

Abstract
In BDC2·5 non-obese diabetic (BDC2·5NOD) mice, a spontaneous model of type 1 diabetes, CD4+ T cells express a transgene-encoded T cell receptor (TCR) with reactivity against a pancreatic antigen, chromogranin. This leads to massive infiltration and destruction of the pancreatic islets and subsequent diabetes. When we reconstituted lethally irradiated, lymphocyte-deficient B6.g7 (I-Ag7+) Rag–/– mice with BDC2·5NOD haematopoietic stem and progenitor cells (HSPC; ckit+Lin–Sca-1hi), the recipients exhibited hyperglycaemia and succumbed to diabetes. Surprisingly, lymphocyte-sufficient B6.g7 mice reconstituted with BDC2·5NOD HSPCs were protected from diabetes. In this study, we investigated the factors responsible for attenuation of diabetes in the B6.g7 recipients. Analysis of chimerism in the B6.g7 recipients showed that, although B cells and myeloid cells were 98% donor-derived, the CD4+ T cell compartment contained ∼50% host-derived cells. These host-derived CD4+ T cells were enriched for conventional regulatory T cells (Tregs) (CD25+forkhead box protein 3 (FoxP3)+] and also for host- derived CD4+CD25–FoxP3– T cells that express markers of suppressive function, CD73, FR4 and CD39. Although negative selection did not eliminate donor-derived CD4+ T cells in the B6.g7 recipients, these cells were functionally suppressed. Thus, host-derived CD4+ T cells that emerge in mice following myeloablation exhibit a regulatory phenoytpe and probably attenuate autoimmune diabetes. These cells may provide new therapeutic strategies to suppress autoimmunity.
Keywords: BDC2·5NOD, CD73, radio-resistant T cells, Tregs, type 1 diabetes
Introduction
Autoimmune diseases (AD) occur when tolerance to self-antigen fails and the immune system initiates attack against self-tissues. Rheumatoid arthritis (RA) and type I diabetes (T1D) are two ADs that are tightly linked to major histocompatibility complex (MHC) class II alleles and mediated by CD4+ T cells that ultimately trigger destruction of the joint tissue and insulin-producing islet β cells, respectively [1,2]. Mouse models of autoimmunity have provided critical information on the manifestations and progression of autoimmune diseases. For example, non-obese diabetic (NOD) mice develop autoimmune diabetes spontaneously and show genetic and pathophysiological characteristics similar to T1D patients [3]. Studies in NOD mice implicate escape from negative selection of autoreactive CD4+ T cells [4], insufficiency of CD4+CD25+forkhead box protein 3 (FoxP3)+ regulatory T cells (Tregs) [5] and resistance of effector T cells (Teffs) to suppression by Tregs in T1D [6]. Genetic ablation and adoptive transfer of Tregs confirm the importance of Tregs in controlling T1D progression [5,7,8].
Recently, we developed a model of autoimmune disease by transfer of haematopoietic stem and progenitor cells (HSPCs) from the K/BxN mouse, a transgenic mouse model for autoimmune arthritis, into MHC-matched, B6.g7Rag–/– recipients [recombinase activating gene (Rag)-deficient B6.g7 (B6.g7Rag–/–) mice lack T and B cells)]. These transplanted mice developed arthritis, similar to that observed in the K/BxN mouse. In contrast, the transfer of the K/BxN HSPCs into B6.g7 recipients resulted in protection from arthritis [9]. Disease in the K/BxN mouse is dependent upon CD4+ T cells carrying a transgenic T cell receptor (TCR) (termed KRN) specific for the autoantigen glucose-6-phosphate isomerase (GPI) and the NOD class II molecule I-Ag7 [10]. Investigating mechanisms of suppression of the donor-derived, GPI-specific effector CD4+ T cells in the B6.g7 recipients, we found that these cells underwent increased negative selection and survivors were functionally suppressed in the periphery. The host-derived CD4+ T cells included a substantial proportion of CD25+FoxP3+ Tregs and a novel population of suppressive CD4+CD25–FoxP3–CD73+folate receptor 4 (FR4)+ cells, both of which appeared to contribute to attenuation of arthritis. Our results are reminiscent of evidence from a syngeneic bone marrow transplant model, where C57BL/6Rag–/– recipients developed graft-versus-host disease (GVHD)-like symptoms, whereas irradiated, C57BL/6 recipients were protected [11,12]. Protection was attributed to radio-resistant, host-derived Tregs.
Here, we ask whether similar mechanisms suppress autoimmunity in B6.g7 recipients receiving HSPCs from a mouse with a diabetogenic TCR (BDC2·5). This TCR recognizes an islet-specific autoantigen, chromogranin, in the context of the NOD MHC, I-Ag7 [13], and unlike the KRN TCR are less susceptible to negative selection [10,14,15]. All BDC2·5NOD mice develop peri-insulitis, but only 20% progress to severe insulitis and diabetes [16]. Breeding BDC2·5NOD mice onto the NOD.SCID background results in accelerated insulitis, showing that the BDC2·5 TCR+ CD4+ T cells can mediate diabetes in the absence of CD8+ T cells and B cells [15]. Using B6.g7Rag–/– mice carrying the MHC I-Ag7 as recipients for BDC2·5NOD HSPC, we find that these mice develop diabetes, while the B6.g7 recipients are protected. These results are similar to our findings in the K/BxN model. We report the investigations of factors responsible for the attenuation of diabetes in these recipients.
Materials and methods
Mice
B6.g7 mice derived from C57BL/6J mice expressing the NOD MHC class II molecules I-Ag7 were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Rag-deficient B6.g7 (B6.g7Rag–/–) mice lack T and B cells (G. B., J. S., unpublished). BDC2·5.NOD mice with transgenic TCR against chromagranin were kindly provided by Dr Garrison Fathman at Stanford University. All animals were bred, housed and cared for in the Stanford Veterinary Service Center under the approval of Administrative Panel for Laboratory Animal Care, protocol number 15867.
Haematopoetic stem and progenitor cell (HSPC) isolation
HSPCs were isolated as described, with some modifications [17]. Bone marrow (BM) cells were harvested and selected for c-Kit+ cells by the magnetic affinity cell sorting (MACS) system (Miltenyi Biotech, Auburn, CA, USA). The c-Kit+ BM cells were then sorted in a BD fluorescence activated cell sorter (FACS)Aria for c-Kit+Sca1hiLin– cells after staining with antibodies listed in Supporting information Table S1. All antibodies were obtained from eBioscience (San Diego, CA, USA). Flow cytometry sorting was performed at the Stanford shared FACS facility at Stanford.
