

Introduction
Despite the plummeting cost of next-generation sequencing, the preparation of sequencing libraries using commercially available kits still remains expensive. This can be prohibitive for large-scale comparative or experimental studies, where hundreds to thousands of samples need to be analyzed. The increasing use of multiplexing dozens to hundreds of samples underscores the urgent need to develop a cost effective and time efficient high-throughput method for library preparation. By optimizing and scaling down the steps in library construction and using commonly available reagents, the protocol described here allows for the preparation of DNA libraries in a 96 well format, using no specialized equipment, at a substantial savings in both reagent cost and personnel hours. Utilizing this optimized high-throughput format results in a 10 fold cost reduction, compared to commercially available kits, making per library or pooled sample costs ~$12.6–14.9 for individually prepared libraries and ~$8.6–10.6 for pooled libraries with individual barcodes; both techniques allow for up to 144 samples to be pooled on a single lane with the barcodes tested herein.
Materials
⅛ in diameter stainless steel ball bearing (Hartford Technologies 034-008-2C)
1.1 ml tube in strips of 8 (USA Scientific 1212-8000)
8-cap strip for 1.1 ml racked tubes USA (Scientific 1294-0800)
96-Well Collection Plate (Zymo Research C2002)
Agarose I (Amresco 0710-100G)
Cell & Lysis Buffer (Epicentre MTC096H)
DNA Clean & Concentrator Kit-5 (Zymo Research D4004)
Klenow exo- (NEB M0212L)
MPC Buffer (Epicentre MMP03750)
NEBuffer 2 (NEB B7002S)
NEBNext dsDNA Fragmentase (NEB M0348S)
O’GeneRuler Plus 100bp Ladder (SM0323)
PCR Sealing Mat for 96 Well PCR Plate (USA Scientific 1400-9605)
Phusion DNA Polymerase (NEB M0530S)
Proteinase K (Qiagen 19133)
Qubit dsDNA HS Assay Kit (Life Technologies Q32851)
Quick Blunting Kit (NEB E0542S)
Quick Ligation Kit (NEB M2200S)
SafeView (abm G108)
TempPlate No-Skirt 0.2mL PCR Plates, Natural (USA Scientific 1402-9596)
-
ZR-96 DNA Clean & Concentrator Kit-5 (Zymo Research D4023 or D4024)
Alternatively, Agencourt AMPure XP beads (Beckman Coulter A63880, A63881, or A63882) can be substituted for all Zymo Research Clean & Concentrator kits. See alternative procedures section for instructions. Library construction costs per sample can increase depending upon bead volume purchased. ZR-96 Quick g-DNA (Zymo Research D3010)
-
Zymoclean Gel DNA Recovery Kit (Zymo Research D4001)
Alternatively, Agencourt AMPure XP beads (Beckman Coulter A63880, A63881, or A63882) can be substituted for gel based size selection. See alternative procedures section for instructions.
Equipment
Qiagen TissueLyzer II (Retsch)
96-well plate centrifuge
Centrifuge
PCR machine
-
Qubit fluorometer (Life Technologies)
Alternatively, the kappa quantification kit (Kapa Biosystems KK4824) can be used to quantify each sample for pooling at step 14 of the protocol. Transilluminator (Clare Chemical)
Method
Interruption of the protocol can occur after completion of Steps 5, 6, 7, 8, 9, 10, 11, 12, and 13, however greater yields have ben observed if libraries are constructed without interruption. We suggest that master mixes or individual reagents are first pipetted into 12-tube strip tubes and a 12-channel multipipettor used for pipetting into wells of a 96-well plate(s). All mixes are given for a single sample in the following steps. All sample cleaning steps using the ZR-96 DNA Clean & Concentrator-5 kit should add an additional 4 μl of elution buffer or PCR grade H2O since 4 μl is typically lost during the elution spin. For steps using DNA Clean & Concentrator-5 kit, add an additional 0.3 μl of elution buffer or H2O. The volumes in the protocol do not reflect the expected loss of H2O.
1) Adaptor Formation
-
Combine the following for:
See table 1 for oligonucleotide sequences. For barcoded adaptor formation the barcoded adaptor oligo’s are substituted for the Multi/Std Adaptor 1 and Multiplexing Adaptor 2.Reagent Volume (μl) Multi/Std Adaptor 1 (100 μM) 45 Multiplexing Adaptor 2 (100 μM) 45 NEBuffer 2 10 dH2O 0
Final Volume 100 -
Mix and cycle using the following temperatures:
After cycling program is finished, incubate at 4°C for 30 minutes (or place on ice). Store at −20°C. Thaw adaptor on ice or at 4°C.95°C 5 min 90°C 1 min 85°C 1 min 80°C 1 min 75°C 30 s Drop 1°C every cycle Go to step 5, for 69 cycles 4°C forever
Table 1.
