

Abstract
The role of non-steroidal anti-inflammatory drugs in inflammatory bowel disease is controversial, as they have been implicated in disease aggravation. Different from other cyclooxygenase inhibitors, acetylsalicylic acid (ASA) enhances the formation of anti-inflammatory and proresolution lipoxins derived from arachidonic acid as well as resolvins from omega-3 polyunsaturated fatty acids such as docosahexaenoic acid (DHA). In this study, we examined the effect of ASA on murine dextran sodium sulfate colitis. A mouse magnetic resonance imaging (MRI) protocol and post mortem assessment were used to assess disease severity, and lipid metabolites were measured using liquid chromatography-coupled tandem mass spectrometry. Decreased colitis activity was demonstrated by phenotype and MRI assessment in mice treated with ASA, and confirmed in postmortem analysis. Analysis of lipid mediators showed sustained formation of lipoxin A4 and an increase of DHA-derived 17-hydroxydocosahexaenoic acid (17-HDHA) after treatment with ASA. Furthermore, in vitro experiments in RAW264.7 murine macrophages demonstrated significantly increased phagocytosis activity after incubation with 17-HDHA, supporting its proresolution effect. These results show a protective effect of ASA in a murine colitis model and could give a rationale for a careful reassessment of ASA therapy in patients with inflammatory bowel disease and particularly ulcerative colitis, possibly combined with DHA supplementation.
1. Introduction
The inflammatory bowel diseases (IBDs) ulcerative colitis (UC), confined to the colon, and Crohn's disease (CD), affecting the whole gastrointestinal tract, are chronic inflammatory disorders with considerable morbidity and, particularly for UC, mortality due to a high risk of colorectal cancer development. Current therapy for IBD focuses on drugs that inhibit inflammation [1–4]. Human data regarding the potential benefits and risks of COX inhibition in patients with IBD are conflicting [5–9]. This is also reflected in animal studies examining COX inhibition in experimental colitis models. While some recent studies found deleterious effects in animals with DSS colitis associated with inhibition of COX [10–13], others demonstrated a benefit of COX-2 inhibitor treatment [14, 15].
Recently, newly discovered lipid mediators have been implicated in the alleviation of colitis [16]. The lipoxins are lipid mediators originating from the omega-6 polyunsaturated fatty acid (n-6 PUFA) arachidonic acid (AA) by enzymatic action of different lipoxygenases or by acetylated COX-2 [17]. Studies have demonstrated potent anti-inflammatory and inflammation dampening properties for lipoxins [18], and stable analogues of lipoxins were shown to alleviate experimental colitis [19, 20]. Similarly, protective lipid mediators (so-called resolvins) can be formed from omega-3 polyunsaturated fatty acids (n-3 PUFAs) such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Our previous work demonstrated formation of resolvins in mice with increased omega-3 fatty acid tissue content in the context of DSS colitis [21], and other studies have shown an anti-inflammatory effect of several of these compounds in the context of experimental colitis models [22–24].
In this study, we analyzed the effect of acetylsalicylic acid (ASA) in chemically induced acute DSS colitis in mice. DSS colitis is characterized by initial injury to the epithelial cells followed by inflammation and has been shown to be a good model to test therapeutics for human inflammatory bowel disease [25]. Many studies have used this model in the past as it is regarded as a good animal model of IBD, and particularly UC, with a predominant site of inflammation in the distal colon [26].
To evaluate disease activity in the different groups, we extended our analysis beyond the conventional phenotype and morphology markers and employed small animal MRI technology. Noninvasive imaging in murine models of colitis has been previously described using CT and MRI technologies [27–29]. For the study presented here, a modified protocol was devised in order to reliably measure and compare colon wall thickness and hyperemia in different mice.
To assess possible mechanisms responsible for the observed protection in ASA-treated mice, mass spectrometric measurements were then employed to determine hydroxylated lipid metabolites and mediators derived from AA (lipoxin A4 and 15-epi-lipoxin A4) and from 17-hydroxydocosahexaenoic acid (DHA).
2. Materials and Methods
2.1. Induction of Colitis
Female C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA) and held until the desired age (6 weeks) and body weight (19–21 g).
For testing of different treatment regimens, mice were divided into 3 groups. One group received 3% DSS dissolved in sterile drinking water and daily intraperitoneal (ip) injections of 0.5 mg acetylsalicylic acid (ASA, Sigma-Aldrich, St. Louis, MO) dissolved in 300 μL of vehicle (3.33% EtOH in 0.9% NaCl) for 5 days (n = 5). Another group received 3% DSS as well as ip injections of the vehicle alone for 5 days (n = 6). Finally, the last group received sterile drinking water and ip injection of vehicle alone and served as a control (n = 3). The first ip injection of ASA or vehicle was administered right before the mice were switched to DSS-containing drinking water. After 5 days, the DSS-containing water was replaced with sterile drinking water.
2.2. Evaluation of Colitis Severity
Body weight measurement (presented as percentage of body weight on day 0) as well as evaluation of stool status was carried out daily. Stool samples from individual mice were evaluated on a three-point scale using a test for occult blood (Hemoccult, Beckman Coulter Inc., Fullerton, CA) as well as macroscopic evaluation (0 = no occult blood; 1 = test for occult blood slightly positive; 2 = test for occult blood strongly positive; and 3 = bloody stool). On day 9, mice were sacrificed, colons were excised, and length was measured. All samples were snap frozen in liquid nitrogen and stored at −80°C for further analysis.