Haematopoietic cell transplantation (HCT)
Three- to 4-month old recipients (B6.g7 or B6.g7Rag–/– mice) were lethally irradiated with a Phillips Unit irradiator (250 kv, 15 milliamp) at 980 Rad (in two doses with 4-h intervals). Irradiated mice were reconstituted with HSPCs from 3–5-month old donors (BDC2·5NOD mice) 3 h after irradiation; 10 000–12 000 HSPC in 200 μl phosphate-buffered saline (PBS) were given to each recipient by tail vein injection. The reconstituted mice were maintained on sulphamethoxazole and trimethoprim (suspension; Hi-Tech Pharmacal Co. Inc., Amityville, NY, USA) in drinking water up to 3 months post-transplant. Irradiation only (without transplantation) control mice died within 2 weeks.
Evaluation of autoimmune disease
Diabetes in BDC2·5.NOD HSPC recipients was monitored by blood glucose level. One drop of blood was obtained from the tail vein and the glucose level was monitored by the One-Touch Ultra glucose meter (Life Scan Inc., Milpitas, CA, USA). Recipient mice with blood glucose concentrations of ≥200 mg/dl were deemed diabetic.
Histology
Mouse pancreatic tissues were collected and fixed in 4% fresh paraformaldehyde overnight at 4°C, The samples were washed, dehydrated and embedded in paraffin, and then 7-μm sections were mounted on glass slides. Haematoxylin and eosin stain was performed and histology images were acquired using the Leica DM2000 histology scope.
Antibody and tetramer staining and flow cytometry analyses
Thymus, spleen and pancreatic lymph nodes were harvested from mice. Surface staining was performed with various antibodies directly conjugated to fluorochromes, as shown in Supporting information Table S2. For the biotin-labelled antibodies, Pacific orange-conjugated streptavidin (Invitrogen, Carlsbad, CA, USA) was added and incubated for an additional 30 min. For intracellular FoxP3 staining, cells were treated with fixation/permeabilization reagent (eBioscience) followed by staining with antibodies (eBioscience). For cytokine stimulation and intracellular staining, spleen cells were suspended in complete Iscove's modified Eagle's medium (IMDM) [10% fetal bovine serum (FBS), 1% glutamine, 0·1% β-mercaptoethanol] and incubated for 4 h at 37°C with or without a leucocyte activation cocktail (BD Biosciences). Cell viability was assessed by staining with live dead aqua (Invitrogen, Grand Island, NY, USA). Subsequently, cells were permeabilized and stained with fluorescein isothiocyanate (FITC)-conjugated anti-interleukin (IL)-17 antibody (TC11-18H10), phycoerythrin (PE)-conjugated anti-interferon (IFN)-γ antibody (XMG1·2) and allophycocyanin (APC)-conjugated anti-FoxP3 antibody (FJK-16s). For tetramer staining, the single cell suspension was blocked with 0·5 mg/ml Fc block for 10 min on ice. Cells were then washed and stained with PE-labelled IAg7/2·5mi tetramer (kindly provided by Dr Luc Teyton) for 1 h at room temperature [18]. PE-labelled IAg7/hen egg lysozyme (HEL)-peptide tetramer was used as negative control in all experiments. Co-staining of surface markers was performed as mentioned above. In all antibody staining experiments, data were collected on a BD LSR II flow cytometer and analysed with FlowJo software (Tree Star, Inc., Ashland, OR, USA), gating on singlet cell populations. All flow cytometry analyses were performed in the Stanford shared FACS facility at Stanford.
Suppression assay
Tregs (CD4+CD25+) and Teffs (CD4+CD25–) from the spleens and LNs of BDC2·5NOD HSPC-reconstituted B6.g7 mice were first isolated using a CD4+CD25+ regulatory T cell isolation kit (Miltenyi Biotech), as per the manufacturer's instructions. Host or donor origin was then determined by sorting with the BD FACSAria cell sorter based on different CD45 alloantigen expression; 2 × 104 Teffs stimulated with anti-CD3 and CD28 antibody-coated beads were cultured with same number of Tregs in triplicate wells of the 96-well plate. The cell number and ratio of Teff to Tregs had been optimized (not shown). After 72 h co-incubation, [3H]-thymidine (2 μCi/well) was added for an additional 8 h followed by measurement of proliferation with a Harvester96 MACH III (Tomtec, Hamden, CT, USA). The mean proliferation values [counts per minute (cpm)] and standard deviations were calculated. The relative proliferation level of each group was normalized to Teffs alone, given a value of 1.
Statistical analysis
The groups were compared using the two-tailed unpaired Student's t-test. Prism software (version 5; GraphPad Software) was used for statistical analysis.
Results
B6.g7 mice reconstituted with HSPCs from BDC2·5NOD mice do not develop diabetes
In order to create a haematopoietic cell transplantation (HCT) model for autoimmune type 1 diabetes, we reconstituted lethally irradiated B6.g7Rag–/– mice with HSPCs (ckit+Lin–Sca1hi) from BDC2·5NOD mice. The recipients were followed for development of diabetes by measuring blood glucose levels every week following HCT. All the recipient B6.g7Rag–/– mice developed diabetes by week 5 and succumbed to hyperglycaemia by week 6 (Fig. 1a). However, when lethally irradiated B6.g7 mice were reconstituted with BDC2·5NOD HSPCs, the recipients showed no hyperglycaemia (Fig. 1a). Histopathology of the pancreas showed severe destruction of the pancreatic islets marked by cellular infiltration in the B6.g7Rag–/– recipients, whereas the pancreas of the B6.g7 recipients showed only a mild infiltration of the islets (Fig. 1b). Thus, transfer of BDC2·5 HSPCs into B6.g7 mice resulted in protection from diabetes, unlike the transfer into B6.g7Rag–/– mice.
Fig. 1.
