

Abstract
Ischemic damage is recognized to cause cardiomyocyte (CM) death and myocardial dysfunction, but the role of cell-matrix interactions and integrins in this process has not been extensively studied. Expression of α7β1D integrin, the dominant integrin in normal adult CMs, increases during ischemia/reperfusion (I/R), while deficiency of β1 integrins increases ischemic damage. We hypothesized that the forced overexpression of integrins on the CM would offer protection from I/R injury. Tg mice with CM-specific overexpression of integrin α7β1D exposed to I/R had a substantial reduction in infarct size compared with that of α5β1D-overexpressing mice and WT littermate controls. Using isolated CMs, we found that α7β1D preserved mitochondrial membrane potential during hypoxia/reoxygenation (H/R) injury via inhibition of mitochondrial Ca2+ overload but did not alter H/R effects on oxidative stress. Therefore, we assessed Ca2+ handling proteins in the CM and found that β1D integrin colocalized with ryanodine receptor 2 (RyR2) in CM T-tubules, complexed with RyR2 in human and rat heart, and specifically bound to RyR2 amino acids 165–175. Integrins stabilized the RyR2 interdomain interaction, and this stabilization required integrin receptor binding to its ECM ligand. These data suggest that α7β1D integrin modifies Ca2+ regulatory pathways and offers a means to protect the myocardium from ischemic injury.
Introduction
Mechanical signals have come to be recognized as fundamentally important in many aspects of cardiac function, but their role in ischemic myocardial damage is poorly understood. Specifically, how cell-matrix interactions and cell surface integrins might modulate this process has not been extensively studied (1, 2).
Integrins are a family of transmembrane heterodimeric receptors composed of α and β subunits that serve as the primary link between ECM ligands and cytoskeletal structures (3–5). They act as adhesive receptors and mechanotransducers that can directly initiate intracellular signaling events. Generally, α and β integrin subunits consist of a large extracellular domain, a transmembrane region, and a relatively short cytoplasmic domain (6). Each integrin heterodimer binds to specific ECM proteins or cell surface counter-receptors. Integrin β1 is recognized to have 4 splice variants. The β1A variant is ubiquitously expressed, the β1B variant is detected only in skin and liver (7), and the β1C variant is found in hematopoietic cells (8). The β1D variant is expressed dominantly in striated muscles, including the heart. While embryonic cardiomyocytes (CMs) mainly express the β1A integrin form, perhaps as required for cardiac morphogenesis and plasticity of the developing heart (9), the β1D integrin form becomes upregulated late in gestation, becoming the dominant β1 form in postnatal CMs. β1D has been shown to have tighter binding affinity than β1A to proteins such as talin, thus potentially leading to a particularly strong sarcolemmal-cytoskeletal linkage of the type required in the continuously contracting CM (10).
Interestingly, work has shown that the myocyte β1D integrin form is downregulated in the myocardium after myocardial infarction (MI), and this process has been suggested to play an important role in the decreased function of the heart after MI by reducing the ability of the CMs to interact appropriately with the ECM (11). Integrin β1 partners with several α integrin subunits in the myocyte, including α1, α5, and α7. Integrin α1β1 forms a collagen binding heterodimer, while integrin α5β1 is the major fibronectin (FN) receptor in the myocyte. Integrin α5 is expressed in the embryonic heart and is reduced postnatally, but it can be induced by stress such as cardiac ischemia (12–14). Integrin α7 partners with β1 to form a major laminin-binding (LN-binding) receptor that is highly expressed on the adult myocyte. Like β subunits, alternative splicing of integrin α7 generates cytoplasmic domain variants, with α7B being the dominant α subunit isoform expressed in normal adult CMs (9, 15, 16). Integrin α7 has been shown to be significantly increased after MI (17). Conversely, mice globally deficient in β1 integrin (heterozygous integrin β1 knockout mice) had worsened myocardial function after MI compared with that in control animals, with increased apoptosis (18).