2.3. Histological Evaluation
Frozen tissue sections from the distal colon of all mice were mounted in OCT medium. Sections were cut at 5 μm thickness and mounted on glass slides. After air drying, sections were fixed in fixing medium (70% ethanol, 4% formaldehyde, and 5% glacial acetic acid in H2O), washed in ethanol (70%) and water, followed by standard hematoxylin and eosin stain. Slides were evaluated in a blinded manner and scored according to severity of inflammation (0 = no inflammation, 1 = mild, 2 = moderate, and 3 = severe), thickness of inflammatory involvement (0 = no inflammation, 1 = mucosa, 2 = mucosa plus submucosa, and 3 = transmural), character of epithelial damage (0 = intact epithelium, 1 = disruption of architectural structure, 2 = erosion, and 3 = ulceration), and extent of lesions (0 = no lesions, 1 = punctuate, 2 = multifocal, and 3 = diffuse). This evaluation protocol is based on a previously described scoring system for DSS colitis [30].
2.4. Magnetic Resonance Imaging (MRI)
Mice were scanned on day 3 and day 8 of the experimental protocol. MRI assessment of colitis was performed using a Bruker 4.7T Avance horizontal bore system (Bruker Avance, Karlsruhe, Germany) equipped with a 200 mm gradient set capable of 30 G/cm gradient strength. During the MR scan, the mice were mounted on a home-built nonmagnetic holding bar equipped with a nose cone for anesthesia delivery and were anaesthetized using 1.5%–2% (vol/vol) isoflurane (Baxter, Deerfield, IL) in 1 L/min oxygen flow. The breathing rate of the mice was monitored using a small animal monitoring and gating system (Small Animal Instruments Inc., NY). The isoflurane flow was adjusted to achieve a breathing rate of 20–30 breaths per minute during the scanning process. In order to standardize the comparisons of wall thickness as well as colon wall signal intensity between different animals, a nonmagnetic (plastic) tube (outer diameter 2.1 mm) was placed approximately 9 mm into the rectum of the anaesthetized mice (Figure 1(a)). For each mouse, 24 T2 weighted 2D axial images were acquired to cover the colon using a fast spin echo sequence with a repetition time (TR) of 2683 ms, an echo time (TE) of 12 ms, a 128 × 128 matrix, and a 3.0 cm × 3.0 cm field of view (FOV). The slice thickness was set to 1 mm, and the number of averages was 8. After image acquisition, the plastic tube was removed, and the animals were transferred to a heating pad until they were fully awake. The images were analyzed using standard imaging software (Adobe Photoshop, Adobe Systems, San Jose, CA).
Figure 1.
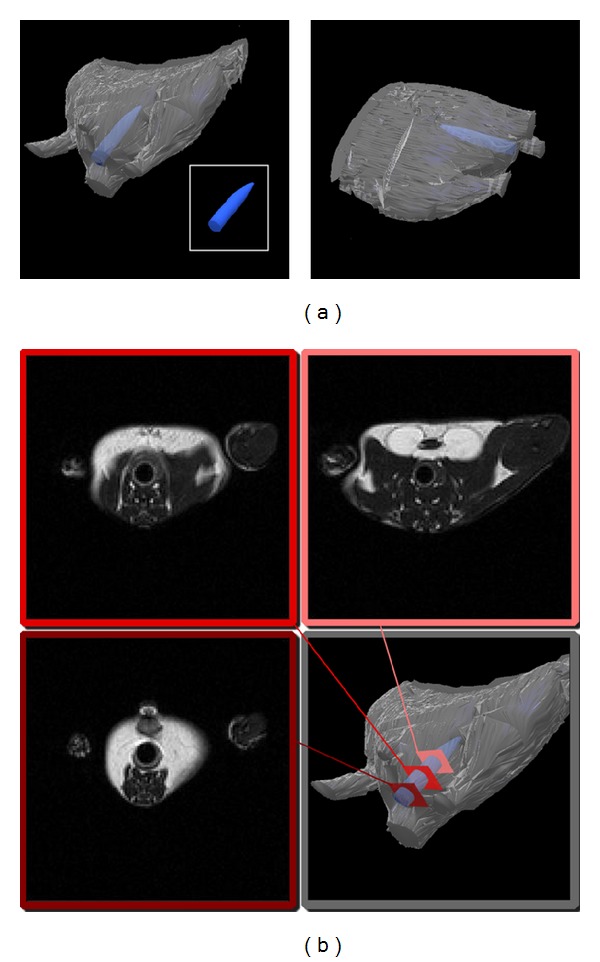
MRI analysis of colitis severity. (a) For MRI analysis of disease severity, a plastic tube with outer diameter of 2.1 mm was carefully placed approximately 9 mm into the rectum of the anaesthetized mouse. (b) Analysis was performed by comparing wall thickness as well as brightness between the different mice in three defined slices.
Colon wall thickness was then measured on the T2 weighted images from three slices of the distal colon with a standardized distance of 3 mm between slices (Figure 1(b)). For each slice, three measurements were performed, and the means for each animal were calculated. For signal intensity analysis, a region of interest (ROI) was drawn within the colon wall. A histogram tool was used to analyze the brightness of the pixels within the ROI. This was repeated for the same three slices that were used for the wall thickness measurements. The pixel brightness means were then normalized by comparison with the nearby fatty tissue around the ovaries of the mice.
