree-based models were particularly good at recovering differences in the relative values of r across species, which varied over a 47-fold range in the caniform data. In the Cervidae, predicted 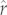 and observed r were significantly rank correlated for the PIC-r model (Spearman's ρ = 0.55; p < 0.03), which is especially noteworthy considering the modest level of variation in r among the Cervidae. However, this did not hold for the cervid PIC-r-mass model, which did not perform as well as the PIC-r model by other metrics, as will be discussed below. Overall, the strong and significant rank correlations emerging from the PIC-r model represented substantial improvements over the corresponding allometric-null models (Spearman's ρ < 0.60 and <0.35 for the Caniformia and the Cervidae, respectively).
and observed r were significantly rank correlated for the PIC-r model (Spearman's ρ = 0.55; p < 0.03), which is especially noteworthy considering the modest level of variation in r among the Cervidae. However, this did not hold for the cervid PIC-r-mass model, which did not perform as well as the PIC-r model by other metrics, as will be discussed below. Overall, the strong and significant rank correlations emerging from the PIC-r model represented substantial improvements over the corresponding allometric-null models (Spearman's ρ < 0.60 and <0.35 for the Caniformia and the Cervidae, respectively).
(c). Comparison of tree-based and alternative null models
The phylogenetically corrected allometric null model tended to yield biased estimates of the observed r (see the electronic supplementary material, figure S1a,b), overestimating r for 41 of 65 caniform species (mean overestimation = 177%). As noted above, the tree-based models tended to overestimate r for small r species, and when such overestimations occurred they were smaller (e.g. median overestimation of 47% and 6% for Caniformia and Cervidae, respectively, using PIC-r; electronic supplementary material, figure S1e–h). For the Caniformia, PIC-r predictions correlated with the corresponding allometric null predictions, but yielded a relationship steeper than 1 : 1 (see the electronic supplementary material, figure S2). Mean prediction errors were always larger for the allometric null models than for the tree-based models, ranging from 2.0 to 2.3× larger for the Caniformia and from 1.1 to 1.3× larger for the Cervidae, all indicating improvement when using the tree-based methods (table 1). For the Brownian motion null model, cross-validation on the null tree yielded very little variation in predicted 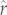 and confidence limits across species (see the electronic supplementary material, figures S3 and S4) but entailed substantial prediction errors (see the electronic supplementary material, figure S1c,d). For example, the Brownian motion null models yielded median prediction errors 3.3× and 2.4× as big as the best performing tree-based models for the Caniformia and the Cervidae, respectively (table 1 and electronic supplementary material, figure S1c–f).
and confidence limits across species (see the electronic supplementary material, figures S3 and S4) but entailed substantial prediction errors (see the electronic supplementary material, figure S1c,d). For example, the Brownian motion null models yielded median prediction errors 3.3× and 2.4× as big as the best performing tree-based models for the Caniformia and the Cervidae, respectively (table 1 and electronic supplementary material, figure S1c–f).
Table 1.
Cross-validation performance for a phylogenetically corrected allometric null model, a Brownian motion null model and two phylogenetically structured models of maximum per capita population growth rate in the Caniformia and Cervidae. (The columns ‘within 1 s.e.’ and ‘within 95% CI/PI’ give, respectively, the proportion of species whose observed r-values fell within 1 s.e. and within the 95% prediction interval PI of the predicted means from the Brownian motion and PIC models (or within the 95% confidence interval (CI) of the allometric null model). The columns min, median, mean and max characterize the precision of the different models, determined across species using the absolute value of 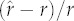 .)
.)
| taxon | model | within 1 s.e. | within 95% CI/PI | prediction errors |
|||
|---|---|---|---|---|---|---|---|
| min | median | mean | max | ||||
| Caniformia | allometric null | 36/65 | 63/65 | 0.004 | 0.77 | 1.21 | 6.49 |
| Brownian motion null | 58/65 | 63/65 | 0.011 | 0.83 | 1.71 | 7.39 | |
| PIC-r | 56/65 | 63/65 | 0.003 | 0.25 | 0.53 | 4.93 | |
| PIC-r-mass | 51/61 | 58/61 | 0.002 | 0.37 | 0.61 | 6.11 | |
| Cervidae | allometric null | 11/15 | 14/15 | 0.018 | 0.08 | 0.16 | 0.57 |
| Brownian motion null | 12/15 | 14/15 | 0.023 | 0.19 | 0.19 | 0.65 | |
| PIC-r | 11/15 | 13/15 | 0.012 | 0.08 | 0.13 | 0.43 | |
| PIC-r-mass | 11/15 | 13/15 | 0.006 | 0.08 | 0.15 | 0.54 | |
The confidence intervals from the allometric null models and the prediction ranges from the Brownian motion null models were typically wide (exceedingly so for the allometric null model whose error structure is determined by the power-law relationship between r and mass; electronic supplementary material, appendix A; electronic supplementary material, figures S3 and S4). These ranges from the null models were much wider than the corresponding ranges from the tree-based models (which are shown in electronic supplementary material, figure S5). For example, for the PIC-r model, 48 of 65 caniform species and 13 of 15 cervid species had prediction ranges smaller than the corresponding Brownian motion null models (average improvement: 28% and 26%, respectively). When using the allometric and Brownian motion null models, nearly all observed values fell within the estimated confidence intervals or prediction intervals (table 1). However, this result is not an indicator of good model performance but rather a consequence of the wide intervals themselves.