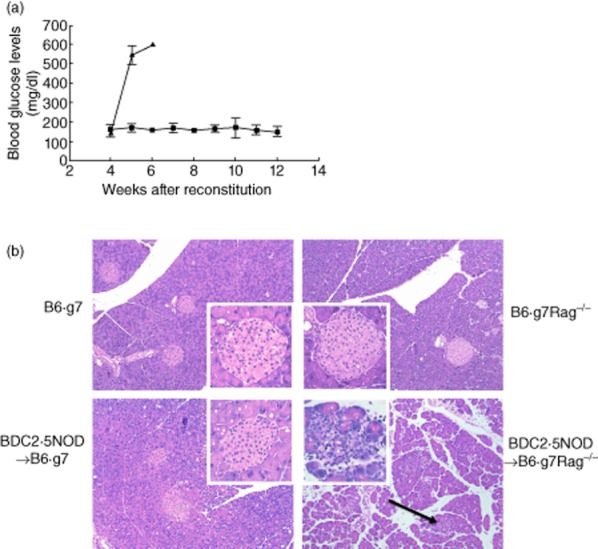
B6.g7 recipients reconstituted with BDC2·5non-obese diabetic (BDC2·5NOD) haematopoietic stem and progenitor cell (HSPCs) do not develop diabetes. (a) HSPCs isolated from BDC2·5NOD bone marrow were transferred into lethally irradiated B6.g7 and B6.g7Rag–/– recipients. Onset and progress of diabetes was recorded by measuring the blood glucose level in the recipient mice using a glucometer (see Materials and methods). The average blood glucose levels ± standard error of the mean in mg/dl are shown (n = 3). Black squares: B6.g7 and black triangles: B6.g7Rag–/– recipients. Data are representative of two experiments with three mice in each group. (b) Islet histology. Haematoxylin and eosin (H&E) staining of the pancreas of indicated mouse strains is shown. A representative pancreatic islet for each strain is shown in the inset. BDC2·5NOD→B6.g7Rag–/– shows pancreas with severe mononuclear cell infiltration (arrow) and destruction of the islets (inset), whereas the other strains show normal histology. Data are representative of three mice in each group.
Radio-resistant host-derived CD4+ T cells persist in the B6.g7 recipients
CD4+ T cells are required for the initiation and manifestation of diabetes in the BDC2·5NOD mice [14]. Thus, to determine if resistance of diabetes in the B6.g7 recipients was due to incomplete reconstitution of the CD4+ T cells, we analysed post-transplant chimerism using the spleen cells of B6.g7 recipients 12 weeks post-HCT. We distinguished the host and donor haematopoietic cells in these mice using the congenic markers CD45·2 and CD45·1 (Supporting information Fig. S1a, represented by B6.g7 and BDC2·5NOD strains, respectively).We found that the B cells (B220+) and myeloid cells (CD11b+) were largely donor-derived (B220: 98·4 ± 0·64%; CD11b: 97 ± 0·95%) (Fig. 2a). However, 52·4 ± 3·9% of splenic CD4+ T cells were host-derived (Fig. 2b). As pathogenic autoreactive CD4+ T cells undergo expansion in the pancreatic lymph nodes (PLN) in the BDC2·5NOD mice, we assessed chimerism in PLN [19]. Similar to the spleen, the pancreatic lymph nodes showed that 50·5 ± 7·4% CD4+ T cells were host-derived (Fig. 2c).
Fig. 2.
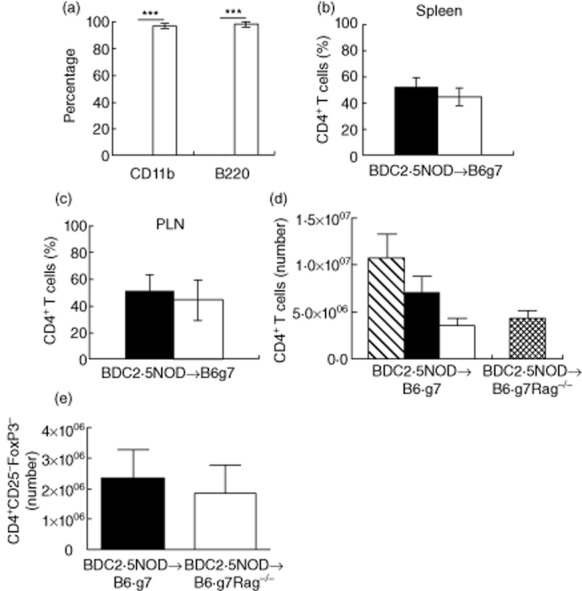
Emergence of host-derived CD4+ T cells in the reconstituted B6.g7 mice. (a) Spleen cells from BDC2·5non-obese diabetic (BDC2·5NOD)→B6.g7 mice at week 12 post-transplant were stained for CD11b (myeloid cells) and B220 (B cells) and analysed to determine host (CD45·2+; black bar) or donor (CD45·1+; white bar) origin by flow cytometry. (b) Frequency of host (black bar) and donor-derived (white bar) splenic CD4+ T cells. (c) Frequency of host (black bar) and donor-derived cells (white bar) in the draining lymph node. The histograms (a,b,c) depict mean percentage of host and donor-CD4+ T cells ± standard error of the mean (s.e.m.) from three animals. ***P < 0·0001 (t-test). Data are representative of two experiments. (d) Absolute numbers of host- and donor-derived splenic CD4+ T cells. Splenocytes from BDC2·5NOD→B6.g7 mice and BDC2·5NOD→B6.g7Rag–/– mice at 6 weeks post-haematopoietic cell transplantation (HCT) were stained for CD4 and CD45·2 (host) or CD45·1 (donor). Absolute numbers were determined by multiplying the total number of cells in each spleen (determined using a Coulter counter) by the percentage of the host and donor-derived CD4+ T cells. The histogram in (d) depicts mean number (± s.e.m., n = 5) of the total CD4+ T cells (striped bar), host-derived (black bar) and donor-derived (white bar) CD4+ T cells in the BDC2·5NOD→B6.g7 mice, and the total number of donor-derived CD4+ T cells (netted bar) in the BDC2·5NOD→B6.g7Rag–/–. The histogram in (e) depicts mean number (± standard error of the mean, n = 5) of the donor-derived CD4+CD25–forkhead box protein 3 (FoxP3)– T cells in the BDC2·5NOD→B6.g7 mice (black bar) and BDC2·5NOD→B6.g7Rag–/– (white bar). Data are pooled from two experiments.
The reduced frequency of donor-derived CD4+ T cells observed in the periphery of the BDC2·5NOD→B6.g7 mice could lead to lack of sufficient donor-derived CD4+ T cells to initiate diabetes. Therefore, we enumerated the absolute numbers of donor-derived CD4+ T cells that were present in the spleens of the BDC2·5NOD→B6.g7 mice and the diabetic BDC2·5NOD→B6.g7Rag–/– mice at 6 weeks post-HSPC transplant. The spleen of BDC2·5NOD→B6.g7 mice showed 32 ± 2·8% donor-derived CD4+ T cells (data not shown). They had 2·85 ± 0·8 × 106 donor-derived CD4+ T cells compared to 3·27 ± 1 × 106 donor-derived CD4+ T cells in the BDC2·5NOD→B6.g7Rag–/– mice (Fig. 2d). This modest difference was not statistically significant. Further, the absolute number of CD4+CD25–FoxP3– cells (Teffs) in the BDC2·5NOD→B6.g7 mice (2·3 ± 0·9 × 106) was similar to that in the BDC2·5NOD→B6.g7Rag–/– mice (1·8 ± 0·9 × 106) (Fig. 2e). These results argue that the number of donor-derived CD4+ Teffs in the periphery is not likely to be a factor in protection against diabetes in the BDC2·5NOD→B6.g7 mice.