Given this, we hypothesized that increased integrin expression or activity in the CM would offer protection from ischemia/reperfusion (I/R) injury. To test this, we generated Tg mice with CM-specific integrin overexpression (OE) and subjected them to I/R. We found that α7β1D integrin can protect CMs from I/R injury. α7β1D OE reduced excessive Ca2+ entry into mitochondria and prevented mitochondrial permeability transition pore (mPTP) opening after hypoxia/reoxygenation (H/R). In addition, muscle-enriched β1D integrin colocalized with and bound to ryanodine receptor 2 (RyR2). When integrins bound, they stabilized the RyR2 channel interdomain interaction. Thus, CM OE of α7β1D integrin offers a potential novel means to protect myocytes and the intact heart from injury.
Results
Dual expression of α and β integrin subunits is necessary for appropriate cell surface localization on the CM.
Functional integrin receptors require interaction between α and β subunits. To determine the requirements to produce increased expression of integrin receptors on the cell surface of adult CMs, we first used an adult rat ventricular myocyte (ARVM) model. Cells were isolated and ARVMs were transduced with recombinant adenoviruses producing β1D integrin, either alone or in combination with α5 integrin or α7B integrin subunits. We chose to use rat as opposed to mouse adult CMs due to the difficulty in maintaining viable, differentiated mouse CMs for the time required for these studies. Protein OE of integrins was confirmed in each group 48 hours after adenoviral infection by analyzing whole cell lysates (Figure 1, A and B). While forced expression of β1D integrin alone significantly increased both the mature and precursor forms of this integrin subunit in the ARVMs compared with those in control infected cells, coexpression of either α5 or α7 along with β1D significantly increased and stabilized expression of the mature β1D integrin subunit as well as the respective α subunit.
Figure 1. Combined expression of integrin α and β subunits is required for highest level of cell surface expression of integrins.

(A) Expression of integrins in ARVMs 48 hours after adenovirus infection. Western blot detecting LacZ, β1D integrin (β1D), α5 integrin (α5), and α7 integrin (α7) indicates increased expression of respective integrin subunits following viral infection. (B) Densitometric quantification of integrin expression in virally infected cells. Note that immature (precursor) form of β1D integrin subunit is increased when the cells are infected with the β1D subunit alone but not when they are infected with both α and β subunits. *P < 0.05 vs. LacZ, n = 6 per group (1-way ANOVA). (C) Representative immunofluorescent images of CMs following adenoviral-mediated OE of LacZ (control), β1D integrin alone, or β1D integrin in combination with α5 integrin or α7B integrin. β1D expression is noted on the cell membrane as well as in the cytoplasm when the β1D integrin subunit is overexpressed alone. When combined OE of α and β subunits occurs, the integrin expression is dominantly at the membrane, with little detected in the cytoplasm. Antibodies detect β1D (green), α7 (red), or α5 (green). Nuclei were stained with DAPI (blue). Scale bar: 20 μm. (D) Western blot analysis of β1D integrin expression in cytosolic and cell membrane fractions in ARVMs. As confirmation of the microscopic data, Western blotting shows that β1D integrin singly infected cells expressed integrin dominantly in the cytosolic fraction, while combined αβ integrin subunit infection allowed dominant cell membrane fraction expression.
Next, to address how the OE of integrin subunits might alter the subcellular localization of integrins in the ARVMs, immunofluorescent microscopy was used (Figure 1C). In control cells, endogenous β1D integrin was expressed on the cell surface. With OE of β1D integrin alone, expression was seen in both the cytoplasm and on the cell surface. With combined infection of α5 or α7B subunits along with β1, integrin expression was predominantly at the cell membrane, not the cytoplasm. We confirmed this visual result using Western blot analyses (Figure 1D). These data indicated that increased cell surface expression of myocyte integrins, necessary for their function, required combined expression of α and β integrin subunits. Based on this finding, we used combined infection and transgenesis of α and β integrin subunits for our studies.
CM-specific integrin OE protects against I/R injury in vivo.