(d). General comparisons between the PIC-r and PIC-r-mass phylogenetic models
Overall, we found better performance by the simple PIC-r model of trait evolution than by the more complex PIC-r-mass model (table 1 and figure 1; electronic supplementary material, figure S1). In particular, we found that the addition of body mass as a covariate did not improve our ability to predict r, and for the Caniformia, the inclusion of the mass covariate actually yielded worse predictions (median prediction error was 48% larger with PIC-r-mass). This stems from a bias-variance trade-off: adding the covariate always decreased bias, such that the predictions were more correct on average, but the variance increased so the predictive error for some species was larger.
For both the Caniformia and the Cervidae (and all subgroups we examined), estimates of σ2, the Brownian motion variance for r, were marginally, but uniformly, smaller in the PIC-r-mass models than in the simpler PIC-r models (see the electronic supplementary material, table S2), suggesting a portion of the evolutionary ‘rate’ signal in r is instead explained by mass or a trait that covaries with mass. For both Caniformia and Cervidae, slope estimates from phylogenetically corrected regressions of PIC-r prediction error against mass are very nearly zero, helping to explain why the more complex PIC-r-mass model does not improve fit (see the electronic supplementary material, appendix A). Another possibility, that the mass-r relationship is ‘noisy’ and therefore masks the benefits of including mass as a covariate in the PIC models, is not supported (see the electronic supplementary material, appendix B).
Regardless of the tree-based model used, the 95% prediction intervals of 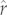 values estimated by the cross-validation procedure were large (and for the Caniformia, often encompassed zero). However, the observed values for 83–86% and 73 per cent of caniform and cervid species, respectively, fell within 1 s.e. of the predicted mean (table 1; electronic supplementary material, figure S5). Prediction intervals were smaller for the PIC-r-mass model than for the PIC-r model in 55 of 61 species-wise comparisons in the Caniformia, but shrank by an average of only 1.3 per cent (±0.5 s.e.).
values estimated by the cross-validation procedure were large (and for the Caniformia, often encompassed zero). However, the observed values for 83–86% and 73 per cent of caniform and cervid species, respectively, fell within 1 s.e. of the predicted mean (table 1; electronic supplementary material, figure S5). Prediction intervals were smaller for the PIC-r-mass model than for the PIC-r model in 55 of 61 species-wise comparisons in the Caniformia, but shrank by an average of only 1.3 per cent (±0.5 s.e.).
(e). Comparative model performance for the Caniformia, a case of moderate data availability
For the Caniformia, the better performing PIC-r model yielded median and mean absolute prediction errors smaller than those of the PIC-r-mass model (25% versus 37% and 53% versus 61%, respectively; table 1). In addition, for the better performing PIC-r model, 75 per cent of caniform species had prediction errors less than 62 per cent. Averaged across species, predictions from the PIC-r-mass model were 9 per cent higher than the corresponding predictions from the PIC-r.
Measured in terms of proportion prediction error (table 1), the PIC-r model did better than the PIC-r-mass model for 37 out of 61 species (four comparisons were not possible because of missing data). This included a majority of species in the Canidae (7 out of 11) and Pinnipedia (13 out of 20). Overall, the improvement in proportional prediction error afforded by the PIC-r model over the PIC-r-mass model correlated positively with two traits: biomass (ρ = 0.36; p < 0.004), and mean lifespan (ρ = 0.38; p < 0.003). These two traits were themselves correlated such that neither was significantly correlated to the decrease in error, once the other was accounted for.
One suite of species (e.g. members of the Lutridae plus species with long tip-lengths relative to other members of their clades, such as Potos flavus and Ailuropoda melanoleuca) tended to have large prediction errors for both tree-based models (see the electronic supplementary material, figure S6). Overall, however, we found no correlation between prediction error and tip length (p = 0.85).