Oligonucleotides
| Oligo ID | Sequencea |
|---|---|
| Non-barcoded Multiplexing (Indexing) Adaptor: | |
| Multi/Std Adapter 1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| Multiplexing Adapter 2 | P-GATCGGAAGAGCACACGTCT |
| Barcoded Multiplexing (Indexing) Adaptor: | |
| PE A1-ACTG | P-CAGTAGATCGGAAGAGCACACGTCT |
| PE A2-ACTG | ACACTCTTTCCCTACACGACGCTCTTCCGATCTACTGT |
| PE A1-ATGC | P-GCATAGATCGGAAGAGCACACGTCT |
| PE A2-ATGC | ACACTCTTTCCCTACACGACGCTCTTCCGATCTATGCT |
| PE A1-AGCT | P-AGCTAGATCGGAAGAGCACACGTCT |
| PE A2-AGCT | ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGCTT |
| PE A1-TACG | P-CGTAAGATCGGAAGAGCACACGTCT |
| PE A2-TACG | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTACGT |
| PE A1-TCGA | P-TCGAAGATCGGAAGAGCACACGTCT |
| PE A2-TCGA | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCGAT |
| PE A1-TGAC | P-GTCAAGATCGGAAGAGCACACGTCT |
| PE A2-TGAC | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTGACT |
| PE A1-CAGT | P-ACTGAGATCGGAAGAGCACACGTCT |
| PE A2-CAGT | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGTT |
| PE A1-CTAG | P-CTAGAGATCGGAAGAGCACACGTCT |
| PE A2-CTAG | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTAGT |
| PE A1-CGTA | P-TACGAGATCGGAAGAGCACACGTCT |
| PE A2-CGTA | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCGTAT |
| PE A1-GATC | P-GATCAGATCGGAAGAGCACACGTCT |
| PE A2-GATC | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGATCT |
| PE A1-GCAT | P-ATGCAGATCGGAAGAGCACACGTCT |
| PE A2-GCAT | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCATT |
| PE A1-GTCA | P-TGACAGATCGGAAGAGCACACGTCT |
| PE A2-GTCA | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTCAT |
| Multiplexing Primers: | |
| Multi/Std PCR Primer 1.0 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC* T |
| Multiplexing PCR Primer 2.0 | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC* T |
| Index primers: | |
| Primer Index 1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTT* C |
| Primer Index 2 | CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTT* C |
| Primer Index 3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTT* C |
| Primer Index 4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTT* C |
| Primer Index 5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTT* C |
| Primer Index 6 | CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTT* C |
| Primer Index 7 | CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGTT* C |
| Primer Index 8 | CAAGCAGAAGACGGCATACGAGATTCAAGTGTGACTGGAGTT* C |
| Primer Index 9 | CAAGCAGAAGACGGCATACGAGATCTGATCGTGACTGGAGTT* C |
| Primer Index 10 | CAAGCAGAAGACGGCATACGAGATAAGCTAGTGACTGGAGTT* C |
| Primer Index 11 | CAAGCAGAAGACGGCATACGAGATGTAGCCGTGACTGGAGTT* C |
| Primer Index 12 | CAAGCAGAAGACGGCATACGAGATTACAAGGTGACTGGAGTT* C |
5′-3′
Represents PTO bond. The use of this modification is necessary for long-term stability of the oligo.
P- Corresponds to a 5′-Phosphate modification
DNA Extraction
Procedure A
2) Zymo Research ZR-96 Quick-gDNA Kit
-
Follow manufacturer’s protocol.
A large concentration per microliter of DNA is preferred (100 ng/μl). Although the Quick g-DNA MiniPrep kit requires 50 μl for elution, we suggest 25 μl or less to guarantee high DNA concentrations. If tissue quantity is limiting, use the Quick-gDNA MicroPrep kit, however column clogging has sometimes been observed. The use of β-mercaptoethanol is highly recommended. Depending upon sample size, single column or 96-well based DNA extraction kits are available.
Procedure B
Optional Pre-Zymo Kit
3) Cell Culture Samples
Grow bacteria in 15 ml Falcon tubes, pellet at 3,000 rpm, and remove supernatant.
Combine 280 μl of Tissue and Cell Lysis Solution (Epicentre) and then add 20 μl Qiagen Proteinase K to each sample. Resuspend and incubate at 55°C for 30 min.
-
Allow samples to cool and then add 200 μl of MPC Buffer (Epicentre). Mix by inverting several times, then spin at maximum speed for 15 min.
A cloudy white precipitate should be seen after mixing. If precipitate is not observed, additional MPC buffer can be added following manufacturer’s suggestions. Without disrupting pellet, transfer supernatant to a new 96-well deep-well plate.
-
Proceed to Zymo Research ZR-96 Quick-gDNA kit protocol (DNA Extraction Procedure A).
ZR-96 Quick-gDNAStep (iii), addition of MPC can be omitted if β-mercaptoethanol is used as described in theprotocol. Otherwise, equal volumes, not 4 times, Zymo Lysis Buffer can be used if excessive sample is not used and MPC step is performed. Quantify using Life Technologies Qubit fluorometer.
4) Tissue Samples
Tissue can be stored long-term before processing at −80°C. Place tissue in 1.1 mL 8-strip tubes with caps and freeze in liquid N2. To crush add a single ⅛″ diameter stainless steel ball bearing to each tube and process samples in a QIAGEN TissueLyzer II (Retsch) at 30Hz for 30s. Samples can be re-frozen in liquid N2 if additional crushing is needed. Spin samples down immediately after crushing.
Combine 280 μl of Tissue and Cell Lysis Solution (Epicentre) and 20 μl Qiagen Proteinase K, per sample, and add 100 μl of mix to each sample and repeat bead beating.
Spin samples at maximum speed for 1 minute. Add remaining 200 μl of lysis/proteinase K mix to each sample and incubate at 55°C for 30 min, periodically inverting or flicking to mix.
-
Spin samples for 1 min at maximum speed and add 200 μl of MPC Buffer (Epicentre), mix and spin at max speed for 15 min.
If excessive sample is used, more than 200 μl of MPC Buffer can be used. A cloudy white precipitate should be seen after mixing. If precipitate is not observed, more can be added following manufacturer’s protocol. Without disrupting pellet, transfer supernatant into a new 96-well deep-well plate.