2.5. Lipid Metabolite Analysis
30 mg ground and frozen colon tissue was mixed with 500 μL water and 500 μL methanol, internal standard consisting of 10 ng LTB4-d4 was added, and the sample was then hydrolyzed with 300 μL of 10 M sodium hydroxide for 30 minutes at 60°C. The solution was neutralized with 60% acetic acid, and the pH was adjusted to 6.0 with sodium acetate buffer. A solid phase extraction was performed as previously described [31]. For elution, an n-hexane:ethyl acetate extraction mixture (25 : 75) with 1% acetic acid was used. The eluate was evaporated on a heating block at 40°C under a stream of nitrogen to obtain a solid residue. Residues were then dissolved in 70 μL acetonitrile and measured using an Agilent 1200 HPLC system coupled to an Agilent 6410 Triple quad mass spectrometer with an electrospray ionization source. Analysis of lipid metabolites was performed with multiple reaction monitoring in negative mode.
2.6. Phagocytosis Assay
RAW 264.7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin. All experiments were performed in a humidified atmosphere under 5% CO2 at 37°C. Cells were plated in 96-well plates at a density of 1 × 105 cells/well and incubated overnight. To quantify phagocytosis, cells were incubated with fluorescein-labelled bioparticles (Vybrant Phagocytosis Assay, Invitrogen, Carlsbad, CA), diluted 1 : 2. Uptake was measured after 4 h in a fluorescence plate reader, essentially following the manufacturer's protocol. To evaluate the effect of 17-HDHA on phagocytosis, cells were pretreated with 1 μM 17-HDHA for 2 h. Positive controls with equivalent volumes of ethanol (vehicle) and negative controls were performed in parallel.
2.7. Statistical Analysis
Statistical analysis was performed with GraphPad Prism 3.02v Software (GraphPad, La Jolla, CA). Comparison was made using the Mann-Whitney U test or as indicated. All values are presented as the mean ± SEM. Statistical significance was set at P < 0.05.
3. Results
Induction of colitis resulted in marked changes in body weight, appearance of fecal blood, and general status. Mice treated with ASA showed significantly less body weight loss (Figure 2(a)) as well as a recovery of body weight within 3 days after the end of DSS exposure, while untreated mice exhibited only a stabilization of body weight after cessation of DSS.
Figure 2.
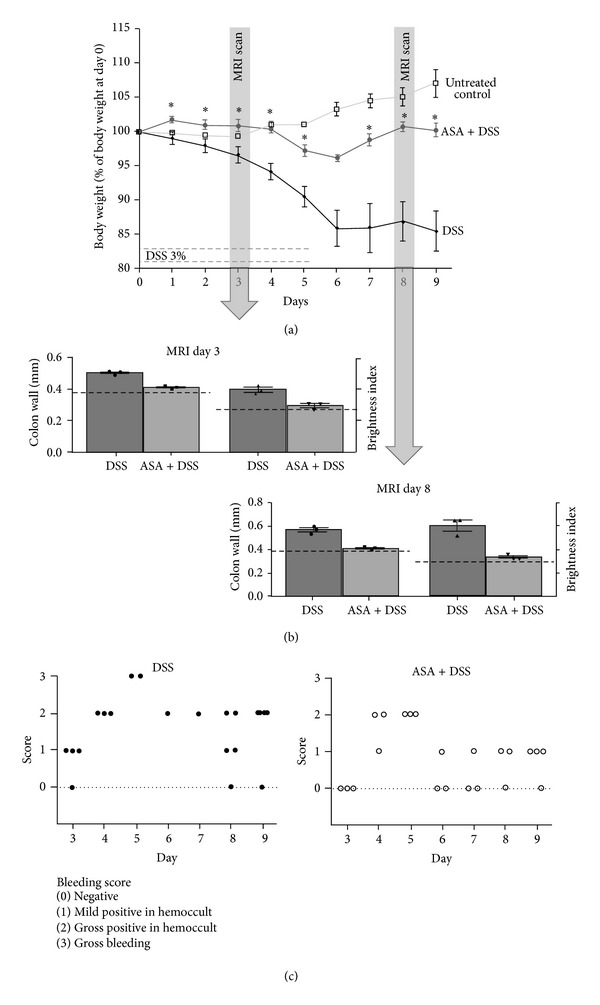
In vivo assessment of colitis severity. (a) Body weight course in the different groups, n = 3 for the control group, n = 5 for the ASA-group, and n = 6 for the DSS group; *P < 0.05 versus DSS. (b) MRI-measured colon wall thickness and brightness on day 3 as well as day 8 of the experimental protocol, n = 3 for each treatment group. In dashed lines, the values of measurements in untreated control mice are shown. (c) Bleeding score evaluation in animals treated with DSS or ASA + DSS. There was no bleeding in control animals (not shown).
Colitis disease activity was evaluated by MRI on day 3 and day 8 of the treatment protocol. The MRI results show a marked difference between the treated groups of mice with less wall thickness and a significantly lower T2 weighted signal intensity (brightness) in the mice treated with ASA, as signs of a lower inflammatory infiltration (Figure 2(b)). Due to technical limitations in the number of MRI scans we were able to perform, only 3 animals per group were assayed, which does not give enough values for statistical normality testing using, for example, the Kolmogorov-Smirnov test. Applying nonparametric Mann-Whitney U testing to these data led to nonsignificant findings. However, when hypothesizing normal distribution—and thus applying Student's t-test—for these data, the differences for thickness as well as brightness were significant on day 3 and day 8 with P < 0.01 when comparing the ASA + DSS group versus DSS alone.
The results of the MRI and body weight analyses confirmed the decreased disease activity in the mice treated with ASA. Furthermore, assessment of bloody stools in the animals showed a trend towards more severe and prolonged bleeding in the DSS group (Figure 2(c)). However, conclusive statistical evaluation of these data could not be performed, as it was not possible to do stool testing on every animal every day of the experimental protocol.