To directly determine whether increased expression of integrin receptors would be cardioprotective, we generated Tg mice with CM-specific OE of α5, α7, or β1D integrins. For each Tg construct, multiple founder lines were identified (α5 [8 lines], α7 [5 lines], and β1D [10 lines]). Of the lines generated, we chose to study those with varied protein expression levels, which were termed “high,” “middle,” and “low,” respectively (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI64216DS1). All of these lines were viable and fertile, and there were no detectable differences in cardiac size or structure between Tg and WT mice (data not shown). Our data from ARVMs indicated the importance of dual α and β subunit expression to produce cell surface expression of integrins. Having these mice, which overexpressed individual integrin subunits, allowed us to interbreed them to produce double Tg (DTg) mice, which had OE of α5β1D integrin or α7β1D integrin in the CMs. DTg mice were created from lines we termed “middle” β1D, “middle” α5, and “high” α7 single Tg mice, so that similar expression levels of β1D were present in the α5β1D and α7β1D DTg mice. This allowed for the best comparisons among groups.
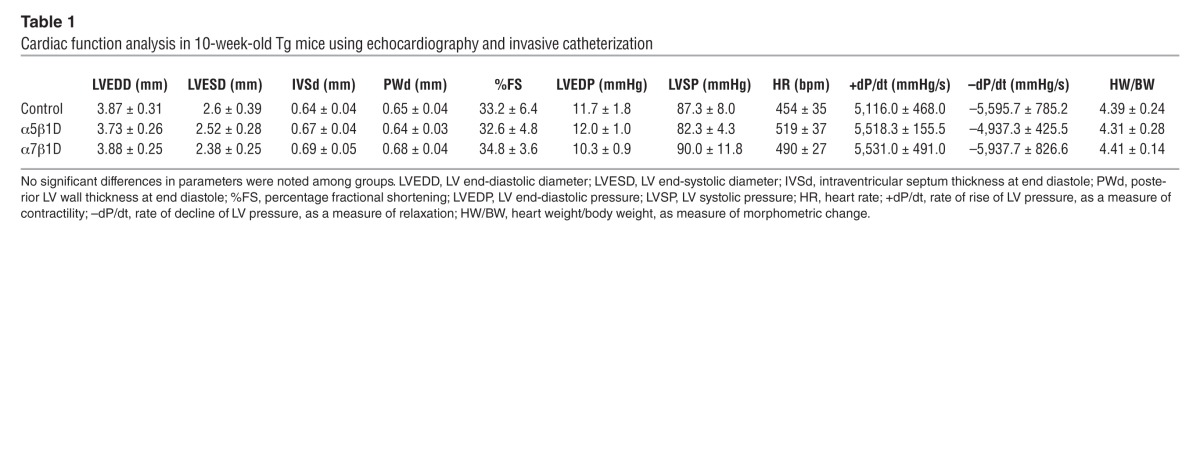
We evaluated the expression of integrins in LV tissue of DTg mice along with that in littermate controls. The mature form of β1D integrin was found to be expressed at 300% of control levels, while the precursor (immature) form of β1D integrin showed decreased expression compared with levels detected in β1D integrin single Tg mice (Figure 2A). Interestingly, β1D expression was increased 2 fold when α subunits were overexpressed in the myocyte. Yet in contrast, forced expression of β1D did not affect α subunit expression level. As in the cultured cells, when both α and β integrin transgenes were expressed in the CM, cell surface expression was detected (Figure 2B). Importantly, there were no significant morphometric or functional differences basally between single subunit Tg or DTg 10-week-old OE male mice as compared with controls (Table 1 and Supplemental Figure 2). Further, there were no differences in integrin-related signaling molecules between DTg mice and controls (Figure 2C).
Figure 2. Functional and morphological analyses of integrin DTg mice.

(A) Expression of integrins in Tg mice. Western blot shows mature β1D integrin expression (top band) was increased 3 fold in both the α5β1D and α7β1D DTg mice. In contrast, β1D precursor (immature, bottom band) was decreased compared with β1D single Tg mice. Integrin β1D expression was increased 2 fold in Tg mice with only α subunit OE. When β1D integrin expression was increased in Tg hearts, no significant change in α subunit expression level was detected. *P < 0.05 vs. control. (B) Immunofluorescent microscopy shows increased expression of integrins on the sarcolemmal membrane in α5β1D and α7β1D DTg hearts. Scale bar: 100 μm. (C) Western blot analyses of integrin-related signaling molecules were performed using lysates extracted from intact hearts. There were no significant differences between control or Tg groups.