-
Proceed to Zymo Research ZR-96 Quick-gDNA protocol
ZR-96 Quick-gDNAStep (iv), addition of MPC can be omitted if β-mercaptoethanol is used as described in theprotocol. Otherwise, equal volumes, not 4 times, Zymo Lysis Buffer can be used if excessive sample is not used and MPC step is performed. Quantify using Life Technologies Qubit fluorometer.
5) Fragmentation: NEB Fragmentase
It is necessary to perform several sample fragmentation trials to optimize the fragment size range for the users specific sample. DNA starting concentration of 1 μg or greater can be used; however, this protocol has been optimized for 500 ng.
-
Set up the digestion reaction in 96-well format. Mix thoroughly.
NEBNext dsDNA Fragmentase should not be added at this point.Reagent Volume (μl) gDNA (500 ng) variable 10X Fragmentase Buffer 1 100X BSA 0.1 dsDNA Fragmentase 1 Sterile dH2O variable
Final Volume 10 Incubate on ice for 5 min.
-
Vortex NEBNext dsDNA Fragmentase and add to the reaction. Mix thoroughly.
Fragmentase is an enzyme mix and must be vortexed. Given the minimal reaction volume, ensuring the addition of exactly 1 μl is critical to the success of this reaction. This is a common step contributing to poor library construction. -
Incubate at 37°C according to the recommended times in manufacturer’s protocol.
We routinely find an incubation time of ~ 20 min to be ideal. If multiple samples are to be fragmented, it is highly recommended to stagger the samples with 20 sec increments, or to perform fragmentation in an 8- or 12- strip tube format for multichannel use. -
After incubation immediately add 6 μl of 0.5 M EDTA to stop the reaction.
As the reaction reaches the optimal fragmentation range, any time beyond the predetermined incubation time significantly alters the success of library construction and the variability between samples. Purify the DNA with ZR-96 DNA Clean & Concentrator-5 kit. Elute in 7.5 μL H2O.
6) Quick Blunting Kit (NEB)
-
Prepare the following reaction mix:
Do not forget E. coli DNA ligase for Fragmentase, found in the NEB dsDNA Fragmentase kit. The addition of ligase is critical for library construction.Reagent Volume (μl) Eluted DNA 7.5 10X Blunt Buffer 1.25 1 mM dNTP mix 1.25 Blunt Enzyme Mix 0.5 E. coli DNA ligase for Fragmentase 0.4 Sterile dH2O 1.6
Final Volume 12.5 -
Incubate the sample at 25°C for 30 min, and hold at 4°C.
Although this kit can be heat inactivated, it is not recommended in order to retain AT-rich genomic fragments. Purify the DNA with ZR-96 DNA Clean & Concentrator-5 kit. Elute in 15 μl H2O.
7) dATP Addition (NEB)
-
Prepare the following reaction mix:
Reagent Volume (μl) Eluted DNA 15 NEBuffer 2 2.5 10 mM dATP 0.5 Klenow exo- (NEB) 1.5 Sterile dH2O 5.5
Final Volume 25 Incubate the sample at 37°C for 30 min, and hold at 4°C.
Purify the DNA with ZR-96 DNA Clean & Concentrator-5 kit. Elute in 6 μl H2O.
Adaptor ligation (NEB Quick Ligation Kit)
8) Procedure A: 96-well Non-pooled, Single Sample Approach
-
Prepare the following reaction mix:
The adaptor should be added directly to each sample prior to making ligase/buffer mix. Ligase and buffer should be combined and vortexed thoroughly. The high viscosity of both the ligase and 2X Buffer can result in poor mixing of these two components. Ligase is added to a tube and then the buffer. The ligase is aspirated from the bottom of the tube into the buffer and then vortex to thoroughly mix. See table 1 for oligonucleotide sequences.Reagent Volume (μl) Eluted DNA 6 2X Quick Ligation Buffer 7.5 Multiplexing (Indexing) Adaptor (45 μM) 1.2 Quick T4 DNA Ligase 0.6
Final Volume 15.3 -
Incubate at 25°C for 15 min.
Although this kit can be heat inactivated, it is not recommended in order to retain AT-rich genomic fragments. -
Purify with ZR-96 DNA Clean & Concentrator-5 kit. Elute in 6 μL H2O.
Cleaning after ligation is critical. Excess adaptor-adaptor product will cause a greater propensity for adaptor-adaptor product to co-migrate with desired product during gel purification and reduce PCR efficiency.
9) Procedure B: 96-well pooled Approach
-
Prepare the following reaction mix: One barcoded adaptor per column, Figure 1.
The adaptor should be added directly to each sample prior to making ligase/buffer mix. Ligase and buffer should be combined and vortexed well. The high viscosity of both the ligase and 2X Buffer can result in homogenization failure. Ligase is added to a tube, followed by buffer. The ligase is aspirated from the bottom of the tube into the buffer and then vortex to thoroughly mix. This mixture is added to the sample and adaptor mix. See table 1 for oligonucleotide sequences.Reagent Volume (μl) Eluted DNA 6 2X Quick Ligation Buffer 7.5 Barcoded Adaptor (45 μM) 1.2 Quick T4 DNA Ligase 0.6
Final Volume 15.3 Incubate at 25°C for 15 min.
Retain 50% of the ligation reaction from each sample for long-term storage at −80°C.
-
Purify the remaining 50% cDNA with Zymo Research DNA Clean & Concentrator kit. Elute in 6 μL H2O.