The in vivo disease activity assessments were validated in the postmortem analysis of the colons, with wall thicknesses similar to those observed in the MRI measurements (Figure 3(a)) and significantly less colon shortening in mice treated with ASA as compared to mice exposed only to DSS (Figure 3(b)). Microscopic grading of the changes in the colon revealed decreased inflammatory activity in mice treated with ASA (summarized scores are shown in Figure 3(c)). However, we were not able to document changes in inflammatory activity biochemically, as serum TNF-α levels, measured by ELISA, were not significantly different between the different treatment groups (data not shown).
Figure 3.
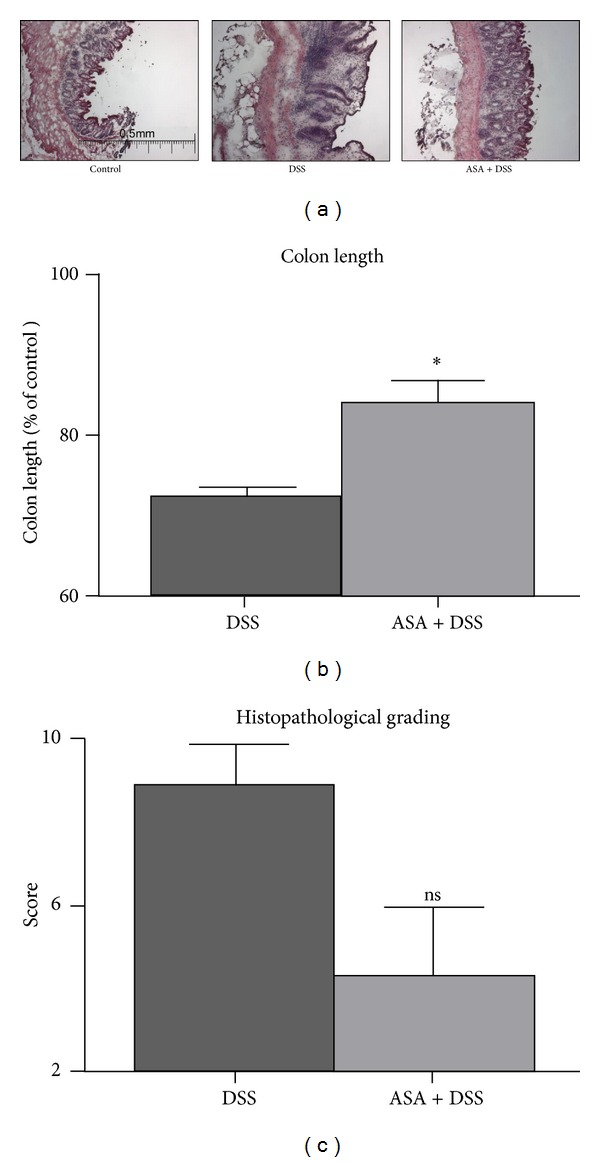
Postmortem assessment of colitis severity. (a) Hematoxylin and eosin stains of transmural colon walls. Representative images of control tissue as compared to animals treated with DSS and ASA + DSS. (b) Colon shortening as an indicator of disease severity was compared between the different experimental groups, n = 5 for the ASA-group and n = 6 for the DSS group; *P = 0.044 versus DSS. (c) Histological grading score, n = 5 for the ASA-group and n = 6 for the DSS group; ns P = 0.054 versus DSS. Untreated controls (not shown) reached a maximum of 2 points in this score.
As lipid mediators formation of AA-derived lipoxins and n-3 PUFA-derived resolvins can be increased by ASA-induced acetylation of the COX-2 enzyme [32], we assessed several of these mediators and their precursors in the colons of mice with DSS-induced colitis with and without treatment with ASA. Due to the presence of AA and DHA but not EPA in the colon tissue of the mice used in this study (data not shown), we focused on the assessment of AA-derived lipoxins and DHA-derived 17-hydroxydocosahexaenoic acid (17-HDHA). Lipoxin A4 and 15-epi-lipoxin A4 concentrations were decreased in colon tissue from mice three days after DSS exposure, but formation of 15-epi-lipoxin A4 was significantly higher in mice treated with ASA as compared to the DSS alone treatment group (Figures 4(a) and 4(b)). Unexpectedly, the DHA-metabolite 17-HDHA was significantly higher in colon tissues from mice three days after DSS exposure (Figure 4(c)) and was increased even more in tissue samples from mice treated with ASA.
Figure 4.