Donor-derived CD4+CD25–FoxP3– cells in BDC2·5→B6.g7 mice are less activated, produce less IFN-γ and IL-17 and proliferate less than those in BDC2·5→B6.g7Rag–/– mice
The donor-derived CD4+CD25–FoxP3– T effector cells carrying the self-reactive BDC2·5 TCR may show signs of stimulation or, if tolerized, may appear to be quiescent. To assess the activation state of these cells in the chimeras, we measured the expression of CD44, an activation marker, and CD62L, a marker of naive CD4+ T cells, on the CD4+CD25–FoxP3– T cells by flow cytometry analysis. We found that in the diabetic BDC2·5→B6.g7Rag–/– mice, 50·85 ± 9·73% of the CD4+CD25–FoxP3– T cells expressed CD44 and 32·93 ± 7·78% were of the naive phenotype expressing CD62L (Fig. 3a). However, the same CD4+ T cell subset in the BDC2·5→B6.g7 mice showed higher expression of CD62L (75·3 ± 3·8%), and only 4·273 ± 1·3% of the cells expressed CD44 (Fig. 3a). The difference in the frequency of cells expressing CD44 between the two chimeras was statistically significant (P = 0·0019). We also analysed the donor-derived CD4+CD25–FoxP3– T cells for production of inflammatory cytokines, IFN-γ and IL-17. The donor-derived CD4+CD25–FoxP3– T cells in the BDC2·5→B6.g7 mice showed a reduced frequency of IFN-γ (BDC2·5→B6.g7: 0·5486 ± 0·056%, BDC2·5→B6.g7Rag–/–: 12·96 ± 3·4%; P = 0·0041) and IL-17 (BDC2·5→B6.g7: 0·2082 ± 0·04%, BDC2·5→B6.g7Rag–/– 3·028 ± 0·80%; P = 0·0053) (Fig. 3b,c). On measuring splenocytes for the frequency of cells expressing a marker of cellular proliferation, Ki67, we found that the donor-derived CD4+CD25– FoxP3– T cells in the BDC2·5→B6.g7 mice exhibited reduced proliferation compared to BDC2·5→B6.g7Rag–/– mice (BDC2·5→B6.g7: 7·042 ± 1·5%, BDC2·5→B6.g7Rag–/–: 20·38 ± 2·9%; P = 0·0031) (Fig. 3d). Thus, the donor-derived CD4+T cells in the BDC2·5→B6.g7 were less activated, less inflammatory and less proliferative than those in the BDC2·5→B6.g7 Rag–/– mice.
Fig. 3.
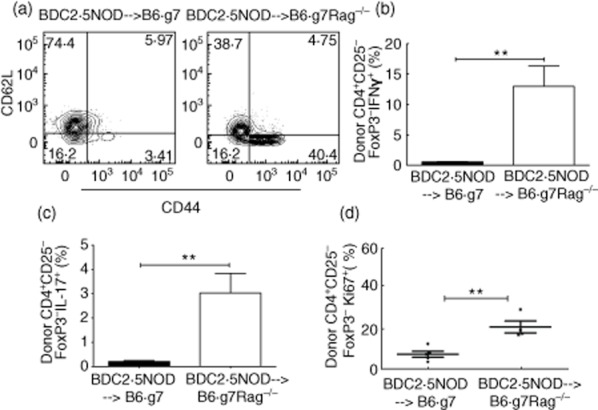
Presence of donor-derived CD4+ T cells with decreased activation and reduced cytokine expression in the BDC2·5non-obese diabetic (BDC2·5NOD)→B6.g7 mice. (a) Flow cytometry plots showing the frequency of CD62L- and CD44-expressing cells in the donor-derived CD4+ CD25–forkhead box protein 3 (FoxP3)– cells in the spleen in the BDC2·5NOD→B6.g7 mice and BDC2·5NOD→B6.g7Rag–/– mice at week 6 post-transplant. (b) Frequency of interferon (IFN)-γ expressing cells in the donor-derived CD25–FoxP3– cells in the BDC2·5NOD→B6.g7 and BDC2·5NOD→B6.g7Rag–/– chimeras. (c) Frequency of interleukin (IL)-17 expressing cells in the donor-derived CD25-FoxP3– cells in the BDC2·5NOD→B6.g7 and BDC2·5NOD→B6.g7Rag–/– chimeras. Data are pooled from of two independent experiments. (d) Frequency of Ki67-expressing cells in the donor-derived CD25-FoxP3– cells in the BDC2·5NOD→B6.g7 and BDC2·5NOD→B6.g7Rag–/– chimeras. Splenocytes from BDC2·5NOD→B6.g7 (n = 5) and BDC2·5NOD→B6.g7Rag–/– (n = 4) mice at week 6 post-transplant were evaluated. Frequency shown as mean ± standard error of the mean. **P < 0·0054 (t-test).
BDC2·5 specificity and selection of donor-derived CD4+ T cells
Negative selective of autoreactive donor-CD4+ T cells in the BDC2·5NOD→B6.g7 mice could also contribute to attenuating autoimmunity. The thymus being the primary lymphoid organ mediating central T cell tolerance, we examined the thymus of the chimeras. In the thymi of the BDC2·5NOD→B6.g7 chimera, unlike the spleen or PLN, the majority of CD4+ single-positive (SP) T cells (CD4SP) were donor-derived (Fig. 4a). However, the absolute number of donor CD4SP thymocytes in the BDC2·5NOD→B6.g7 was reduced compared to that in the BDC2·5NOD→B6.g7Rag–/– mice (Fig. 4b, P < 0·01).
Fig. 4.