Assuming 12 samples are to be pooled. Prior to purification after ligation, pool samples in the following fashion. Pool samples A1–A6, A7–A12, B1–B6, B7–B12 and so on for each row for a total of 16 pooled samples. Use these 16 samples to load on a gel. Alternatively, pooling samples down a column is also an option; however never pool more than 6 samples. Cleaning after ligation is critical. Excess adaptor-adaptor product will cause a greater propensity for adaptor-adaptor product to co-migrate with desired product during gel purification. This will result in variable and reduced amplification efficiency in the final indexing PCR step. Pooling of more than 6 ligation reactions at a time results in clogging of the purification column and should not be attempted.
Figure 1. Barcoding and Indexing Schematic.
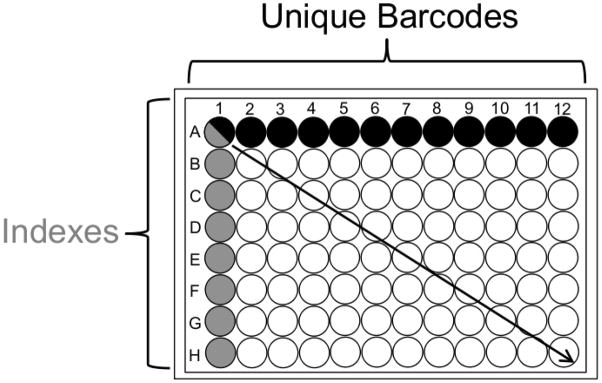
Example 96 well format combinatorial barcoded adaptors/indexing primers.
Each well contains a unique sample. Each column has one of 12 barcoded adaptors ligated to it. Each row is then pooled for eight indexing PCR reactions.
Gel purification of cDNA Templates (Zymo Research)
10) Procedure A: 96-well Non-pooled, Single Sample Approach
-
Prepare a 1.5–2% 1X TBE agarose gel.
Use a small width comb size because product will be barely visible. Volume of gel in the cast should not be excessive, but sufficient for sample to be loaded. Excess gel thickness increases incubation times and leads to greater product loss. Load individual samples with a ladder between each sample. Include blank lanes between each sample and ladder to prevent cross-contamination. Load sample and loading dye with a 100 bp ladder in the flanking well.
Run gel at 120V for 30–60 min until sufficient separation of the 100bp and 200bp bands of the DNA ladder occurs.
-
Cut a gel slice at 350/400 bp (+/− 20 bp) to 800 bp (depending upon sequencing length) and purify the DNA with Zymo Research Zymoclean Gel DNA Recovery Kit. Elute the DNA into 7.5 μL of H2O.
Use a transilluminator from Clare Chemical to prevent UV mediated DNA damage during gel excision. Incubate gel in ADB at no greater than 30°C. The Zymoclean Gel kit typically results in a loss of 0.3 μl during the elution step.
11) Procedure B: 96-well Pooled Sample Approach
-
Prepare a 1.5–2% 1X TBE agarose gel.
Use a small width comb size because product will be barely visible. Volume of gel in the cast should not be excessive, but sufficient for sample to be loaded. -
Load sample and loading dye with a 100 bp ladder in the flanking well.
Load row pools (2 per row of 96 well plate) adjacent to one another, i.e. ladder, A1–A6, A7–A12, ladder, B1–B6, B7–B12, ladder, etc. When cutting the samples from the gel, cut both pooled row samples in the same slice in order to gel purify an entire row in a single column. Do not separate the 2 samples between each row. Run gel at 120V for 30–60 min until sufficient separation of the 100bp and 200bp bands of the DNA ladder occurs.
-
Cut a gel slice at 350/400 bp (+/− 20 bp) to 800 bp (depending upon sequencing length). Purify with Zymo Research Zymoclean Gel DNA Recovery Kit. Elute the DNA into 15 μL of H2O.
Use a transilluminator from Clare Chemical to prevent UV mediated DNA damage during gel excision. Incubate gel in ADB at no greater than 30°C. The Zymoclean Gel kit typically results in a loss of 0.3 μl during the elution step.
Indexing PCR
12) Procedure A: 96-well Non-pooled, Single Sample Approach
-
Prepare the following reaction mix: One index primer per sample.
See table 1 for oligonucleotide sequences.Reagent Volume (μl) 5X Phusion Buffer 5 Multi/Std PCR Primer 1.0 (25 μM) 0.5 Multiplexing PCR Primer 2.0 (0.5 μM) 0.5 PCR Primer Index 1 (or others) (25 μM) 0.5 dNTP mix (10 mM) 0.75 Phusion DNA polymerase 0.5 Gel Extracted Sample 7.5 Sterile dH2O 9.75
Final Volume 25 -
Run the following PCR cycling program for a total of 18 cycles:
98°C 1 min 98°C 10 s 65°C 30 s 72°C 30 s Cycle to step 2 for 17 rounds 72°C 5 min 4°C hold -
Purify the DNA with ZR-96 DNA Clean & Concentrator-5 kit. Elute in 16–26 μl Qiagen EB Buffer.
After indexing PCR, single sample libraries can be cleaned either as pools and then quantified or cleaned as single samples, quantified, and then pooled.
13) Procedure B: 96-well pooled Approach
-
Prepare the following reaction mix: One index primer per pooled row, Figure 1.
