

Abstract
Amyloid is identified microscopically as an amorphous extracellular hyaline material that exhibits “apple-green” birefringence with Congo red stains. Amyloid is not a chemically distinct entity, and currently available molecular methods are capable of identifying over 20 amyloidogenic precursor proteins. Some of the more common diseases associated with amyloidosis include plasma cell dyscrasias, chronic inflammatory disorders, hereditary-familial mutations involving transthyretin, Alzheimer's disease, and so-called “senile” or age-related amyloidosis. The amyloid deposits in these various diseases may be isolated to a single organ such as the heart or brain, or the amyloidosis may be systemic. The senile types of cardiac amyloidosis can result from overproduction of atrial natriuretic factor or from accumulation of otherwise normal or wild-type transthyretin. We present the case of an 83-year-old hospitalized woman with known atrial fibrillation and previous pacemaker implantation who had cardiac arrest unresponsive to attempted resuscitation. Autopsy disclosed prominent amyloidosis involving the left atrium, and subsequent molecular studies identified the amyloidogenic material as alpha atrial natriuretic factor. Since the clinical management and genetic implications of the various diseases associated with amyloidosis are markedly different, we stress the importance of molecular classification whenever possible.
Although identified microscopically as an extracellular amorphous eosinophilic material with “apple-green” birefringence by Congo red staining, amyloid is not a chemically distinct entity (1). Furthermore, the tissue distribution of amyloid among the various subtypes may be systemic or isolated to a single organ such as the heart or brain, producing completely different clinical syndromes. The more common disorders associated with amyloid deposition include immunocyte or plasma cell dyscrasias, chronic inflammatory disorders, genetic (familial) disorders, Alzheimer's dementia, and so-called “senile” or age-related amyloid deposition. Also, within the “senile” amyloidosis category, involvement may be systemic or isolated to the heart, and two distinct subtypes of senile cardiac amyloidosis have been described. In isolated atrial amyloidosis (IAA), amyloid accumulates due to an overproduction of alpha-atrial natriuretic factor or protein (alpha-ANF, alpha-ANP) (2), whereas in senile systemic amyloidosis, the amyloid is derived from native or wild-type transthyretin, a transport protein produced by the liver (3). Since amyloidosis may cause abnormal cardiac function, including restrictive heart disease, conduction disorders, arrhythmias, and death, the clinical diagnosis and pathologic identification of this disorder are important (1). Furthermore, amyloid subtyping may inform therapy, and molecular typing may have profound implications for family members (4).
CASE STUDY
An 83-year-old woman was admitted to Baylor University Medical Center at Dallas with new-onset dyspnea and bilateral pulmonary infiltrates on chest radiograph. In the past a pacemaker had been implanted for “permanent” atrial fibrillation, and she was on warfarin with a recorded international normalized ratio of 4.4. Also, she was a long-term rehabilitation facility resident, and her problems included osteoarthritis, a hip fracture a year earlier, and poor mobility. On examination there was lower-extremity edema and muscle weakness, and she fatigued quickly. Her body mass index was 21.4 kg/m2.
The patient was admitted to the telemetry floor and given diuretics. The night of admission, acute hypercapnic respiratory failure prompted admission to the intensive care unit, and bilevel positive airway pressure support was initiated. Her blood pressure was 130/70 mm Hg; heart rate, 75 beats/minute; and respiratory rate, 34 breaths per minute. A grade 2/6 holosystolic murmur was audible at the cardiac apex. Computed tomographic examination of the chest disclosed right atrial enlargement, biapical pulmonary opacities (pneumonia versus edema), and pleural effusions (left > right). A brain natriuretic peptide was 314 pg/mL (normal ≤100 pg/mL), and an alanine transaminase was 67 U/L (normal 9–60 U/L). Troponin I was 0.03 ng/mL (normal <0.05 ng/mL). Transthoracic echocardiogram disclosed a normal left ventricular cavity size and normal systolic function with an ejection fraction of 55%; a dilated right atrium and right ventricle with preserved right ventricular systolic function; mild to moderate tricuspid regurgitation; an estimated right ventricular systolic pressure of 46 mm Hg; a dilated left atrium; mild aortic regurgitation; and a small pericardial effusion.
Shortly after intubation and placement of a central line, she developed ventricular fibrillation and pulseless electrical activity and died.