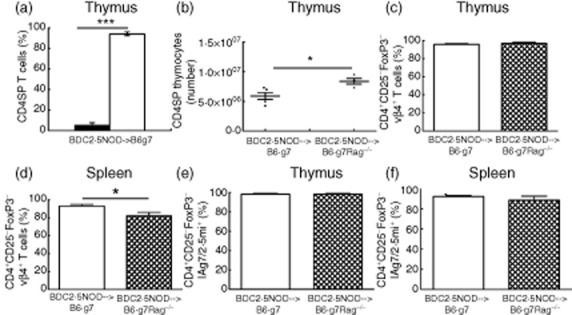
BDC2·5 specificity and selection of donor-derived CD4+ T cells. (a) Frequency of host-derived (black bar) and donor-derived (white bar) cells in the thymus. The histograms show the mean frequency of host- and donor-CD4+ T cells ± standard error of the mean (s.e.m.) from five animals; ***P < 0·0005 (t-test). (b) Absolute numbers of donor-derived CD4SP thymocytes in the thymi of BDC2·5non-obese diabetic (NOD)→B6.g7 (n = 5) and BDC2·5NOD→B6.g7Rag–/– (n = 4) mice at 6 weeks post-haematopoietic cell transplantation (HCT). The histograms show the mean number of donor-CD4+ T cells ± s.e.m., *P < 0·01 (t-test). (c,d) Expression of T cell receptor Vβ4 on the donor-derived CD4+ T cells. Frequency of Vβ4 expressing cells in the donor-derived CD4+ CD25–forkhead box protein 3 (FoxP3)– cells in the thymus (c) and spleen (d) in the BDC2·5NOD→B6.g7 mice and BDC2·5NOD→B6.g7Rag–/– mice at week 6 post-transplant. (e,f) Percentage of donor-derived CD4+CD25-FoxP3– CD4+ T cells specific for BDC2·5 mimotope peptide, 2·5mi, was determined by flow cytometry staining of thymocytes (e) and splenocytes (f) with IAg7/2·5mi tetramer. Frequency of IAg7/2·5mi tetramer expressing cells is shown as mean ± standard error of the mean from four BDC2·5NOD→B6.g7Rag–/– and five BDC2·5NOD→B6.g7 mice. Data are pooled from two independent experiments.
To further evaluate selection of CD4+ T cells in the chimeras, we took two approaches. First, as the β chain of the BDC2·5 TCR in the BDC2·5NOD mice is Vβ4, selection of the BDC2·5 CD4+ T cells in the chimeric mice can be evaluated by expression of Vβ4 on the donor-derived CD4+ T cells in the thymus and periphery [14]. The BDC2·5NOD→B6.g7 mice did not show a significant difference in the Vβ4 expression on CD4SP thymocytes when compared to the BDC2·5NOD→B6.g7Rag–/– (96·05 ± 0·89% versus 97·10 ± 1·35%; Fig. 4c). Indeed, the frequency of donor-derived CD4+ T cells expressing Vβ4 in the spleen of BDC2·5NOD→B6.g7 mice (93·18 ± 1·6%) was higher than the BDC2·5NOD→B6.g7Rag–/– mice (82 ± 3·9%) (P = 0·0381) (Fig. 4d). To analyse further the circulating cells expressing the BDC2·5 TCR based on their recognition of I-Ag7 molecules with bound BDC2·5 peptide [18], we used a tetramer (IAg7/2·5mi, see Methods) to stain CD4+ T cells in the thymus and spleen from BDC2·5NOD→B6.g7 and BDC2·5NOD→B6.g7Rag–/– mice at 6 weeks post-HSPC transplant. The frequency of the donor-derived CD4+CD25–FoxP3– cells in the thymus of the two chimeras expressing IAg7/2·5mi+ was similar (98·34 ± 0·9% versus 98·38 ± 0·77%; Fig. 4e). We also observed a similar frequency of donor-derived, CD4+CD25–FoxP3– Ag7/2·5mi+ cells in the spleen of the BDC2·5NOD→B6.g7 mice (92·28 ± 0·87%) compared to that in the BDC2·5NOD →B6.g7Rag–/– mice (88·43 ± 4·03%) (Fig. 4f). Taken together, these results imply that there are sufficient potential effector CD4+ T cells in the BDC2·5NOD→B6.g7 mice.
Host-derived CD4+ T cells are enriched for CD25+FoxP3+ regulatory T cells
The persistence of host-derived CD4+ T cells in the BDC2·5NOD→B6.g7 chimera indicates a possible role of these cells in attenuating autoimmunity. To investigate if these cells are regulatory in nature, we assessed these cells for expression of the regulatory T cell marker FoxP3. We found a higher frequency (17·3 ± 1·76% versus 2·8 ± 0·29%; P < 0·0004) of CD4+CD25+FoxP3+ Tregs among host-derived compared to donor-derived CD4+ T cells in the spleen (Fig. 5a). The frequency of CD4+CD25+FoxP3+ Tregs in the host-derived fraction also was higher than that observed in normal B6.g7 mice (8 ± 0·5%, Fig. 5a).
Fig. 5.
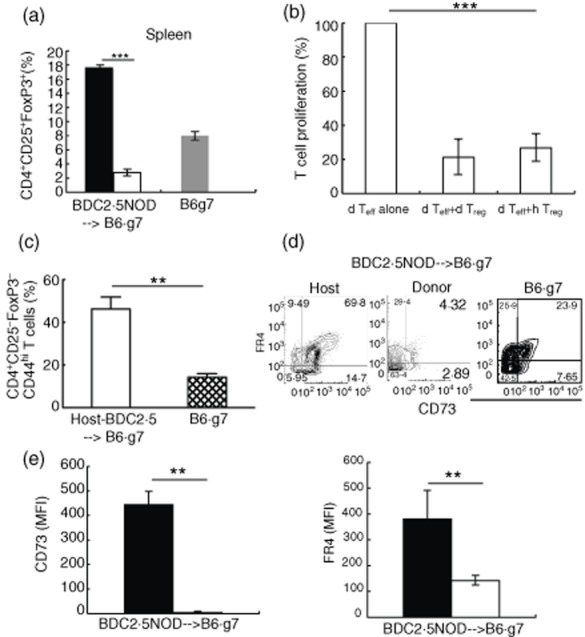
Forkhead box protein 3 (FoxP3)– expression in CD4+ CD25+ T cells in the reconstituted mice and memory-like phenotype (CD44+) in host-derived CD4+CD25-FoxP3– cells. (a) Frequency of CD4+CD25+FoxP3+ cells in the host (black bar) and donor-derived (white bar) splenocytes of the BDC2·5NOD→B6.g7 chimeras at 12 weeks post-transplant in comparison to B6.g7 mice. Frequency shown as mean ± standard error of the mean (s.e.m.), n = 3; ***P < 0·0004 (t-test). (b) Donor-derived T effectors (CD4+CD25–) and host- or donor-derived regulatory T cells (Tregs) (CD4+CD25+) sorted from BDC2·5NOD→B6.g7 mice were mixed at a ratio of 1 : 1 and co-cultured for 72 h in the presence of CD3/CD28 co-stimulation. Proliferation (thymidine incorporation) was determined after additional 8 h incubation with [3H]-thymidine. Normalized proliferation of each group was compared to dTeffs alone sample, represented as 100%. Frequency shown as mean ± s.e.m., n = 2; ***P < 0·0001 (t-test). (c) CD44 expression on CD4+CD25–FoxP3– cells in the spleens of B6.g7 and host-derived CD4+CD25-FoxP3– cells in the spleens of the BDC2·5NOD→B6.g7 chimeric mice at 12 weeks post-reconstitution. Frequency is shown as mean ± s.e.m. from three BDC2·5NOD→B6.g7 and three B6.g7 mice; **P < 0·0037 (t-test). (d) Expression of FR4 and CD73 on spleen cells in the CD25–FoxP3– population. Representative profiles from one of three mice are shown. Average frequencies from all three mice ± s.e.m.; B6.g7 CD73+FR4+: 26·17 ± 6%; BDC2·5→B6.g7: host CD73+FR4+: 66 ± 7·5%, donor CD73+FR4+: 5·3 ± 2%. Statistical significance (t-test) BDC2·5NOD→B6.g7 host versus BDC2·5NOD→B6.g7 donor P < 0·0021; BDC2·5NOD→B6.g7 host versus B6.g7 P < 0·03. (e) CD73 and FR4 mean fluorescence intensity (MFI) from the host (black bar) and donor-derived (white bar) CD4+CD25–FoxP3– cells in the two chimeras. MFI is shown as mean ± s.e.m. from three mice. **P < 0·004; **P < 0·0194 (t-test), for MFI CD73 and FR4 respectively. Representative data from one of two independent experiments.