(f). Comparative model performance for the Cervidae, a case of very limited data availability
For the Cervidae, the PIC-r and PIC-r-mass models performed comparably in terms of accuracy and precision (table 1). Even with few observations of r available for cervid species, approximately 87 per cent of the observed values fell within the 95% prediction interval of the predicted values using either tree-based model (table 1 and electronic supplementary material, figure S5). For both models, median prediction error was approximately 8 per cent, and 80 per cent of species had prediction errors less than 22 per cent (table 1). Prediction errors were comparable between the PIC-r and PIC-r-mass models across cervid species (see the electronic supplementary material, figure S7). Cervid PIC-r prediction errors were positively correlated with inter-litter interval, and negatively correlated with observed r and mass, but exhibited no correlations with other life-history traits nor with tip length (see the electronic supplementary material, appendix B).
(g). Predictions
Reconstruction of 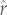 for ancestral nodes and prediction for tip species in both clades using the best-fit PIC-r models are shown in figure 2 and reported in electronic supplementary material, table S1.
for ancestral nodes and prediction for tip species in both clades using the best-fit PIC-r models are shown in figure 2 and reported in electronic supplementary material, table S1.
Figure 2.

Phylogenetic relationships within the (a) Caniformia and (b) Cervidae giving predicted node and tip reconstruction of 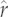 for the PIC-r models. The circles at each tip or node have diameter proportional to values of r ranging from −0.647 to 2.75 (a) and between 0.22 and 0.55 (b). Orange circles represent tips with observed r-values, green circles show reconstructed nodes from which tips were predicted and blue circles represent unknown, predicted tips. For green and blue, the darker inner and lighter outer circles show the estimated
for the PIC-r models. The circles at each tip or node have diameter proportional to values of r ranging from −0.647 to 2.75 (a) and between 0.22 and 0.55 (b). Orange circles represent tips with observed r-values, green circles show reconstructed nodes from which tips were predicted and blue circles represent unknown, predicted tips. For green and blue, the darker inner and lighter outer circles show the estimated 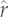 -value minus or plus the s.e., respectively.
-value minus or plus the s.e., respectively.
4. Discussion
Knowledge of species' potential population growth rates is critical for understanding population dynamics and informed conservation decision-making [44]. Because of this, researchers have estimated growth rate parameters using demographic traits in concert with allometric regressions and related approaches [8,13,17]. By contrast, our approach leveraged species shared evolutionary history to predict potential population growth rates, and it performed well even when only limited life-history data were available to inform the predictions. The tree-based methods we adopted routinely yielded credible predictions of r within each of two dissimilar mammalian groups, thereby improving over null models (table 1; electronic supplementary material, figure S1). Importantly, application of the modelling approach to the smaller, less diverse cervid clade proved robust to both limited observed r data and incomplete phylogenetic resolution, two problems likely to appear in other taxa.
(a). Relevance to conservation biology and life-history theory
By leveraging data from better known species to inform understanding of poorly known species, phylogenetic comparative methods help fill a gap in the toolkit of quantitative conservation biology, providing conservation practitioners with a method for predicting species' capacities for population growth when no species-specific trait data are available (figure 2; electronic supplementary material, table S1). Without the appropriate suite of life-history trait data, it is not possible to parametrize equations (1.2)–(1.3) for a focal species. Previously, this left conservation practitioners without much guidance as to that species' capacity for population growth or recovery (but see [45]). By contrast, using phylogenetic comparative approaches, researchers can estimate r for poorly known species reasonably accurately, and with an assessment of uncertainty.
Estimating r via tree-based prediction methods may be especially advantageous to managers seeking ways of comparing species with regard to their needs or risks. For example, in landscape-specific comparisons across several species, information on r, whether observed or predicted, may be viewed as an index of species' vulnerabilities to extinction processes owing to a common threat [46,47]. The widespread agreement between observed r and mean predicted 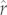 -values we found here (figure 1) highlights the potential use of this PIC approach for multispecies prioritization efforts. Moreover, the strong rank agreement that we observe offers planners reassurance that species predicted to be especially vulnerable because they have low
-values we found here (figure 1) highlights the potential use of this PIC approach for multispecies prioritization efforts. Moreover, the strong rank agreement that we observe offers planners reassurance that species predicted to be especially vulnerable because they have low 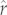 will actually have low maximum population growth rates compared with other species. Ranking species' vulnerabilities using phylogenetically predicted estimates of
will actually have low maximum population growth rates compared with other species. Ranking species' vulnerabilities using phylogenetically predicted estimates of 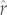 would be most useful in data-poor situations where a suite of species faced a common external threat, as opposed to the (much rarer) data-rich situations in which formal assessments of extinction risk via population viability analyses are possible.
would be most useful in data-poor situations where a suite of species faced a common external threat, as opposed to the (much rarer) data-rich situations in which formal assessments of extinction risk via population viability analyses are possible.