At necropsy, the pericardial sac contained 100 mL of clear fluid. The pacemaker leads were well seated. The heart weighed 460 g. The subepicardial adipose tissue was markedly increased, causing flotation of the heart in formalin. The epicardial surface was smooth and devoid of adhesions. The heart was examined through parasagittal incisions after fixation. The left atrial appendage was markedly dilated. Both atrial cavities were quite large and roughly dilated to similar degrees. Brown discolorations were present in the left atrial endocardium (Figure 1). The right ventricular cavity and the coronary sinus were dilated.
Figure 1.
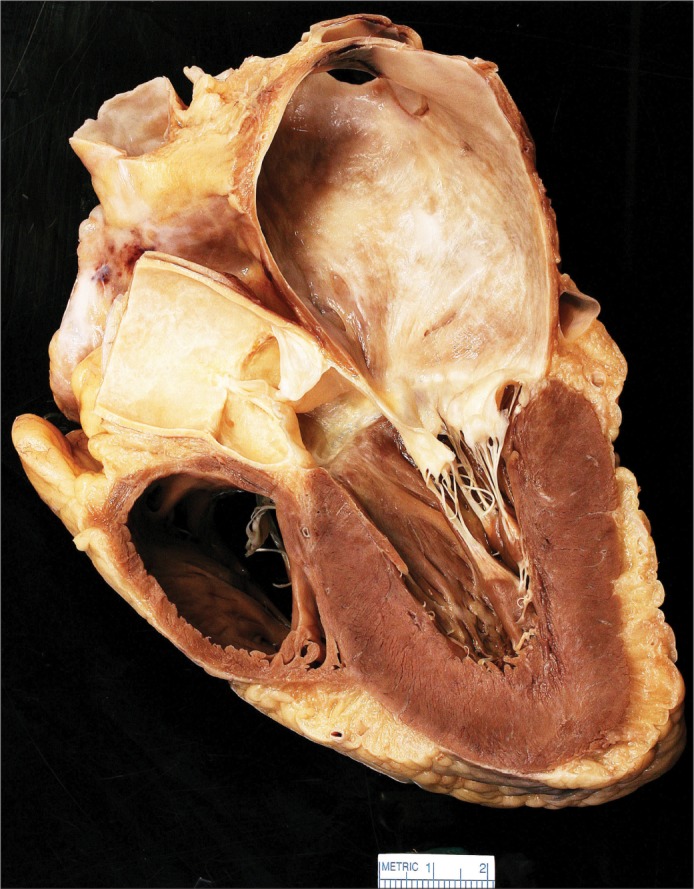
Sagittal section of the heart showing a markedly dilated left atrium with brown endocardial discolorations.
Microscopically, there were amyloid deposits within the walls of both atria (confirmed by Congo red stains), but these deposits were much more prominent within the left atrial wall (Figure 2). Congo red stains of the ventricular walls were negative.
Figure 2.
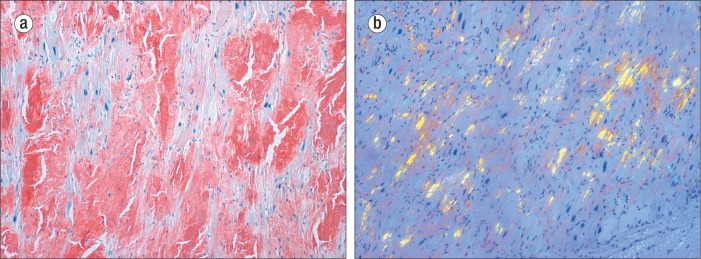
The left atrium showing heavy amyloid deposits. (a) Congo red stain, ×100. (b) “Apple-green” birefringence apparent on Congo red stain (polarized), ×100.
CD138 immunostain and kappa and lambda in situ hybridization studies of the spinal bone marrow did not reveal plasmacytosis or cellular clonality. Postmortem serum immunofixation electrophoresis was also negative for monoclonal peaks.
To further characterize the amyloid deposits, paraffin blocks were submitted to the Mayo Clinic Laboratories, where molecular amyloid subtyping by liquid chromatography-tandem mass spectrometry was performed on peptides extracted from Congo red–positive/microdissected areas of paraffin-embedded left atrial tissue. The resultant diagnosis was amyloidosis, atrial natriuretic factor type, in the left atrium of the heart.
The remainder of the autopsy revealed moderate cerebral and aortic atherosclerosis, centrilobular hepatic congestion, mild arteriolar nephrosclerosis with prominent medullary congestion, and a previous right thyroidectomy with residual left lobe nodular goiter. A small leiomyoma was present at the gastroesophageal junction, and several small gastrointestinal stromal tumors of the stomach wall were seen. Acute and chronic bronchitis was present microscopically, but there was no bronchopneumonia.