In order to test the immunosuppressive capacity of these host-derived Tregs, we performed a standard T cell co-culture suppression assay. Host (CD45·2+) and donor (CD45·1+)-derived Tregs (CD4+CD25+) and donor-derived Teffs (CD45·1+CD4+CD25-) were sorted by flow cytometry from BDC2·5.NOD→B6.g7 chimeras. In-vitro co-cultures of Teffs and Tregs were set up with each cell type from both donor (d) and host (h). As shown in Fig. 5b, both hTregs and dTregs were efficient at inhibiting the proliferation of dTeffs, induced by CD3/CD28 co-stimulation.
Host-derived CD4+CD25–FoxP3– cells have a memory-like phenotype and express the suppression markers, CD73 and FR4
Radio-resistant CD4+ T cells in irradiated hosts have a memory-like phenotype, characterized by high expression of CD44 [20]. These memory-like CD4+ T cells have suppressive effects on T cell proliferation [21]. With this in mind, we assessed host-derived CD4+CD25–FoxP3– T cells for evidence of a regulatory phenotype. We found that a higher proportion of the host-derived CD4+CD25–FoxP3– cells from the spleen of the B6.g7 chimeras express CD44 (46·3 ± 5·6%) compared to this T cell subset in B6.g7 mice (14·3 ± 1·58%; P < 0·0037) (Fig. 5c).
FR4 and CD73 are associated with regulatory function [22,23]. Interestingly, we found that the host-derived splenic CD4+CD25–FoxP3– cells in BDC2·5NOD→B6.g7 mice showed a significantly increased frequency of cells expressing both FR4 and CD73 compared to the donor-derived CD4+CD25–FoxP3– cells (host CD73+FR4+: 66 ± 7·5%; donor CD73+FR4+: 5·3 ± 2%; P < 0·0021) or those in normal B6.g7 mice (26·17 ± 6%) (Fig. 5d). The host-derived CD4+CD25– cells also showed higher expression (MFI) of CD73 and FR4 than the donor-derived CD4+CD25– T cells (Fig. 5e). CD73 is an ecto-nucleotidase that mediates suppression by generating adenosine in concert with another surface marker of suppression CD39 [24]. We found that 31·5 ± 6·5% of the host-derived CD4+CD25–FoxP3–CD73+FR4+ cells expressed CD39 (data not shown), arguing for a suppressive function for this subset.
Discussion
Transfer of HSPCs from the BDC2·5NOD mice into the B6.g7Rag–/– mice results in autoimmune diabetes, characterized by high blood glucose levels and severe insulitis. However, the transfer of HSPCs into the B6.g7 mice results in attenuation of these features of diabetes. Here we have investigated factors that regulate autoimmunity in the reconstituted mice. Pathogenic CD4+ T cell clones expressing BDC2·5 TCR drive diabetes in the transgenic BDC2·5NOD.SCID mice [14,15]. Thus defects in the donor-derived CD4+ T cells bearing the BDC2·5 TCR in the reconstituted B6.g7 recipients could lead to attenuation of diabetes. We found that only about half of peripheral CD4+ T cells in the BDC2·5→B6.g7 mice were donor-derived, while the other half were host-derived CD4+ T cells that emerge despite lethal irradiation. This pattern was confined to CD4+ T cells, as B cells and myeloid cells were almost completely of donor origin. However, diminished frequency of donor-derived cells among the total CD4+ T cells in the BDC2·5→B6.g7 mice is unlikely to be the key cause of reduced diabetes; the absolute number of donor-derived CD4+ T cells in the BDC2·5NOD→B6.g7 mice was similar to that in the BDC2·5→B6.g7Rag–/– mice.
Immunosuppression of the donor-derived CD4+ T effector cells appears to play a major role in attenuation of disease in the BDC2·5→B6.g7 mice. These mice had only a small proportion of activated donor-derived CD4+ T cells, as determined by their expression of CD44, when compared to the BDC2·5→B6.g7Rag–/– mice. Further, they showed reduced levels of IFNγ and IL-17, two cytokines important for the development of diabetes [25,26]. A key contributor to this suppression is probably the conventional Treg population of CD25+FoxP3+ cells that are enriched among the host-derived CD4+ T cells in the BDC2·5→B6.g7 mice chimeras and persist even at 3 months post-transplant. Consistent with our results, residual, host-derived Tregs allow survival of myeloablated C57BL/6 recipient mice following syngenic BM transfer, whereas C57BL/6Rag–/– recipients succumb to lethal syngeneic GVHD [10]. Furthermore, during transplantation of scurfy (FoxP3-deficient) bone marrow cells into wild-type hosts, radio-resistant host-derived Tregs can restore Treg population in the absence of Treg production from scurfy donor BM cells [27].