See table 1 for oligonucleotide sequences.Reagent Volume (μl) 5X Phusion Buffer 10 Multi/Std PCR Primer 1.0 (25 μM) 1 Multiplexing PCR Primer 2.0 (0.5 μM) 1 PCR Primer Index 1 (or others) (25 μM) 1 dNTP mix (10 mM) 1.5 Phusion DNA polymerase 1 Gel Extracted Sample 15 Sterile dH2O 19.5
Final Volume 50 -
Run the following PCR cycling program for a total of 18 cycles:
98°C 1 min 98°C 10 s 65°C 30 s 72°C 30 s Cycle to step 2 for 17 rounds 72°C 5 min 4°C hold Purify the DNA with Zymo Research DNA Clean & Concentrator-5 kit. Elute in 16–26 μl Qiagen EB Buffer.
14) Library Quantification and Dilution
-
Quantify with Invitrogen QuBit fluorometer, High Sensitivity DNA kit. Dilute the samples with EB Buffer to 10 nM in the presence of 0.1% Tween-20. Pool the desired number of samples per lane of sequencing.
Alternatively, the kappa quantification kit can be used to quantify each sample for pooling, however per sample costs will increase. -
Sequence libraries on any Illumina platform, or store at −80°C.
The final quantified library (step 14) is the best indication of a successful library construction. Final values of 5 ng/μl in 16–26 μl following Procedure A or 10 ng/μl in 16–26 μl following Procedure B routinely results in high quality sequence with minute adaptor contaminate. Values of < 2 ng/μl indicate that the library preparation was not efficient and will generally result in a poor sequencing run, as a result of excessive adaptor sequence and/or low cluster densities.
Alternative Procedures
1) AMPure Bead XP based sample cleanup
Alternative for Zymo Research DNA Clean & Concentrator kits. Prepare fresh 70% ethanol prior to each use of beads. If using a robot, bring samples up to 40 μl volume with H2O.
-
Resuspend beads and add 1 volume of beads per volume of sample and mix well.
The beads are viscous. Watch for residual beads retained on the outside of pipet tip or inside pipet tip after adding to sample. Incubate the mix for 5 min at room temperature.
Place samples on a magnet for 1 min. Once beads have been pulled to the side of the tube, remove the supernatant.
While samples are on magnet, add 100 μl of 70% ethanol. Wait 30 sec and remove the ethanol. Repeat this step one additional time. Be certain that all ethanol is removed prior to elution step.
Remove from magnet and add desired volume of H2O. Incubate at room temperature for 2 minutes.
Place on magnet for 1 min and remove supernatant containing purified sample.
2) AMPure Bead XP based sample size selection
Alternative for Zymoclean Gel DNA Recovery Kit and gel size selection. Prepare fresh 70% ethanol prior to each use of beads. If using a robot, bring samples up to 40 μl volume with H2O. For size selection after adaptor ligation, bring samples up to 40 μl. This protocol will select for fragments > ~150–200 bp.
-
Resuspend beads and add 0.7X volume of beads per volume of sample and mix well.
The beads are viscous. Watch for residual beads retained on the outside of pipet tip or inside pipet tip after adding to sample. Incubate the mix for 5 min at room temperature.
Place samples on a magnet for 1 min. Once beads have been pulled to the side of the tube, remove the supernatant.
While samples are on magnet, add 100 μl of 70% ethanol. Wait 30 sec and remove the ethanol. Repeat this step one additional time. Be certain that all ethanol is removed prior to elution step.
Remove from magnet and add desired volume of H2O. Incubate at room temperature for 2 minutes.
Place on magnet for 1 min and remove supernatant containing purified sample.
Troubleshooting
Problem: Fragmentation smear, observed on a 1.5% agaorse gel, deviates from those pictured in the manufacturer’s protocol.
[Step 5]
Solution: Likely due to incomplete fragmentation (a high molecular weight DNA band with a smear and limited DNA in target range) or excessive fragmentation (a faint 50–1000bp smear). Prior to starting this library construction protocol, the fragmentation step should always be optimized with a subset of the samples for desired smear size and intensity. We have determined the two major causes of fragmentation failure these being; 1) the quality of extracted DNA; and 2) NEBNext dsDNA Fragmentase pipetting error. DNA quality is primarily a function of the kit used for extraction. We have provided two extraction methods that select for high molecular weight DNA, removal of RNA and post fragmentation results that consistently yield the desired fragmentation size range and intensity. The second issue occurs as a result of error while pipetting small volumes. As the reaction volume is only 10 μl the volume of NEBNext dsDNA Fragmentase needed is miniscule, therefore any excess reagent added to the reaction contributes significantly to the overall reaction dynamics. Careful attention to detail i.e. specifically pipetting off the surface of the reagent rather than submerging the tip and proper pipetting technique will ensure that the desired fragmentation range is achieved.
Problem: Gel of fragmented sample looks normal, however library concentration is < 1 ng/μl.
[Step 6]
Solution: Due to the presence of nicked DNA after fragmentation it is critical to add E. coli DNA ligase (supplied with NEBNext dsDNA Fragmentase kit) prior to downstream steps to ensure that the DNA is repaired. Failure to add this enzyme will result in poor amplification from adaptors during the indexing PCR step.
Discussion
Genomic sequencing is becoming increasingly popular in both model and non-model systems. This is primarily due to a continuously decreasing cost per base sequenced across sequencing platforms (Stein LD, 2010). With Illumina sequencing in particular it is the result of greater cluster densities, increased sequencing lengths, sample pooling per lane, and paired-end sequencing. Unfortunately, the excessive cost of library construction using commercially available kits, $50 – $110 per sample, has, to date, been unable to parallel the reduction in sequencing cost.