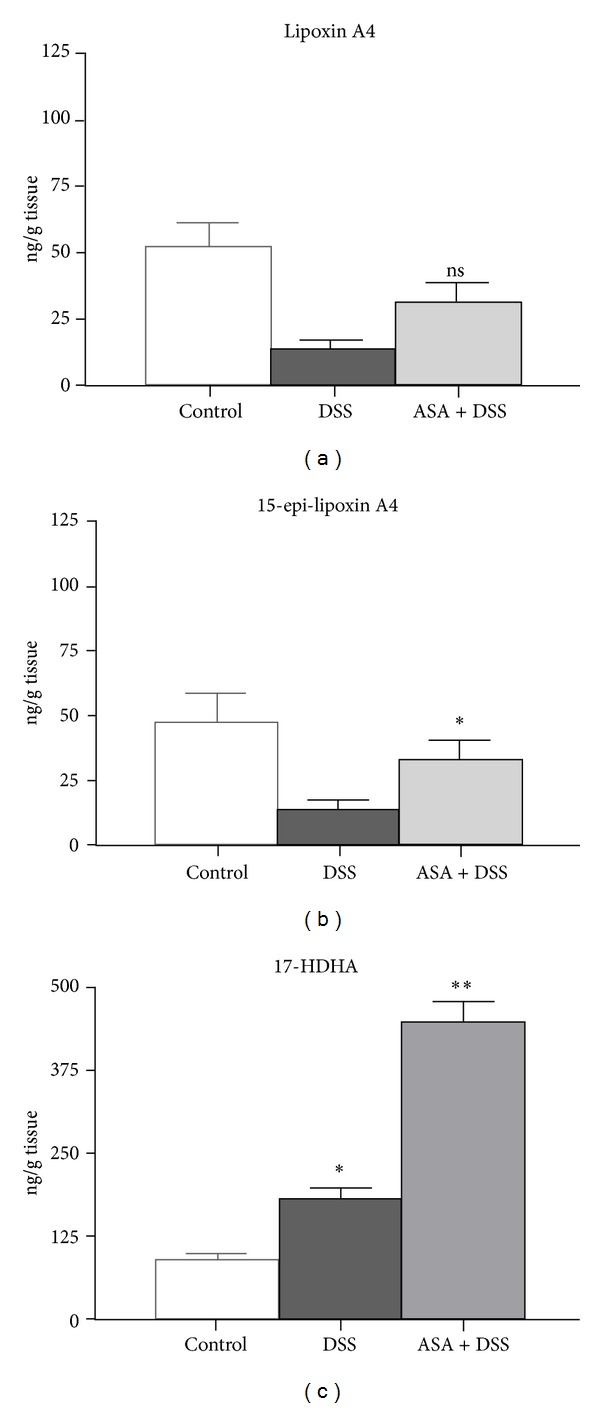
Assessment of lipid mediators. Shown are the arachidonic acid-derived lipoxin A4 (a) and 15-epi-lipoxin A4 (b) with n = 3 for the control group, n = 5 for both the DSS alone and the DSS plus ASA groups; ns P = 0.056 versus DSS (not significant); *P = 0.016 versus DSS. (c) The levels of 17-HDHA, with n = 3 for the control group, n = 5 for both the DSS alone and the DSS plus ASA groups; *P = 0.036 versus controls; **P = 0.008 versus DSS (b).
In order to further elucidate the mechanism by which 17-HDHA might contribute to inflammation dampening and resolution, the in vitro ability of 17-HDHA to increase phagocytosis was tested. In these experiments, an assay system with fluorescently labeled E. coli K-12 bioparticles was used as a tool to study overall phagocytotic activity of macrophages [33, 34]. A significant (1.25-fold) increase in phagocytosis activity after 1 μM 17-HDHA preincubation in the RAW 264.7 murine macrophage cell line was demonstrated (Figure 5(a)).
Figure 5.
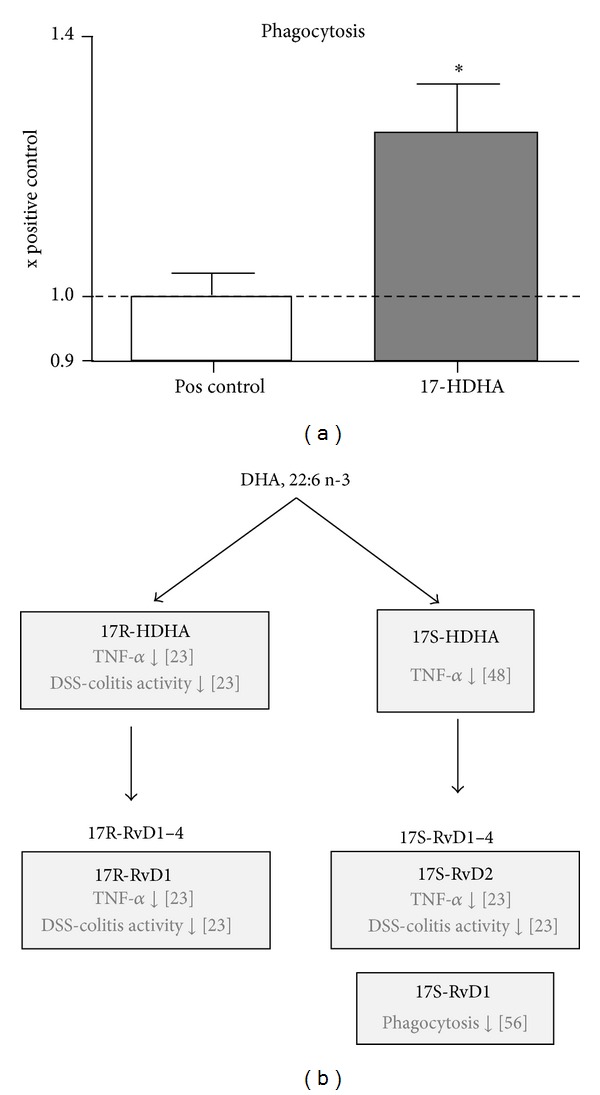
Effect of 17-HDHA on phagocytosis and overview of DHA-derived lipid mediators. (a) Effect of 17R/S-HDHA on phagocytosis in RAW 264.7 murine macrophages (n = 12, *P = 0.026). (b) Synthesis pathways of DHA-derived anti-inflammatory lipid mediators arising from 17R- and 17S-HDHA, and the resulting mediators are called the resolvins D1-4. Included also are the references to the published effects of the DHA-derived mediators on TNF-α secretion from macrophages [23, 48], phagocytosis [56], and DSS-colitis activity [23].