Beyond conservation-relevant results, our efforts have the additional benefit of introducing a joint empirical–theoretical framework for explicitly modelling key aspects of the ‘ecogenetic loop’ that links life-history traits, demography and evolution [48,49]. Specifically, future work could compare how well these macroevolutionary models perform for various life-history quantities, such as those in equations (1.2)–(1.3), both relative to one another, and relative to r as a synthetic life-history trait (see the electronic supplementary material, appendix B). Continued development of macroevolutionary models for r and other life-history traits should yield insights into the limits of demographic plasticity across species and, at the same time, increase our understanding of species resilience [50].
(b). Statistical considerations
Just how much information (e.g. observed r-values, covariates) is necessary to accurately estimate r will depend on several factors, including the size and resolution of the available phylogenies, the relative positions of the well-studied and poorly known species within those phylogenies, and interspecific variability in r. In broad terms, the same kinds of ‘data density’ considerations that are important in using PIC to predict other traits (e.g. colour, morphology and behaviour) will be important in applications that use PIC to predict r [23,24,30]. For example, achieving more accurate and more precise predictions of r for ‘unknown’ species depends upon the location of the closest node with at least two ‘known’ descendants, and one should obtain better predictions from shallower rather than deeper nodes. Put another way, efforts to improve accuracy and narrow prediction intervals for unknown species in poorly sampled clades would benefit more from new data on closely related species than they would from observations elsewhere on the tree. For instance, note that the median and mean cervid prediction errors were three to four times smaller than the caniform errors, even though the Caniformia included over four times as many observed species.
Branch length may also influence the robustness of the predicted 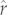 -values. Most obviously, longer branch lengths are associated with less precise estimates because variance under the Brownian motion model increases linearly with elapsed time. In addition, long branch lengths may also lead to less accurate predictions. Any evolutionary deviation from the Brownian motion model of trait evolution (e.g. a sustained trend) would, in theory, lead to a statistical deviation from the value predicted under Brownian motion. Such deviations would be amplified on long branches where the mean, as well as the variance, of the true process could depart from predictions.
-values. Most obviously, longer branch lengths are associated with less precise estimates because variance under the Brownian motion model increases linearly with elapsed time. In addition, long branch lengths may also lead to less accurate predictions. Any evolutionary deviation from the Brownian motion model of trait evolution (e.g. a sustained trend) would, in theory, lead to a statistical deviation from the value predicted under Brownian motion. Such deviations would be amplified on long branches where the mean, as well as the variance, of the true process could depart from predictions.
Overall, this suite of potential statistical difficulties implies that it may prove especially difficult to use PIC-based approaches to estimate r for species that are phylogenetically distinct because of a long period of evolutionary isolation. This is potentially problematic because such phylogenetically distinctive species are often priorities for conservation efforts simply because of their distinctiveness and the unique evolutionary histories they embody [51,52]. However, as noted above, we found no systematic correlation between tip length and error magnitude, but only hints of such difficulties in our cross-validations of particular species with long tip lengths (e.g. P. flavus and A. melanoleuca; electronic supplementary material, figure S6).
Another very different source of prediction errors involves our confidence in the phylogenetic trees themselves. Although we did not consider the influences of phylogenetic uncertainty on model prediction, our methods could be applied to each tree in a sample from the posterior distribution obtained during phylogeny estimation. Prediction intervals from analyses on single trees could then be compared with variability in the estimated r-values across multiple trees to determine the contribution of phylogenetic uncertainty.
(c). Future directions
We expected the PIC-r-mass model to improve upon the results from the simpler PIC-r model. However, with the exception of some narrower prediction intervals (see the electronic supplementary material, figure S5), such improvement was lacking (table 1 and figure 1; electronic supplementary material, figure S1). These results stem, at least in part, from the largely similar performance of the PIC-r model across species irrespective of differences in mass. These results echo findings in Lynch & Fagan [11] where mass was not a good predictor of mammalian r as estimated from life table data, but other traits, such as trophic level and diet were. Relationships among r, prediction errors from the PIC-r model, mass and individual life-history traits are detailed in electronic supplementary material, appendix B.