Table 1.
Cardiac function analysis in 10-week-old Tg mice using echocardiography and invasive catheterization
Next, we tested whether increased integrin expression could protect CMs from I/R injury. For this, male (9- to 12-week-old) mice were subjected to in vivo I/R. We exposed α5β1D integrin OE, α7β1D integrin OE, and littermate Tg-negative controls to 30 minutes of cardiac ischemia, followed by 24 hours of reperfusion (Figure 3A). The area at risk was not different among groups (control: 49.4% ± 8.6%; α5β1D OE: 46.8% ± 7.4%; integrin α7β1D OE: 50.5 ± 2.0; Figure 3B). α7β1D OE mice showed a substantial reduction in infarct size compared with either that of the α5β1D OE or littermate control mice (control: 43.3% ± 4.6%; α5β1D OE: 45.8% ± 2.0%; α7β1D OE: 28.6% ± 2.3%, P < 0.01 vs. littermate control, n = 6; Figure 3C). Therefore, increased CM expression of integrins appears to protect against ischemic damage.
Figure 3. I/R injury in the integrin OE mice.

(A) Representative sections of TTC-stained hearts following I/R. (B and C) Quantification was performed by normalizing the myocardial infarct area (MI) to the area at risk (AAR). *P < 0.05 vs. littermate control, n = 8 per group (1-way ANOVA).
Integrin β1 deficiency aggravates I/R injury.
To further emphasize how important integrin expression is in protecting the CM, we performed in vivo I/R studies on mice with induced knockout of β1 integrin, specifically in CMs (19). For this, integrin-deficient and control mice were subjected to in vivo I/R. Mice with a “floxed” β1 integrin gene were bred to the tamoxifen-inducible α-myosin heavy chain Mer-Cre-Mer Tg mice (20, 21) to produce α-myosin heavy chain Mer-Cre-Mer/Itgb1fl/fl mice (termed MCM-Itgb1fl/fl mice). When injected with tamoxifen, these mice allow for inducible deletion of β1 integrin in CMs specifically. Diluent-injected mice served as one control group, while Itgb1fl/fl mice (without Cre) injected with tamoxifen served as a second control group. Mice were injected with tamoxifen or control diluent for 5 days. Then, they were examined 4 weeks after injection, a time when we have previously shown that cardiac function is normal (19). Tamoxifen-injected MCM-Itgb1fl/fl mice developed substantial reduction in β1 integrin following Cre induction (Figure 4, A and B). In these knockout mice, percentage MI area/area at risk was increased significantly compared with that in controls (Figure 4, C and D). The area at risk was the same in all groups (Figure 4D). These results support the importance of CM integrins in I/R injury.
Figure 4. I/R injury is increased in mice with CM-specific reduction of β1 integrins.

(A and B) Representative Western blot and immunofluorescent analyses for β1D integrin protein in the MCM-Itgb1fl/fl mice 4 weeks after tamoxifen injection show significant reductions in integrin expression compared with MCM-Itgb1fl/fl mice injected with diluent only. Scale bar: 100 μm. (C) Representative sections of TTC-stained hearts. (D) Quantification performed by normalizing the area at risk to LV area and myocardial infarct area to the area at risk. *P < 0.05 vs. littermate control, n = 6 per group (1-way ANOVA).
Integrins protect CMs against reoxygenation injury.