4. Discussion
The data presented here indicate that DSS-induced colon inflammation can be alleviated by acetylsalicylic acid. Although a number of previous studies have examined the effectiveness of COX inhibitors in the context of DSS colitis, the outcomes of these studies were inconsistent or conflicting [10–15], and, to our knowledge, ASA was never used in experimental DSS colitis. This may be due to the fact that ASA causes ulcerations, erosions, and gastrointestinal bleeding in the stomach as well as the small bowel, and many clinicians believe it therefore to be too GI toxic to be of any value in the control of colonic inflammation.
At the same time, patients with IBD, and particularly UC, remain at increased risk for colorectal cancer [35, 36]. Long-term ASA treatment has been shown to be effective for colon cancer prevention [37–40]. In patients with UC, although data in this field is scarce, a similar risk reduction of colorectal cancer could be shown [41], arguing for ASA use also in these patients.
We set out to study the effect of ASA supplementation on the course of DSS-induced colitis in mice, hypothesizing an effect of aspirin treatment on the formation of hydroxylated lipid mediators such as the lipoxins as well as DHA-derived compounds. These were characterized as potent anti-inflammatory compounds also in the context of DSS colitis in previous studies [19, 23].
In our experiments, we aimed for an ASA dose comparable to low-dose ASA administration in humans. Previously published data on low-dose ASA administration in mice with intestinal or other tumors used doses of 40 mg/kg [42] and 50 mg/kg [43]. High-dose ASA dosage was at 400 mg/kg in a previous tumor prevention study in mice [44]. Two studies in intestinal tumor models used a dose of 25 mg/kg [45, 46], which the authors calculated to correspond to a dose of 80–110 mg/day in humans on a 2000 kcal diet following nutrient density calculations [47], and we decided to use this dose in our experiment to mimic low-dose ASA administration. A limitation of the data presented here is that no experiments with ASA only treatment were performed. However, the previous study by Mahmoud et al. has established the safety of long-term administration of ASA at a dose of 25 mg/kg in mice [45].
In the study presented here, we found significant changes in the tissue concentrations of anti-inflammatory lipoxins, with suppression of lipoxin A4 and 15-epi-lipoxin A4 in the mice with DSS colitis. As compared to this, the lipoxin levels in the ASA-treated animals were higher, which could contribute to the protection from colitis seen in these animals. In contrast, the amount of the DHA-derived 17-HDHA increased in animals after DSS treatment and even more so with concomitant ASA treatment. While higher levels of 17-HDHA were present in the colons from mice after DSS colitis induction as compared to controls, the treatment with ASA led to even higher 17-HDHA tissue concentrations.
17-HDHA is the precursor for the formation of anti-inflammatory D resolvins, and several previous studies from other groups have demonstrated biological effects of the different stereoisomers of this compound, as summarized in (Figure 5(b)). It was shown that TNF-α secretion from murine macrophages can be suppressed by 17S-HDHA [48]. Furthermore, it has been demonstrated that 17S-HDHA is a precursor of the D(S)-resolvins, which are potent anti-inflammatory mediators [49–51]. Similarly, the 17-HDHA epimer 17R-HDHA as well as the D(R)-resolvins were recently shown to be anti-inflammatory in the context of experimental colitis, peritonitis, and arthritis [23, 51, 52].
The liquid chromatograph tandem mass spectrometry (LC-MS/MS) setup used in the study presented here was not able to differentiate between the 17S- and 17R-HDHA epimers and could not reliably identify resolvin D1 in tissue samples. However, as both epimers of 17-HDHA are precursors of anti-inflammatory D-resolvins, the increase in total 17(R/S)-HDHA observed in this study is an indicator of significantly increased formation of these anti-inflammatory lipid mediators derived from DHA.
Indeed, our in vitro results presented previously showed a 17-HDHA-triggered decrease in TNF-α secretion [53]. Together with the increased phagocytosis activity by 17-HDHA shown here (Figure 5), this indicates that a mix of the 17R/S-HDHA epimers itself carries anti-inflammatory activity. Data for lipoxin A4 [54, 55] and resolvin D1 [56] have established the concept of increased phagocytosis as a marker of inflammation dampening and resolution. Therefore, the data shown here for 17-HDHA add to the hypothesis that increasing the phagocytosis activity of macrophages is an important effect of the hydroxylated anti-inflammatory and proresolution lipid mediators derived from either AA or DHA.
Concerning the anti-inflammatory effect of ASA, the results presented here support additional anti-inflammatory mechanisms of ASA as compared to the mere inhibition of prostaglandin synthesis, by enhancing the formation of 17-HDHA in murine colitis. Together with the established effect of ASA in colon cancer prevention, the findings presented here might thus be able to contribute to interest in a careful reevaluation of ASA therapy in patients with UC with its high risk of colon cancer development, possibly in combination with DHA supplementation.
Conflict of Interests
The authors have no relevant financial and personal interests to declare.
Acknowledgment
This work was supported by a grant from the German Research Foundation (DFG) to Karsten H. Weylandt.