Given the interdependencies among life-history traits and population growth (see the electronic supplementary material, appendices A and B; see also [45]), improved PIC approaches may be possible using non-mass traits as covariates of the evolutionary model. That is, even if we lack sufficient life-history data to calculate r for one of the poorly known species, we may have data on one or more of the life-history parameters that enter equations (1.2)–(1.3). Traits such as age at first reproduction or average litter size covary with measures of population growth rate across diverse species [8,45], and including them as covariates in our macroevolutionary models might sharpen predictions of how r evolves across species. The potential importance of such a future extension is made clear by the cross-validation errors that occurred in isolated cases where certain species had r much different than their neighbours on the phylogeny. For example, compared with closely related species, the short-tailed weasel Mustela erminea (Caniformia: Mustelidae) had an unusually large r-value that was driven largely by a young age at first reproduction (e.g. females of this species are often mated before being weaned [53]; electronic supplementary material, figure S5a).
Another future direction would be to include more complex models of character evolution. In particular, a complicating factor not accounted for in our analysis is the potential effect of r on extinction or speciation rates. Population growth rates have previously been used as a proxy for evolutionary fitness and have been implicated as potential drivers of diversity [54,55] and diversification rate [56]. Because species with low r are less able to recover from low population size, they may therefore be more prone to extinction. Similarly, correlations between r, generation time and rates of molecular evolution [6,57,58] may lead to a positive association between r and speciation rate. Effects of traits on diversification rate are not naturally incorporated in the PIC framework, but a recent phylogenetic model of the evolution of a continuously valued character that affects diversification [59] presents an alternative approach to model the evolution of population growth rate while disentangling its effects on speciation and extinction rates.
Acknowledgements
Primary support came from the US DOD SERDP Award SI 1475 to W.F.F. E.E.G. was supported by NSF grants DEB-0919089 and DEB-1120279. A.E.N. was supported by NSF Emerging Frontiers grant 0827460 to A. Hastings. We are indebted to J. Calabrese, C. Cosner, J. Drake, E. Holmes, J. Gilbert, D. Skelly, M. Donoghue, S. Levin and anonymous reviewers for helpful comments. I. Agnarsson provided access to the Caniformia phylogeny. J. Billiet, P. Casanovas, R. Harper and J. Rivkin helped assemble data. A. Harris and T. Mueller helped with graphics. We thank M. Neel and A. Leidner for discussions leading to this paper. W.F.F. designed research and wrote the paper (bfagan@umd.edu); Y.E.P. designed and conducted research, helped write the paper (yanthe.pearson@gmail.com); E.A.L. compiled and analysed data (ealarsen@umd.edu); H.J.L. and S.B. made statistical and graphical contributions (hlynch@life.bio.sunysb.edu and sharon_bewick@hotmail.com); J.B.T. and H.S. compiled data (jessica.b.turner@gmail.com and hilary.staver@yale.edu); A.E.N. made mathematical contributions (andrewenoble@gmail.com); E.E.G. designed research; made mathematical, statistical and graphical contributions (eeg@uic.edu).
References
- 1.Caughley G. 1980. Analysis of vertebrate populations. New York, NY: John Wiley & Sons. [Google Scholar]
- 2.Mace GM, Collar NJ, Gaston KJ, Hilton-Taylor C, Akcakaya HR, Leader-Williams N, Miliner-Gulland EJ, Stuart SN. 2008. Quantification of extinction risk: IUCN's system for classifying threatened species. Conserv. Biol. 22, 1424–1442. 10.1111/j.1523-1739.2008.01044.x (doi:10.1111/j.1523-1739.2008.01044.x) [DOI] [PubMed] [Google Scholar]