In order to analyze the mechanism(s) by which integrins protect against CM I/R injury, we used recombinant adenoviruses to produce increased amounts of integrin protein in an ARVM model. ARVMs were cultured with adenoviruses for 48 hours and then subjected to hypoxia by placement in a chamber filled with N2/CO2 (95:5) for 1 hour. Subsequently, the hypoxic cells were reoxygenated, and control cells continued incubation for 1 additional hour in a normoxic environment. Cell samples were then analyzed using trypan blue staining and lactic dehydrogenase (LDH) release as markers of cell damage. Cells with an increased amount of α7β1D integrin were protected against reoxygenation injury to a greater extent, compared with control (LacZ) cells or ones in which α5β1D integrin was increased (Figure 5, A and B). The significant differences among groups were apparent only after H/R and not after hypoxia alone. To determine a potential mechanism for this cell death, we evaluated apoptosis, but no significant differences were found among groups (Supplemental Figure 3). Further, we inquired whether this protective effect might occur by modulating important integrin-related signaling pathways. No differences were detected among groups in the activation states of major integrin signaling molecules, including focal adhesion kinase (FAK), integrin linked kinase (ILK), PYK2, Src, and related downstream kinases, p38 and Akt (Supplemental Figure 4). Therefore, as we found in vivo, increased expression of α7β1D integrin protected CMs against reoxygenation injury.
Figure 5. Integrin heterodimers protect CMs from reoxygenation injury via prevention of mitochondrial membrane potential reduction.

(A and B) Cell survival was improved by α7β1D integrin OE. (A) Survival rate of cells assayed by trypan blue staining after H/R injury. α7β1D integrin OE inhibited cell death compared with that in control cells. In contrast, α5β1D integrin had no beneficial effect against H/R. HPF, high-power field. (B) Cell survival evaluated by LDH release after H/R confirmed data in A. *P < 0.05 vs. LacZ following reoxygenation (1-way ANOVA). (C) α7β1D integrin OE allows for maintenance of mitochondrial membrane potential during reoxygenation injury. Mitochondrial membrane potential was measured with the membrane potential–sensitive dye JC-1 after H/R. The excitation ratio (red/green) indicates the mitochondrial membrane potential. Cells in which α7β1D was overexpressed maintained the mitochondrial membrane potential during reoxygenation injury, in comparison with cells with control virus (LacZ) or α5β1D integrin. *P < 0.05 vs. LacZ (1-way ANOVA). (D and E) α7β1D integrin OE allows for maintenance of mitochondrial [Ca2+] during reoxygenation. Mitochondrial [Ca2+] was measured by pericam. (D) During normoxia, there was no significant difference among groups. (E) During reoxygenation, α7β1D allowed for maintenance of mitochondrial [Ca2+] in comparison with cells overexpressing LacZ or α5β1D in which mitochondrial [Ca2+] increased. *P < 0.05 vs. LacZ (1-way ANOVA).
Integrin β1D OE protects mitochondrial membrane potential in ARVMs subjected to H/R injury.
Mitochondria play an important role not only in energy production, but also in Ca2+ homeostasis and cell survival/death of CMs. The combination of Ca2+ overload and oxidative stress can open a nonspecific pore in the inner mitochondrial membrane known as the mPTP. The mPTP is normally held in the fully closed state, but, under some pathological conditions, it can open, collapse mitochondrial membrane potential, and predispose cells to death. To test whether integrin OE modified mitochondrial membrane potential (Ψμm) during reperfusion injury, we measured it using the membrane potential–sensitive dye, 5,5′,6,6′–tetrachloro-1,1′,3,3′-tetraethylbenzimidazoly-carbocyanine iodide (JC-1). As shown in Figure 5C, OE of α7β1D integrin prevented reduction of mitochondrial membrane potential that occurred in the reoxygenated cells expressing either the control LacZ virus or α5β1D integrin.
We next measured mitochondrial [Ca2+] using mitochondrial pericam (22, 23). During normoxia there were no significant differences among groups (Figure 5D). In contrast, during reoxygenation α7β1D OE inhibited increases in mitochondrial [Ca2+] that occurred in control cells as well as α5β1D integrin–overexpressing cells (Figure 5E). These results suggested that increased expression of α7β1D inhibited excessive Ca2+ entry into mitochondria and prevented mPTP opening after H/R injury.