References
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. The Lancet. 2007;369(9573):1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. The Lancet. 2007;369(9573):1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 3.Podolsky DK. Inflammatory bowel disease. The New England Journal of Medicine. 2002;347(6):417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 4.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi K, Smale S, Premchand P, et al. Prevalence and mechanism of nonsteroidal anti-inflammatory drug-induced clinical relapse in patients with inflammatory bowel disease. Clinical Gastroenterology and Hepatology. 2006;4(2):196–202. doi: 10.1016/s1542-3565(05)00980-8. [DOI] [PubMed] [Google Scholar]
- 6.Bloomfeld RS. Are COX-2 inhibiotrs safe for anyone. Inflammatory Bowel Diseases. 2006;12(9):922–923. doi: 10.1002/ibd.3780120913. [DOI] [PubMed] [Google Scholar]
- 7.El Miedany Y, Youssef S, Ahmed I, El Gaafary M. The gastrointestinal safety and effect on disease activity of etoricoxib, a selective Cox-2 inhibitor in inflammatory bowel diseases. The American Journal of Gastroenterology. 2006;101(2):311–317. doi: 10.1111/j.1572-0241.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 8.Matuk R, Crawford J, Abreu MT, Targan SR, Vasiliauskas EA, Papadakis KA. The spectrum of gastrointestinal toxicity and effect on disease activity of selective cyclooxygenase-2 inhibitors in patients with inflammatory bowel disease. Inflammatory Bowel Diseases. 2004;10(4):352–356. doi: 10.1097/00054725-200407000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Feagins LA, Cryer BL. Do non-steroidal anti-inflammatory drugs cause exacerbations of inflammatory bowel disease? Digestive Diseases and Sciences. 2010;55(2):226–232. doi: 10.1007/s10620-009-1042-7. [DOI] [PubMed] [Google Scholar]
- 10.Okayama M, Hayashi S, Aoi Y, Nishio H, Kato S, Takeuchi K. Aggravation by selective COX-1 and COX-2 inhibitors of dextran sulfate sodium (DSS)-induced colon lesions in rats. Digestive Diseases and Sciences. 2007;52(9):2095–2103. doi: 10.1007/s10620-006-9597-z. [DOI] [PubMed] [Google Scholar]
- 11.Tsubouchi R, Hayashi S, Aoi Y, et al. Healing impairment effect of cyclooxygenase inhibitors on dextran sulfate sodium-induced colitis in rats. Digestion. 2007;74(2):91–100. doi: 10.1159/000097657. [DOI] [PubMed] [Google Scholar]
- 12.Morteau O, Morham SG, Sellon R, et al. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. Journal of Clinical Investigation. 2000;105(4):469–478. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace JL, Keenan CM, Gale D, Shoupe TS. Exacerbation of experimental colitis by nonsteroidal anti-inflammatory drugs is not related to elevated leukotriene B4 synthesis. Gastroenterology. 1992;102(1):18–27. doi: 10.1016/0016-5085(92)91779-4. [DOI] [PubMed] [Google Scholar]
- 14.Martín AR, Villegas I, Alarcón de la Lastra C. The COX-2 inhibitor, rofecoxib, ameliorates dextran sulphate sodium induced colitis in mice. Inflammation Research. 2005;54(4):145–151. doi: 10.1007/s00011-004-1337-2. [DOI] [PubMed] [Google Scholar]
- 15.Kankuri E, Vaali K, Korpela R, Paakkari I, Vapaatalo H, Moilanen E. Effects of a COX-2 preferential agent nimesulide on TNBS-induced acute inflammation in the gut. Inflammation. 2001;25(5):301–310. doi: 10.1023/a:1012860509440. [DOI] [PubMed] [Google Scholar]
- 16.Weylandt KH, Kang JX, Wiedenmann B, Baumgart DC. Lipoxins and resolvins in inflammatory bowel disease. Inflammatory Bowel Diseases. 2007;13(6):797–799. doi: 10.1002/ibd.20109. [DOI] [PubMed] [Google Scholar]
- 17.Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(17 I):5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkinson JF. Lipoxin and synthetic lipoxin analogs: an overview of anti-inflammatory functions and new concepts in immunomodulation. Inflammation and Allergy. 2006;5(2):91–106. doi: 10.2174/187152806776383125. [DOI] [PubMed] [Google Scholar]
- 19.Gewirtz AT, Collier-Hyams LS, Young AN, et al. Lipoxin A4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. Journal of Immunology. 2002;168(10):5260–5267. doi: 10.4049/jimmunol.168.10.5260. [DOI] [PubMed] [Google Scholar]
- 20.Fiorucci S, Wallace JL, Mencarelli A, et al. A β-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(44):15736–15741. doi: 10.1073/pnas.0404722101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudert CA, Weylandt KH, Lu Y, et al. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(30):11276–11281. doi: 10.1073/pnas.0601280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(21):7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. Journal of Immunology. 2011;187(4):1957–1969. doi: 10.4049/jimmunol.1101305. [DOI] [PubMed] [Google Scholar]
- 24.Ishida T, Yoshida M, Arita M, et al. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflammatory Bowel Diseases. 2010;16(1):87–95. doi: 10.1002/ibd.21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melgar S, Karlsson L, Rehnström E, et al. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. International Immunopharmacology. 2008;8(6):836–844. doi: 10.1016/j.intimp.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 26.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nature Protocols. 2007;2(3):541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 27.Fredin MF, Hultin L, Hyberg G, et al. Predicting and monitoring colitis development in mice by micro-computed tomography. Inflammatory Bowel Diseases. 2008;14(4):491–499. doi: 10.1002/ibd.20343. [DOI] [PubMed] [Google Scholar]
- 28.Larsson AE, Melgar S, Rehnstrom E, et al. Magnetic resonance imaging of experimental mouse colitis and association with inflammatory activity. Inflammatory Bowel Diseases. 2006;12(6):478–485. doi: 10.1097/00054725-200606000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Melgar S, Gillberg PG, Hockings PD, Olsson LE. High-throughput magnetic resonance imaging in murine colonic inflammation. Biochemical and Biophysical Research Communications. 2007;355(4):1102–1107. doi: 10.1016/j.bbrc.2007.02.090. [DOI] [PubMed] [Google Scholar]