DISCUSSION
Although more than 20 amyloidogenic precursor proteins have been identified by current molecular methods (5), some forms are more common than others. Furthermore, the subtypes of amyloidosis have markedly different clinical implications. For example, AL amyloidosis, a common systemic form, is due to overproduction of immunoglobulin components associated with immunocyte dyscrasias, and effective treatment may require chemotherapy. AA amyloidosis, in contrast, may be associated with chronic inflammatory disorders, and therapy involves control of the underlying condition. Hereditary-familial forms of amyloidosis may be associated with mutations involving production of an abnormal form of transthyretin (prealbumin), and management may include genetic counseling and consideration for liver and heart transplantation.
The so-called senile or age-related forms of cardiac amyloidosis can be further divided into two major groups. In one form an otherwise normal or wild type of transthyretin molecule accumulates to form amyloid deposits for generally unknown reasons (6). In so-called IAA, amyloid deposition is limited to the cardiac atria (predominantly the left) as a result of overproduction of atrial natriuretic factor. This subtype of amyloidosis has been cited as one of the most common forms of amyloidosis. The clinical implications of this disorder remain controversial. IAA is frequently associated with atrial fibrillation, but whether this accumulation is its cause is not known. One theory postulates a vicious circle in which atrial dilatation and fibrillation lead to overproduction of atrial natriuretic factor and amyloid deposition, which further exacerbates fibrillation and dilatation (7).
Overall, cardiac amyloidosis remains the primary determinant of prognosis in patients with systemic amyloidosis (3). However, regardless of the subtype of cardiac amyloidosis, this diagnosis may be clinically elusive, and management of these patients may be difficult. The gold standard for diagnosis of cardiac amyloidosis remains endocardial biopsy, but indirect evidence for diagnosis includes echocardiographic evidence of amyloidosis and histologic confirmation of amyloid in noncardiac tissues.
Diagnostic efforts can be rewarding, as shown by the case above, since a diagnosis of amyloidosis may help explain functional cardiac disturbances and a patient's failure to respond to therapeutic and supportive measures, even if amyloid is only one contributor to the problem. Furthermore, as previously stated, the clinical implications and management of the various subtypes of amyloidosis are profoundly different; therefore, we encourage molecular subtyping of these disorders whenever possible. Fortunately, such testing is now reasonably available; however, research is still needed in order to improve our understanding of the clinical implications and optimal management of the various forms of cardiac and systemic amyloidosis as we proceed into this era of precision medicine. Furthermore, this case illustrates, once again, the relevance of traditional autopsy, especially when combined with modern and precise molecular tools to elucidate the true nature of a patient's disease.
Acknowledgment
We thank Jong M. (Jamie) Ko, BA, for photographic and technical assistance.
References
- 1.Kumar V, Abbas A, Fausto N, Aster J. Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia: Saunders/Elsevier; 2010. [Google Scholar]
- 2.Steiner I, Hájková P. Patterns of isolated atrial amyloid: a study of 100 hearts on autopsy. Cardiovasc Pathol. 2006;15(5):287–290. doi: 10.1016/j.carpath.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Kapoor P, Thenappan T, Singh E, Kumar S, Greipp PR. Cardiac amyloidosis: a practical approach to diagnosis and management. Am J Med. 2011;124(11):1006–1015. doi: 10.1016/j.amjmed.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Gertz MA. The classification and typing of amyloid deposits. Am J Clin Pathol. 2004;121(6):787–789. doi: 10.1309/TR4L-GLVR-JKAM-V5QT. [DOI] [PubMed] [Google Scholar]
- 5.Loo D, Mollee PN, Renaut P, Hill MM. Proteomics in molecular diagnosis: typing of amyloidosis. J Biomed Biotechnol. 2011;2011:754109. doi: 10.1155/2011/754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picken M, Dogan A, Herrera G. Amyloid and Related Disorders, Surgical Pathology and Clinical Correlations. New York: Humana Press; 2012. [Google Scholar]
- 7.Goette A, Röcken C. Atrial amyloidosis and atrial fibrillation: a gender-dependent “arrhythmogenic substrate”? Eur Heart J. 2004;25(14):1185–1186. doi: 10.1016/j.ehj.2004.04.014. [DOI] [PubMed] [Google Scholar]