- 3.Sibly RM, Hone J. 2002. Population growth rate and its determinants: an overview. Phil. Trans. R. Soc. Lond. B 357, 1153–1170. 10.1098/rstb.2002.1117 (doi:10.1098/rstb.2002.1117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coulson T, Mace GM, Hudson E, Possingham H. 2001. The use and abuse of population viability analysis. Trends Ecol. Evol. 16, 219–221. 10.1016/S0169-5347(01)02137-1 (doi:10.1016/S0169-5347(01)02137-1) [DOI] [PubMed] [Google Scholar]
- 5.Cole LC. 1954. The population consequences of life history phenomena. Q. Rev. Biol. 29, 103–137. 10.1086/400074 (doi:10.1086/400074) [DOI] [PubMed] [Google Scholar]
- 6.Blueweiss L, Fox H, Kudzma V, Nakashima D, Peters R, Sams S. 1978. Relationships between body size and some life history parameters. Oecologia 37, 257–272. 10.1007/BF00344996 (doi:10.1007/BF00344996) [DOI] [PubMed] [Google Scholar]
- 7.Charnov EL. 1993. Life history invariants: some explorations of symmetry in evolutionary ecology. Oxford, UK: Oxford University Press. [Google Scholar]
- 8.Saether B, Bakke O. 2000. Avian life history variation and contribution of demographic traits to the population growth rate. Ecology 81, 642–653. [Google Scholar]
- 9.Pereira HM, Daily GC, Roughgarden J. 2004. A framework for assessing the relative vulnerability of species to land-use change. Ecol. Appl. 14, 730–742. 10.1890/02-5405 (doi:10.1890/02-5405) [DOI] [Google Scholar]
- 10.Savage VM, Gillooly JF, Brown JH, West GB, Charnov EL. 2004. Effects of body size and temperature on population growth. Am. Nat. 163, 429–441. 10.1086/381872 (doi:10.1086/381872) [DOI] [PubMed] [Google Scholar]
- 11.Lynch HJ, Fagan WF. 2009. Survivorship curves and their impact on the estimation of maximum population growth rates. Ecology 90, 1116–1124. 10.1890/08-0286.1 (doi:10.1890/08-0286.1) [DOI] [PubMed] [Google Scholar]
- 12.Fagan WF, Lynch HJ, Noon BR. 2010. Pitfalls and challenges of estimating population growth rate from empirical data: consequences for allometric scaling relations. Oikos 119, 455–464. 10.1111/j.1600-0706.2009.18002.x (doi:10.1111/j.1600-0706.2009.18002.x) [DOI] [Google Scholar]
- 13.Hone J, Duncan RP, Forsyth DM. 2010. Estimates of maximum annual population growth rates (rm) of mammals and their application in wildlife management. J. Appl. Ecol. 47, 507–514. 10.1111/j.1365-2664.2010.01812.x (doi:10.1111/j.1365-2664.2010.01812.x) [DOI] [Google Scholar]
- 14.Keyfitz N. 1985. Applied mathematical demography, 2nd edn Berlin, Germany: Springer. [Google Scholar]
- 15.Kot M. 2001. Elements of mathematical ecology, p. 453 Cambridge, UK: Cambridge University Press. [Google Scholar]
- 16.Salguero-Gomez R, Jones OR, Vaupel JR, Baudisch A, Scheuerlein A, Conde Ovando DA, Colchero F. 2013. The comADRe demographic database See http://www.demogr.mpg.de/en/laboratories/evolutionary_biodemography_1171/projects/compadre_iii_and_comadre_demographic_databases_1867.htm
- 17.Pereira HM, Daily GC. 2006. Modeling biodiversity dynamics in countryside landscapes. Ecology 87, 1877–1885. 10.1890/0012-9658(2006)87[1877:MBDICL]2.0.CO;2 (doi:10.1890/0012-9658(2006)87[1877:MBDICL]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 18.Robinson JG, Redford KH. 1986. Intrinsic rate of natural increase in Neotropical forest mammals: relationship to phylogeny and diet. Oecologia 68, 516–520. 10.1007/BF00378765 (doi:10.1007/BF00378765) [DOI] [PubMed] [Google Scholar]
- 19.Duncan RP, Forsyth DM, Hone J. 2007. Testing the metabolic theory of ecology: allometric scaling exponents in mammals. Ecology 88, 324–333. 10.1890/0012-9658(2007)88[324:TTMTOE]2.0.CO;2 (doi:10.1890/0012-9658(2007)88[324:TTMTOE]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 20.Roff DA. 2002. Life history evolution. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 21.Promislow DEL, Harvey PH. 1990. Living fast and dying young: a comparative analysis of life-history variation among mammals. J. Zool. 220, 417–437. 10.1111/j.1469-7998.1990.tb04316.x (doi:10.1111/j.1469-7998.1990.tb04316.x) [DOI] [Google Scholar]