Procedures A and B of our method expand upon standard single sample library construction approaches (Margulies et al. 2005, Mortazavi et al. 2008), by protocol optimization that reduces construction costs by an order of magnitude in a flexible high-throughput manner. This reduction in cost is directly attributable to the optimization of reagent usage, a 50% reduction in starting material, and the incorporation of unique pooling stages, while utilizing commonly used and highly trusted reagents. Procedure A was tested for whole genome sequencing in eukaryotes (Drosophila, the flatworm Schmidtea mediterranea, and the model legume Medicago truncatula) as well as in prokaryotes (Mesorhizobium and Ensifer bacteria), ranging from a few to 96 individual samples simultaneously processed in our high-throughput manner. For example, Figure 2A, C, and E shows summary statistics from sequencing and de novo assembly of a collection of 48 Mesorhizobium strains. These strains were individually processed following Procedure A, and sequenced to ~20x coverage in 76bp paired end format and draft genomes were assembled using a5 pipeline (Tritt et al. 2012); 190,810,672 reads (95.1%) were used out of 200,550,351 total reads (S. S. Porter, P. L. Chang, C. A. Conow, J. P. Dunham, M. L. Friesen, in preparation). To demonstrate the flexibility and reduction in cost and time achieved by pooled library construction (Procedure B) as compared to individual library construction (Procedure A),18 Ensifer medicae Illumina libraries were constructed, Figure 2B, D, F. De novo assembly was performed using the a5 pipeline (Tritt et al. 2012), and it was determined that each sample contained <1% adaptor sequence and >90% of the bases had a quality score of 20 or greater. Specifically, Figure 2A shows the total number of sequencing reads produced per individual sample. Twelve samples were pooled per lane based upon the final quantification of each library, and consistently resulted in equal coverage across all 48 samples. By comparison, Procedure B also shows equal coverage after sequencing even when samples are pooled at early stages, thus indicating no obvious source of biased library construction or biased amplification within pooled libraries, Figure 2B. Amongst the sequence produced per sample using either Procedure A or B, >95% of the sequence used for de novo assembly was of high quality, Figures 2C and D. Figures 2E and F illustrate that both procedures consistently produce high contig lengths between samples, suggesting little genome coverage bias in using either Procedures A and B. Furthermore the number of de novo contigs per sample as well as the maximum contig length was consistent across samples, Figures 2E and F.
Figure 2. Sequence and Assembly Quality.

Read outputs and assembly statistics for bacterial genomes sequenced with Procedures A and B. Total number of reads from libraries prepared using the individual sample protocol (A: 48 strains) and pooled protocol (B: 18 strains). The fraction of reads used in the de novo assembly was above 95% for both experiments (C, D). Both protocols yielded high quality draft genomes (E, F) with total assemblies ranging from 5.8–7.8 Mbp and 5.8–7.3 Mbp (blue), maximum contigs ranging from 297–2,511 Kbp and 42–697 Kbp (gold), and N50 values ranging from 129–911 Kbp and 5.4–262 Kbp (pink).
Procedures A and B are both high-throughput options that can be used to construct 96 (8 pools of 12 indexes) or 144 (12 indexes, 12 barcodes per index pool) libraries with user flexibility in mind. The first deviation from standard protocols occurs after the barcoded adaptor ligation, step 9, in which approximately 50% of each ligation product is removed for long-term storage (Procedure B). For instance, after removal of 50% of each of the 18 Ensifer samples, they were pooled into three groups of six samples each, size selected, and subsequently index PCR amplified for final library construction. Sample pooling can consist of any size, i.e. 12, eight, or even fewer samples. Variation in library construction efficiency between samples can result in competition for coverage during sequencing within pools. Therefore, in the event of poor coverage the stored ligation product, step 9, can be used for a second round of library construction and sequencing. Either Procedure A or B can be used to complete the libraries for these samples.
It has become common to use barcoded adaptors or Illumina’s primer based indexing approaches to differentiate samples (Craig, et al. 2008), however these methods have not been routinely used in combination. Procedure B uses unique pooling stages, as a result of the method’s combinatorial scheme of barcoding and indexing for sample differentiation, Figure 1. Utilizing these pooling stages drastically reduces the number of samples for size selection, which remains one of the most tedious and time-consuming steps of library construction. Using Procedure B, 96 samples are reduced to a manageable 16 samples for size selection and further reduced to eight gel extractions. This is assuming each row of 12 samples per row will be combined for indexing PCR (2 pools per row for gel extraction, six samples per pool). This example demonstrates pooling across a row, however columns can also be pooled according to the needs of the user, Figure 1. Pool size, either Steps 10 and 11 of Procedure B or Step 14 of Procedure A, is dependent upon the scale of the project and the desired coverage per sample in each lane. The user must consider two options to obtain the maximum per sample coverage given the number of pooled samples and genome size: 1) pool a small number of samples to obtain the maximum coverage within a single lane, or 2) distribute a large pool of samples across multiple lanes to achieve the desired per sample coverage. Thus further demonstrating the flexibility, scalability and cost reduction of Procedures A and B.