- 30.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clinical and Experimental Immunology. 1998;114(3):385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomolka B, Siegert E, Blossey K, Schunck W-H, Rothe M, Weylandt KH. Analysis of omega-3 and omega-6 fatty acid-derived lipid metabolite formation in human and mouse blood samples. Prostaglandins and other Lipid Mediators. 2011;94(3-4):81–87. doi: 10.1016/j.prostaglandins.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circulation Research. 2010;107(10):1170–1184. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Scherl A, Medina F, et al. Impaired phagocytosis in caveolin-1 deficient macrophages. Cell Cycle. 2005;4(11):1599–1607. doi: 10.4161/cc.4.11.2117. [DOI] [PubMed] [Google Scholar]
- 34.Wan CP, Park CS, Lau BHS. A rapid and simple microfluorometric phagocytosis assay. Journal of Immunological Methods. 1993;162(1):1–7. doi: 10.1016/0022-1759(93)90400-2. [DOI] [PubMed] [Google Scholar]
- 35.Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Alimentary Pharmacology and Therapeutics, Supplement. 2003;18(supplement 2):1–5. doi: 10.1046/j.1365-2036.18.s2.2.x. [DOI] [PubMed] [Google Scholar]
- 36.Lutgens MW, Oldenburg B, Siersema PD, et al. Colonoscopic surveillance improves survival after colorectal cancer diagnosis in inflammatory bowel disease. British Journal of Cancer. 2009;101(10):1671–1675. doi: 10.1038/sj.bjc.6605359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Curhan GC, Fuchs CS. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. Journal of the American Medical Association. 2005;294(8):914–923. doi: 10.1001/jama.294.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg L, Louik C, Shapiro S. Nonsteroidal antiinflammatory drug use and reduced risk of large bowel carcinoma. Cancer. 1998;82(12):2326–2333. doi: 10.1002/(sici)1097-0142(19980615)82:12<2326::aid-cncr5>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 39.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. The Lancet. 2007;369(9573):1603–1613. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 40.Rothwell PM, Wilson M, Elwin C-E, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. The Lancet. 2010;376(9754):1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 41.Velayos FS, Loftus EV, Jr., Jess T, et al. Predictive and protective factors associated with colorectal cancer in ulcerative colitis: a case-control study. Gastroenterology. 2006;130(7):1941–1949. doi: 10.1053/j.gastro.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 42.Stark LA, Reid K, Sansom OJ, et al. Aspirin activates the NF-κB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28(5):968–976. doi: 10.1093/carcin/bgl220. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Wang Z, Zhang Y, Jia Q, Wu L, Zhang W. Impact of acetylsalicylic acid on tumor angiogenesis and lymphangiogenesis through inhibition of VEGF signaling in a murine sarcoma model. Oncology Reports. 2013;29(5):1907–1913. doi: 10.3892/or.2013.2339. [DOI] [PubMed] [Google Scholar]
- 44.Mcilhatton MA, Tyler J, Kerepesi LA, et al. Aspirin and low-dose nitric oxide-donating aspirin increase life span in a Lynch syndrome mouse model. Cancer Prevention Research. 2011;4(5):684–693. doi: 10.1158/1940-6207.CAPR-10-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahmoud NN, Dannenberg AJ, Mestre J, et al. Aspirin prevents tumors in a murine model of familial adenomatous polyposis. Surgery. 1998;124(2):225–231. [PubMed] [Google Scholar]
- 46.Tian Y, Ye Y, Gao W, et al. Aspirin promotes apoptosis in a murine model of colorectal cancer by mechanisms involving downregulation of IL-6-STAT3 signaling pathway. International Journal of Colorectal Disease. 2011;26(1):13–22. doi: 10.1007/s00384-010-1060-0. [DOI] [PubMed] [Google Scholar]
- 47.Newmark HL. Nutrient density: an important and useful tool for laboratory animal studies. Carcinogenesis. 1987;8(7):871–873. doi: 10.1093/carcin/8.7.871. [DOI] [PubMed] [Google Scholar]
- 48.González-Périz A, Planagumà A, Gronert K, et al. Docosahexaenoic acid (DHA) blunts liver injury by conversion to protective lipid mediators: protectin D1 and 17S-hydroxy-DHA. The FASEB Journal. 2006;20(14):2537–2539. doi: 10.1096/fj.06-6250fje. [DOI] [PubMed] [Google Scholar]
- 49.Spite M, Norling LV, Summers L, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461(7268):1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spite M, Summers L, Porter TF, Srivastava S, Bhatnagar A, Serhan CN. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. British Journal of Pharmacology. 2009;158(4):1062–1073. doi: 10.1111/j.1476-5381.2009.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Y-P, Oh SF, Uddin J, et al. Resolvin D1 and its aspirin-triggered 17R epimer: stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. Journal of Biological Chemistry. 2007;282(13):9323–9334. doi: 10.1074/jbc.M609212200. [DOI] [PubMed] [Google Scholar]
- 52.Lima-Garcia JF, Dutra RC, Da Silva KABS, Motta EM, Campos MM, Calixto JB. The precursor of resolvin D series and aspirin-triggered resolvin D1 display anti-hyperalgesic properties in adjuvant-induced arthritis in rats. British Journal of Pharmacology. 2011;164(2):278–293. doi: 10.1111/j.1476-5381.2011.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weylandt KH, Krause LF, Gomolka B, et al. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α . Carcinogenesis. 2011;32(6):897–903. doi: 10.1093/carcin/bgr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. Journal of Immunology. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 55.Prescott D, McKay DM. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. International Journal of Colorectal Disease. 2011;2301(3):G487–G497. doi: 10.1152/ajpgi.00042.2011. [DOI] [PubMed] [Google Scholar]
- 56.Krishnamoorthy S, Recchiuti A, Chiang N, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]