In addition to the increased frequency of CD25+FoxP3+ cells, the host-derived CD4+ T cells expressed higher levels of CD44 in their CD4+CD25–FoxP3– compartment. CD44 is an activation and memory marker, and in BM transplantation of syngenic Rag-deficient animals it has been demonstrated that memory-like (CD44hi) T cells are enriched among host-derived CD4+ T cells under lymphopenic conditions [12,20,28]. A subset of memory-like CD4+CD44hi T cells can restrain activation and proliferation of naive T cells [22,23]. Importantly, in the B6.g7 chimeras, the CD4+CD25–FoxP3– cells expressed other markers of suppressive capacity, CD73 and FR4. CD73, a GPI-linked surface protein with ecto-5-nucleotidase activity, mediates suppression by CD25+FoxP3+ T cells [22,29]. CD39, an ecto-ATP diphosphohydrolase that is also expressed by Tregs and neutrophils, rapidly converts circulating ATP and ADP to 5′-AMP [24]. CD73 converts 5′-AMP to adenosine [30]. Adenosine mediates its immunoregulatory activities through various adenosine receptors expressed on T cells, B cells, neutrophils and macrophages [31–33]. A subset of memory-like CD4+ T cells expresses high levels of CD73 and is suppressive by producing adenosine [22]. FR4 is one of the receptor subtypes for folic acid. FR4 is expressed at high levels on both natural and TGF-β-induced Tregs. In the BDC2·5NOD→B6.g7 chimeras, a significant portion (∼30%) of the CD73+FR4+ cells also expressed CD39, indicating that these cells could also be immunomodulators in these mice.
We then compared the frequency of Tregs in the donor-derived CD4+T cells between the BDC2·5NOD→B6.g7 and BDC2·5NOD→B6.g7Rag–/– mice. At 6 weeks post-transplant, the spleen of the BDC2·5NOD→B6.g7 mice had a smaller proportion of donor-derived Tregs (CD4+CD25+FoxP3+) cells (2·8 ± 0·29%) to the spleen of the BDC2·5NOD→B6.g7Rag–/– mice (10·8 ± 1·7%, data not shown). Interestingly, the BDC2·5NOD→B6.g7Rag–/– mice still developed diabetes, indicating that donor-derived Tregs could not suppress disease in vivo in these mice. Further, the BDC2·5NOD→B6.g7 mice had 5·3 ± 2% donor-derived CD4+CD25–FoxP3–CD73+FR4+cells versus 15·4 ± 3·4% (data not shown) of these cells in the BDC2·5NOD→B6.g7Rag–/– mice. A minority of donor-derived CD4+CD25–FoxP3– cells in both chimeras express CD73 and FR4, whereas 66 ± 7·5% of host-derived CD4+CD25–FoxP3– cells express these markers. These results also strengthen the conclusion that protection against autoimmunity in the BDC2·5NOD→B6.g7 model is mediated by host-derived CD4+T cells.
The donor-derived CD4+ T cells in BDC2·5→B6.g7 mice did not show reduced frequency of vβ4+ cells or tetramer-positive cells compared to this cell subset in BDC2·5→B6.g7Rag–/– mice, indicating that increased negative selection does not mediate tolerance in the BDC2·5→B6.g7 mice. Previous studies in TCR transgenic mice that showed that CD4+ T cells bearing TCR specific for a peripherally expressed self-antigen were not subject to intrathymic or extrathymic deletion [14,34,35]. However, the lower number of CD4SP thymocytes in the BDC2·5→B6.g7 in comparison to the BDC2·5→B6.g7Rag–/– mice is currently unexplained. A difference in thymocyte egress between the two chimeras could be a possible explanation, associated with structural differences in the thymi of the two recipients at the time of HCT. The B6.g7 recipients have a functional thymus (irradiated during HCT conditioning), unlike the B6.g7Rag–/– recipients, where the thymus does not develop normally.
In the reconstitution of B6.g7 recipients with HSPC from arthritic K/BxN mice, we found that K/BxN→B6.g7 mice were protected from arthritis, while K/BxN→B6.g7Rag–/– mice were not [9]. Similar to the BDC2·5NOD→B6.g7 mice, the K/BxN→B6.g7 mice showed persistent host-derived CD4+ T cells that were enriched for CD25+FoxP3+ Treg and CD25–FoxP3–CD73+FR4+ cells, and these cell types were suppressive by adoptive transfer experiments [16]. The observation of host-derived CD4+ T cells enriched with regulatory cells in both the BDC2·5NOD→B6.g7 and K/BxN→B6.g7 mice shows that the emergence of these cells is not strain-specific. Notably, in the arthritis model, we also found evidence for increased negative selection of the donor-derived CD4+ T cells. This difference could be due to increased susceptibility of the KRN TCR to negative selection compared to the BDC2·5 TCR [10,14,15].
Host-derived CD4+ T cells also are present in the thymus of the BDC2·5NOD→B6.g7 mice (∼ 5%), and they exhibit a mature phenotype (CD44hi) (Supporting information Fig. S1b). This is uncharacteristic of CD4SP thymocytes, which usually acquire the CD44hi phenotype in the periphery after encountering antigen. Further, the host-derived CD4SP cells express a higher frequency of CD25+FoxP3+ cells than donor-derived cells (Supporting information Fig. S1c). The host-derived CD4SP cells in the thymus could be CD4+ T cells that have re-entered the thymus from the periphery. CD4+ T cells of the activated phenotype and Tregs are capable of re-entering the thymus from the periphery under pathological and lymphopenic conditions [21,36–39]. An alternative source of the thymic host-derived CD4SP cells could be a transient wave of differentiation from radio-resistant DN2 precursors within the thymus, evidence for which has been provided in previous studies with syngenic bone marrow transplant [12,40].
The frequency of host-derived CD4+ T cells is higher in the spleen compared to the thymus in the BDC2·5NOD→B6.g7 mice. Both host and donor-derived CD4+ T cells showed similar expression of Ki67 at 6 weeks post-transplant within the respective organs (data not shown). However, it is possible that radio-resistant host-derived cells expand in the periphery in the weeks before T cell re-constitution is complete. Further investigation is required to determine the mechanistic basis of the different proportions of host-derived CD4+ T cells in these organs.
Fatal infections due to the prolonged immunosuppression or insufficient recovery of T cells following autologous HCT or allogenic HCT limit the use of these methods for treating T1D [41]. Use of in-vitro expanded CD4+ Tregs has been proposed as a means to induce tolerance and control autoimmunity [42]. Our model will facilitate further study into the origin and development of radio-resistant, immunosuppressive CD4+T cell subsets and their precursors and may suggest new therapeutic strategies for autoimmune diseases.
Acknowledgments
We thank Dr Luc Teyton for kindly providing us with the IAg7/2·5mi tetramer tetramers; Dr Andreas Hadjinicolaou for his assistance in flow cytometry analysis; and Jeanette Baker and Remi Creusot (Stanford University, Stanford, CA) for their useful discussions. This study was supported by grants from Arthritis Foundation (to N. R.), Juvenile Diabetes Research Foundation (to N. W.), American College of Rheumatology Research and Education Foundation, the Juvenile Diabetes Research Foundation and NIH AI075253 and DK079163 (to E. D. .M.), the H. L. Snyder Medical Foundation Fellowship Award and Stinehart/Reed Diabetes Research (to J. A. S.) and NIH DK067559 (G. F. B. and J. A. S.). The work was also supported in part by the Stanford NIH/NCRR CTSA award number UL1 RR025744, and by the Lucile Packard Foundation for Children's Health.