In addition to protocol flexibility, the cost per sample can pose a significant barrier to the desired scale of an experiment. The library construction costs vary for both procedures, and was determined in two ways: 1) total cost of reagents for a given number of samples; or 2) per unit enzyme cost (the number of samples prepared per kit). Estimating sample preparation costs from the total cost of reagents purchased results in a higher overall cost per sample. However, since each kit within the protocol can process a different number of samples, total reagent cost is an inaccurate estimate of per sample cost. Furthermore, these two approaches of sample cost calculation can vary greatly depending upon sample size. Tables 2A–C and 3A–C display the cost breakdown for Procedures A and B, respectively, reflecting both total kit costs and per enzymatic costs for sample sizes 100, 200, and 400. Using Procedure A the user can easily prepare 96 or more libraries given a sequencing pool size of eight or 12 samples per lane and an enzymatic cost of ~$12.6 – 14.9, depending upon sample size. However the total cost per kit given a particular sample size costs ~$13.4 – $15.86. Alternatively, due to the combinatorial approach of Procedure B, per enzymatic costs are further reduced to ~$8.5 – $10.5, while total kit costs are ~$9.3 – $12.35. Our estimation of per sample cost for all sample sizes was determined to minimize both per sample enzymatic cost as well as total reagent cost. Our protocol uses commonly used and trusted reagents, and although using other reagents could potentially reduce costs further, the reproducibility and efficiency of these reagents relative to this protocol would require additional testing and optimization. Also, it is common for library construction sample costs to neglect including the cost of DNA extraction. Although our cost estimates are based solely upon library construction, we also provide our DNA extraction methods and associated costs, Table 4, as library construction efficiency is highly dependent upon DNA quality.
Table 2A.
Procedure A - 100 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 100 | NEB | 100 | M0348S | 1 | 96 | 96 | 0.96 |
| ZR-96 Pur Kit | 100 | Zymo | 4×96 | D4024 | 1 | 387 | 387 | |
| ZR-96 Pur Kit | 100 | Zymo | 2×96 | D4023 | 1 | 199 | 199 | 5.86 |
| Gel Recovery | 100 | Zymo | 100 | D4001 | 2 | 76 | 152 | 1.52 |
| Quick Blunt | 100 | NEB | 120 | E1201S | 3 | 77 | 231 | 1.92 |
| Klenow exo- | 100 | NEB | 133.3 | M0212L | 1 | 228 | 228 | 1.71 |
| Quick Ligation | 100 | NEB | 100 | M2200S | 2 | 95 | 190 | 1.9 |
| Phusion | 100 | NEB | 100 | M0530S | 1 | 103 | 103 | 1.03 |
| Total Cost: | 1586 | Enzyme Total: | 7.52 | |||||
| Per Sample: | 15.86 | Column Total: | 7.38 | |||||
| Total Unit Cost Per Sample: | 14.90 |
Table 2C.
Procedure A - 400 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 400 | NEB | 500 | M0348L | 1 | 384 | 384 | 0.76 |
| ZR-96 Pur Kit | 400 | Zymo | 4×96 | D4024 | 5 | 387 | 1935 | 4.84 |
| Gel Recovery | 400 | Zymo | 400 | D4002 | 2 | 278 | 556 | 1.39 |
| Quick Blunt | 400 | NEB | 400 | E1201L | 2 | 308 | 616 | 1.54 |
| Klenow exo- | 400 | NEB | 399.9 | M0212L | 3 | 228 | 684 | 1.71 |
| Quick Ligation | 400 | NEB | 500 | M2200L | 2 | 380 | 760 | 1.52 |
| Phusion | 400 | NEB | 500 | M0530L | 1 | 412 | 412 | 0.82 |
| Total Cost: | 5347 | Enzyme Total: | 6.36 | |||||
| Per Sample: | 13.36 | Column Total: | 6.23 | |||||
| Total Unit Cost Per Sample: | 12.59 |
The protocols described herein are applicable to small to moderate sized labs, as typified by most research labs. Thus as a result we did not include robot-based automation as a source of labor cost reduction. However, the determination of labor costs will be dependent upon the construction time of the method, hourly pay of the personnel, and the ability to automate the protocol. The time to complete our procedures is 8–13 hours for two 96-well plates, and is dependent upon the experience of the user. Procedure A does not deviate drastically from traditional library construction protocols and therefore would not significantly increase the labor costs associated with the per sample cost of library construction. While procedure B increases the rate of library construction by allowing for pooling stages that decreases the time spent on gel based size selection. AMPure XP beads is an alternative approach for size selection that typically reduces handling time by 90%, however this procedure was not considered here as associated per sample costs can be prohibitive for smaller labs as the maximum bead volume must be purchased to make beads cost effective.
To our knowledge this is the first optimized cost effective high-throughput protocol for library construction. The uniqueness of this approach lies in the combinatorial barcode and indexing, which not only allows for flexibility in library construction, but also allows for unique pooling stages. This in turn leads to a cost effective method of library construction in which per sample library construction costs are ~$8.5–10.5.
Table 2B.
Procedure A - 200 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 200 | NEB | 200 | M0348S | 2 | 96 | 192 | 0.96 |
| ZR-96 Pur Kit | 200 | Zymo | 4×96 | D4024 | 3 | 387 | 1161 | 5.80 |
| Gel Recovery | 200 | Zymo | 200 | D4002 | 1 | 278 | 278 | 1.39 |
| Quick Blunt | 200 | NEB | 200 | E1201L | 1 | 308 | 308 | 1.54 |
| Klenow exo- | 200 | NEB | 266.6 | M0212L | 2 | 228 | 456 | 1.71 |
| Quick Ligation | 200 | NEB | 250 | M2200L | 1 | 380 | 380 | 1.52 |
| Phusion | 200 | NEB | 200 | M0530S | 2 | 103 | 206 | 1.03 |
| Total Cost: | 2981 | Enzyme Total: | 6.76 | |||||
| Per Sample: | 14.90 | Column Total: | 7.19 | |||||
| Total Unit Cost Per Sample: | 13.95 |
Table 3A.