- 22.Bielby J, Mace GM, Bininda-Emonds ORP, Cardillo M, Gittleman JL, Jones KE, Orme CDL, Purvis A. 2007. The fast–slow continuum in mammalian life history: an empirical reevaluation. Am. Nat. 169, 748–757. 10.1086/516847 (doi:10.1086/516847) [DOI] [PubMed] [Google Scholar]
- 23.Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. 2002. Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 33, 475–505. 10.1146/annurev.ecolsys.33.010802.150448 (doi:10.1146/annurev.ecolsys.33.010802.150448) [DOI] [Google Scholar]
- 24.Jombart T, Pavoine S, Devillard S, Pontier D. 2010. Putting phylogeny into the analysis of biological traits: a methodological approach. J. Theoret. Biol. 264, 693–701. 10.1016/j.jtbi.2010.03.038 (doi:10.1016/j.jtbi.2010.03.038) [DOI] [PubMed] [Google Scholar]
- 25.Purvis A, Agapow P, Gittleman JL, Mace GM. 2000. Nonrandom extinction and the loss of evolutionary history. Science 288, 328–330. 10.1126/science.288.5464.328 (doi:10.1126/science.288.5464.328) [DOI] [PubMed] [Google Scholar]
- 26.Cardillo M, Mace GM, Gittleman JL, Jones KE, Bielby J, Purvis A. 2008. The predictability of extinction: biological and external correlates of decline in mammals. Proc. R. Soc. B 275, 1441–1448. 10.1098/rspb.2008.0179 (doi:10.1098/rspb.2008.0179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper N, Freckleton RP, Jetz W. 2011. Phylogenetic conservatism of environmental niches in mammals. Proc. R. Soc. B 278, 2384–2391. 10.1098/rspb.2010.2207 (doi:10.1098/rspb.2010.2207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenstein J. 1985. Phylogenies and the comparative method. Am. Nat. 125, 1–15. 10.1086/284325 (doi:10.1086/284325) [DOI] [Google Scholar]
- 29.Garland T, Jr, Midford PE, Ives AR. 1999. An introduction to phylogenetically based statistical methods with a new method for confidence intervals on ancestral values. Am. Zool. 39, 374–388. [Google Scholar]
- 30.Garland T, Jr, Ives AR. 2000. Using the past to predict the present: confidence intervals for regression equations in phylogenetic comparative methods. Am. Nat. 155, 346–357. 10.1086/303327 (doi:10.1086/303327) [DOI] [PubMed] [Google Scholar]
- 31.Walker EP, Nowak RM, Paradiso JL. 1983. Walker’s mammals of the world. Baltimore, MD: Johns Hopkins University Press. [Google Scholar]
- 32.Ernest SKM. 2003. Life history characteristics of placental nonvolant mammals. Ecology 84, 3402. 10.1890/02-9002 (doi:10.1890/02-9002) [DOI] [Google Scholar]
- 33.Jones KE, et al. 2009. PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology 90, 2648. 10.1890/08-1494.1 (doi:10.1890/08-1494.1) [DOI] [Google Scholar]
- 34.Forsyth DM, Duncan RP. 2001. Propagule size and the relative success of exotic ungulate and bird introductions to New Zealand. Am. Nat. 157, 583–595. 10.1086/320626 (doi:10.1086/320626) [DOI] [PubMed] [Google Scholar]
- 35.Swenson NG. 2009. Phylogenetic resolution and quantifying the phylogenetic diversity and dispersion of communities. PLoS ONE 4, e4390. 10.1371/journal.pone.0004390 (doi:10.1371/journal.pone.0004390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agnarsson I, Kuntner M, May-Collado LJ. 2010. Dogs, cats, and kin: a molecular species-level phylogeny of Carnivora. Mol. Phylogenet. Evol. 54, 726–745. 10.1016/j.ympev.2009.10.033 (doi:10.1016/j.ympev.2009.10.033) [DOI] [PubMed] [Google Scholar]
- 37.Sanderson MJ. 2002. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol. Biol. Evol. 19, 101–109. 10.1093/oxfordjournals.molbev.a003974 (doi:10.1093/oxfordjournals.molbev.a003974) [DOI] [PubMed] [Google Scholar]
- 38.Bininda-Emonds ORP, et al. 2007. The delayed rise of present-day mammals. Nature 446, 507–512. 10.1038/nature05634 (doi:10.1038/nature05634) [DOI] [PubMed] [Google Scholar]
- 39.Blomberg SP, Garland T, Ives AR. 2003. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745. [DOI] [PubMed] [Google Scholar]
- 40.Pagel M. 1999. Inferring the historical patterns of biological evolution. Nature 401, 877–884. 10.1038/44766 (doi:10.1038/44766) [DOI] [PubMed] [Google Scholar]
- 41.Revell LJ. 2010. Phylogenetic signal and linear regression on species data. Methods Ecol. Evol. 1, 319–329. 10.1111/j.2041-210X.2010.00044.x (doi:10.1111/j.2041-210X.2010.00044.x) [DOI] [Google Scholar]