Disclosure
Authors declare no competing financial interests.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1. (a) Expression of congenic markers CD45·1 and CD45·2 in mice. Spleen cells from B6.g7 (left) and BDC2·5non-obese diabetic (NOD) mice (right) were stained with antibodies specifically against CD45·2 or CD45·1 and analysed by flow cytometry. (b) CD44 expression on host (black bar) and donor-derived (white bar) CD4+CD25–forkhead box protein 3 (FoxP3)– cells in the thymus of BDC2·5NOD→B6.g7 chimeric mice killed 12 weeks post-reconstitution. (c) Frequency of CD4+CD25+FoxP3+ cells in the host (black bar) and donor-derived (white bar) thymocytes of the BDC2·5NOD→B6.g7 chimeras killed 12 weeks post-transplant. Frequency shown as mean ± standard error of the mean, n = 3; **P < 0·006 (t-test).
Table S1. Antibody panel for haematopoietic stem and progenitor cell (HSPC) isolation.
Table S2. Antibody panel for fluorescence activated cell sorter (FACS) analysis.
References
- 1.McInnes IB, O'Dell JR. State-of-the-art: rheumatoid arthritis. Ann Rheum Dis. 2010;69:1898–1906. doi: 10.1136/ard.2010.134684. [DOI] [PubMed] [Google Scholar]
- 2.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 3.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 4.Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol. 2012;12:721–728. doi: 10.1038/nri3294. [DOI] [PubMed] [Google Scholar]
- 5.Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 6.Gregori S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol. 2003;171:4040–4047. doi: 10.4049/jimmunol.171.8.4040. [DOI] [PubMed] [Google Scholar]
- 7.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 8.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 9.Rajasekaran N, Wang N, Truong P, et al. Host-derived CD4+ T cells attenuate stem cell-mediated transfer of autoimmune arthritis in lethally irradiated C57BL/6.g7 mice. Arthritis Rheum. 2013;65:681–692. doi: 10.1002/art.37800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 11.Benard A, Ceredig R, Rolink AG. Regulatory T cells control autoimmunity following syngeneic bone marrow transplantation. Eur J Immunol. 2006;36:2324–2335. doi: 10.1002/eji.200636434. [DOI] [PubMed] [Google Scholar]
- 12.Bosco N, Swee LK, Benard A, Ceredig R, Rolink A. Auto-reconstitution of the T-cell compartment by radioresistant hematopoietic cells following lethal irradiation and bone marrow transplantation. Exp Hematol. 2010;38:222–32e2. doi: 10.1016/j.exphem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Stadinski BD, Delong T, Reisdorph N, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 15.Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of diabetes progression. Immunity. 1997;7:873–883. doi: 10.1016/s1074-7613(00)80405-7. [DOI] [PubMed] [Google Scholar]
- 17.Beilhack GF, Scheffold YC, Weissman IL, et al. Purified allogeneic hematopoietic stem cell transplantation blocks diabetes pathogenesis in NOD mice. Diabetes. 2003;52:59–68. doi: 10.2337/diabetes.52.1.59. [DOI] [PubMed] [Google Scholar]
- 18.Stratmann T, Martin-Orozco N, Mallet-Designe V, et al. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J Clin Invest. 2003;112:902–914. doi: 10.1172/JCI18337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosco N, Agenes F, Rolink AG, Ceredig R. Peripheral T cell lymphopenia and concomitant enrichment in naturally arising regulatory T cells: the case of the pre-Talpha gene-deleted mouse. J Immunol. 2006;177:5014–5023. doi: 10.4049/jimmunol.177.8.5014. [DOI] [PubMed] [Google Scholar]
- 22.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Kobie JJ, Mosmann TR. CD73 and Ly-6A/E distinguish in vivo primed but uncommitted mouse CD4 T cells from type 1 or type 2 effector cells. J Immunol. 2005;175:6458–6464. doi: 10.4049/jimmunol.175.10.6458. [DOI] [PubMed] [Google Scholar]
- 24.Eltzschig HK, Ibla JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang B, Andre I, Gonzalez A, et al. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:13844–13849. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bending D, De la Pena H, Veldhoen M, et al. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119:565–572. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu N, Hori S. Full restoration of peripheral Foxp3+ regulatory T cell pool by radioresistant host cells in scurfy bone marrow chimeras. Proc Natl Acad Sci USA. 2007;104:8959–8964. doi: 10.1073/pnas.0702004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 29.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285(Pt 2):345–365. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J Immunol. 2001;167:4026–4032. doi: 10.4049/jimmunol.167.7.4026. [DOI] [PubMed] [Google Scholar]
- 32.McColl SR, St-Onge M, Dussault AA, et al. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J. 2006;20:187–189. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panther E, Idzko M, Herouy Y, et al. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001;15:1963–1970. doi: 10.1096/fj.01-0169com. [DOI] [PubMed] [Google Scholar]
- 34.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 35.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 36.Agus DB, Surh CD, Sprent J. Reentry of T cells to the adult thymus is restricted to activated T cells. J Exp Med. 1991;173:1039–1046. doi: 10.1084/jem.173.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michie SA, Kirkpatrick EA, Rouse RV. Rare peripheral T cells migrate to and persist in normal mouse thymus. J Exp Med. 1988;168:1929–1934. doi: 10.1084/jem.168.5.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chau LA, Rohekar S, Wang JJ, et al. Thymic re-entry of mature activated T cells and increased negative selection in vascularized allograft recipients. Clin Exp Immunol. 2002;127:43–52. doi: 10.1046/j.1365-2249.2002.01717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCaughtry TM, Wilken MS, Hogquist KA. Thymic emigration revisited. J Exp Med. 2007;204:2513–2520. doi: 10.1084/jem.20070601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kadish JL, Basch RS. Thymic regeneration after lethal irradiation evidence for an intra-thymic radioresistant T cell precursor. J Immunol. 1975;114:452–458. [PubMed] [Google Scholar]
- 41.Fotino C, Ricordi C, Lauriola V, Alejandro R, Pileggi A. Bone marrow-derived stem cell transplantation for the treatment of insulin-dependent diabetes. Rev Diabet Stud. 2010;7:144–157. doi: 10.1900/RDS.2010.7.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bluestone JA, Tang Q. Therapeutic vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc Natl Acad Sci USA. 2004;101(Suppl. 2):14622–14626. doi: 10.1073/pnas.0405234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.