Procedure B - 100 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 100 | NEB | 100 | M0348S | 1 | 96 | 96 | 0.96 |
| ZR-96 Pur Kit | 100 | Zymo | 4×96 | D4024 | 1 | 387 | 387 | 3.87 |
| Quick Blunt | 100 | NEB | 120 | E1201S | 3 | 77 | 231 | 1.92 |
| Klenow exo- | 100 | NEB | 133.3 | M0212L | 1 | 228 | 228 | 1.71 |
| Quick Ligation | 100 | NEB | 100 | M2200S | 2 | 95 | 190 | 1.9 |
| Phusion | 100 | NEB | 50 | M0530S | 1 | 103 | 103 | 2.06 |
| Phusion-Pool 8 | 0.25 | |||||||
| Phusion-Pool 12 | 0.17 | |||||||
| Pool of 8 | Pool of 12 | |||||||
| Enzyme Cost: | 6.75 | 6.66 | ||||||
| Total Cost: | 1235 | Column Cost: | 3.87 | 3.87 | ||||
| Per Sample: | 12.35 | Total Unit Cost Per Sample: | 10.62* | 10.53* |
The cost of gel extraction columns and polymerase costs are not included in the cost estimation of Procedure B because the enzymatic unit cost is negligible when applied across 96 or more samples.
Table 3B.
Procedure B - 200 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 200 | NEB | 200 | M0348S | 2 | 96 | 192 | 0.96 |
| ZR-96 Pur Kit | 200 | Zymo | 4×96 | D4024 | 1 | 387 | 387 | |
| ZR-96 Pur Kit | 200 | Zymo | 2×96 | D4023 | 1 | 199 | 199 | 2.93 |
| Quick Blunt | 200 | NEB | 200 | E1201L | 1 | 308 | 308 | 1.54 |
| Klenow exo- | 200 | NEB | 266.6 | M0212L | 2 | 228 | 456 | 1.26 |
| Quick Ligation | 200 | NEB | 250 | M2200L | 1 | 380 | 380 | 1.71 |
| Phusion | 200 | NEB | 50 | M0530S | 1 | 103 | 103 | 2.06 |
| Phusion-Pool 8 | 0.25 | |||||||
| Phusion-Pool 12 | 0.17 | |||||||
| Pool of 8 | Pool of 12 | |||||||
| Enzyme Cost: | 5.7345 | 5.64 | ||||||
| Total Cost: | 2025 | Column Cost: | 2.93 | 2.93 | ||||
| Per Sample: | 10.12 | Total Unit Cost Per Sample: | 8.66* | 8.57* |
The cost of gel extraction columns and polymerase costs are not included in the cost estimation of Procedure B because the enzymatic unit cost is negligible when applied across 96 or more samples.
Table 3C.
Procedure B - 400 Samples
| Reagent | Total Samples | Company | Total Preps | Catalog # | Quantity | Price | Total Price | Per Unit Cost |
|---|---|---|---|---|---|---|---|---|
| Fragmentase | 400 | NEB | 500 | M0348L | 1 | 384 | 384 | 0.768 |
| ZR-96 Pur Kit | 400 | Zymo | 4×96 | D4024 | 3 | 387 | 1161 | 2.90 |
| Quick Blunt | 400 | NEB | 400 | E1201L | 2 | 308 | 616 | 1.54 |
| Klenow exo- | 400 | NEB | 399.9 | M0212L | 3 | 228 | 684 | 1.71 |
| Quick Ligation | 400 | NEB | 500 | M2200L | 2 | 380 | 760 | 1.52 |
| Phusion | 400 | NEB | 50 | M0530S | 1 | 103 | 103 | 2.06 |
| Phusion-Pool 8 | 0.12 | |||||||
| Phusion-Pool 12 | 0.08 | |||||||
| Pool of 8 | Pool of 12 | |||||||
| Enzyme Cost: | 5.66 | 5.62 | ||||||
| Total Cost: | 3708 | Column Cost: | 2.90 | 2.90 | ||||
| Per Sample: | 9.27 | Total Unit Cost Per Sample: | 8.56* | 8.52* |
The cost of gel extraction columns and polymerase costs are not included in the cost estimation of Procedure B because the enzymatic unit cost is negligible when applied across 96 or more samples.
Table 4.
DNA Extraction for 96 samples
| Reagent | Company | Preps/vol* | Catalog # | Quantity | Price | Total Price | Volume (ul)/sample | Per sample cost |
|---|---|---|---|---|---|---|---|---|
| Cell Lysis | Epicentre | 600 mL | MTC096H | 1 | 213 | 213 | 290 | 0.09 |
| MPC | Epicentre | 50 mL | MMP03750 | 1 | 59 | 59 | 200 | 0.23 |
| Proteinase K | Qiagen | 10 mL | 19133 | 1 | 267 | 267 | 10 | 0.26 |
| Zymo Quick-gDNA MiniPrep kit | Zymo | 50 preps | D3006 | 2 | 76 | 152 | 1.52 |
Volumes of each reagent for DNA purification can vary depending upon the amount of tissue.
Acknowledgments
This work was supported by NIH RO1 EUREKA GM098741, NSF PGRP 08-20846, and GM102227 to S. Nuzhdin. The authors would like to thank S. Porter for the use of her Mesorhizobium data, as well as S. Nuzhdin and H. Simmons for their comments and suggestions.
References
- Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA, et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5:887–893. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Stein LD. The case for cloud computing in genome informatics. Genome Biology. 2010;11:207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritt A, Eisen JA, Facciotti MT, Darling AE. An Integrated Pipeline for de Novo Assembly of Microbial Genomes. PLoS ONE. 2012;7(9):e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]