- 42.Boettiger C, Coop G, Ralph R. 2012. Is your phylogeny informative? Measuring the power of comparative methods. Evolution 66, 2240–2251. 10.1111/j.1558-5646.2011.01574.x (doi:10.1111/j.1558-5646.2011.01574.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ives AR, Midford PE, Garland T. 2007. Within-species variation and measurement error in phylogenetic comparative methods. Syst. Biol. 56, 252–270. 10.1080/10635150701313830 (doi:10.1080/10635150701313830) [DOI] [PubMed] [Google Scholar]
- 44.Mace GM, Lande R. 1991. Assessing extinction threats: toward a reevaluation of IUCN threatened species categories. Conserv. Biol. 5, 148–157. 10.1111/j.1523-1739.1991.tb00119.x (doi:10.1111/j.1523-1739.1991.tb00119.x) [DOI] [Google Scholar]
- 45.Heppell SS, Caswell H, Crowder LB. 2000. Life histories and elasticity patterns: perturbation analysis for species with minimal demographic data. Ecology 81, 654–665. 10.1890/0012-9658(2000)081[0654:LHAEPP]2.0.CO;2 (doi:10.1890/0012-9658(2000)081[0654:LHAEPP]2.0.CO;2) [DOI] [Google Scholar]
- 46.Fagan W, Meir E, Prendergast J, Folarin A, Karieva P. 2001. Characterizing population vulnerability for 758 species. Ecol. Lett. 4, 132–138. 10.1046/j.1461-0248.2001.00206.x (doi:10.1046/j.1461-0248.2001.00206.x) [DOI] [Google Scholar]
- 47.Cortés E. 2002. Incorporating uncertainty into demographic modeling: application to shark populations and their conservation. Conserv. Biol. 16, 1048–1062. 10.1046/j.1523-1739.2002.00423.x (doi:10.1046/j.1523-1739.2002.00423.x) [DOI] [Google Scholar]
- 48.Kokko H, Lopez-Sepulcre A. 2007. The ecogenetic link between demography and evolution: can we bridge the gap between theory and data? Ecol. Lett. 10, 773–782. 10.1111/j.1461-0248.2007.01086.x (doi:10.1111/j.1461-0248.2007.01086.x) [DOI] [PubMed] [Google Scholar]
- 49.Coulson T, Tuljapurkar S, Childs D. 2010. Using evolutionary demography to link life history theory, quantitative genetics and population ecology. J. Anim. Ecol. 79, 1226–1240. 10.1111/j.1365-2656.2010.01734.x (doi:10.1111/j.1365-2656.2010.01734.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reed TE, Schindler DE, Waples R. 2010. Interacting effects of phenotypic plasticity and evolution on population persistence in a changing climate. Conserv. Biol. 25, 56–63. 10.1111/j.1523-1739.2010.01552.x (doi:10.1111/j.1523-1739.2010.01552.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. 10.1016/0006-3207(92)91201-3 (doi:10.1016/0006-3207(92)91201-3) [DOI] [Google Scholar]
- 52.Johnson CN. 1998. Species extinction and the relationship between distribution and abundance. Nature 394, 272–274. 10.1038/28385 (doi:10.1038/28385) [DOI] [Google Scholar]
- 53.Harris S, Yalden D. 2008. Mammals of the British Isles: handbook, 4th edn Southampton, UK: The Mammal Society. [Google Scholar]
- 54.Huston M. 1979. A general hypothesis of species diversity. Am. Nat. 113, 81–101. 10.1086/283366 (doi:10.1086/283366) [DOI] [Google Scholar]
- 55.Geritz SAH, Metz JAJ, Kisdi E, Meszéna G. 1997. Dynamics of adaptation and evolutionary branching. Phys. Rev. Lett. 78, 2024– 2027 10.1103/PhysRevLett.78.2024 (doi:10.1103/PhysRevLett.78.2024) [DOI] [Google Scholar]
- 56.Liou LW, Price TD. 1994. Speciation by reinforcement of premating isolation. Evolution 48, 1451–1459. 10.2307/2410239 (doi:10.2307/2410239) [DOI] [PubMed] [Google Scholar]
- 57.Martin AP, Palumbi SR. 1993. Body size, metabolic rate, generation time, and the molecular clock. Proc. Natl Acad. Sci. USA 90, 4087–4091. 10.1073/pnas.90.9.4087 (doi:10.1073/pnas.90.9.4087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith SA, Donoghue MJ. 2008. Rates of molecular evolution are linked to life history in flowering plants. Science 322, 86–89. 10.1126/science.1163197 (doi:10.1126/science.1163197) [DOI] [PubMed] [Google Scholar]
- 59.FitzJohn RG. 2010. Quantitative traits and diversification. Syst. Biol. 59, 619–633. 10.1093/sysbio/syq053 (doi:10.1093/sysbio/syq053) [DOI] [PubMed] [Google Scholar]