In previous reports, it was shown that oxidative stress and/or mitochondrial Ca2+ overload could provoke mPTP opening (24–26). Therefore, we next tested whether integrin OE would modulate oxidative stress protein expression. Western blot analyses were performed for iNOS, 12-LO, ECSOD, Cu/ZnSOD, MnSOD, and Cox-2. There was no significant difference among any treatment group under normoxic conditions or following H/R (Supplemental Figure 5A). Likewise, superoxide anion quantification was performed after H/R injury, and no differences were noted among the various groups (Supplemental Figure 5B).
Effect of integrins on myocyte Ca2+-handling proteins in H/R — β1D integrin colocalizes with and binds to RyR2.
To assess how α7β1D could reduce mPTP opening and control cytosolic and mitochondrial [Ca2+], we evaluated how integrin OE might affect Ca2+-handling proteins. First, we performed Western blot analyses to evaluate expression of Ca2+-handling proteins, including L-type Ca2+ channel (LTCC), sodium-Ca2+ exchanger-1 (NCX-1), SERCA2a, phospholamban (PLB), and phospho-PLB proteins, in ARVMs under normoxic conditions and also following H/R. No significant differences were detected in expression of any of these proteins among groups (Supplemental Figure 6A). Further, to examine whether β1D integrin might interact with any of these proteins, coimmunoprecipitations (co-IPs) were performed, but no interactions were detected (Supplemental Figure 6B).
In contrast to the other proteins examined, co-IPs showed that RyR2 interacted with β1D integrin under endogenous conditions. This interaction was detected in human heart tissue as well as samples obtained from rats (Figure 6, A and B). In addition, binding was increased in CMs when this integrin receptor was overexpressed (Figure 6, C–E).
Figure 6. Integrins interact with RyR2 in human and rat heart.

(A) Human and (B) rat heart protein was immunoprecipitated using anti-β1D integrin or RyR2 antibodies and then immunodetected with the reciprocal antibody. (C–E) RyR2 binding and β1D integrin were increased by integrin OE. (C) IP with a RyR2 antibody and detection with an anti-β1D integrin antibody. (D) No differences in RyR2 were detected in the various input lysates. (E) Densitometry of immunoprecipitated RyR2-β1D integrin product. LacZ control = 1. Other samples are displayed relative to control. *P < 0.05 vs. LacZ, n = 6 per group (1-way ANOVA).
Using immunofluorescent microscopy, we next showed that integrins are clearly located on T-tubules (Figure 7, A and B). Colocalization of β1D integrin and junctophilin-2 or wheat-germ agglutinin, both markers of membranes/T-tubules, was found in isolated ARVMs (27, 28). Similarly, microscopy also showed that β1D integrin and RyR2 were colocalized in the T-tubules of CMs (Figure 7C). Moreover, not only was the β1D subunit localized to T-tubules in CMs, but we also detected α5 and α7 integrins in T-tubules when these α integrin subunits were overexpressed (Supplemental Figure 7, A–C). β1D colocalization with RyR2 was also confirmed and correlated using 3D rendering techniques (Figure 7D) (Pearson coefficient of 0.83 ± 0.09 [n = 10]). This result was supported by companion studies using immunogold electron microscopy in which colocalization of β1D and RyR2 was also observed (Figure 7, E–G). These results show that β1D integrins are positioned in T-tubules in which they colocalize and complex with RyR2.
Figure 7. Integrins colocalized with RyR2 in T-tubules.

(A and B) β1D integrin localizes in T-tubules. Integrin (green) with (A) wheat germ agglutinin (WGA, red) and (B) junctophilin-2 (JP-2, red). Scale bar: 20 μm. (C–G) β1D integrin colocalizes with RyR2 in CMs. (C) Immunofluorescent microscopy: β1D integrin (green) and RyR2 (red) colocalized. Scale bar: 20 μm. (D) Confocal 3D analysis shows β1D integrin (green) and RyR2 (red) are tightly colocalized (Pearson coefficient of 0.83 ± 0.09 [n = 10]). Scale bar: 20 μm. (E–G) Immunoelectron microscopy shows colocalization of β1D integrin and RyR2. (E) Immunoelectron microscopy of ARVM T-tubule. Scale bar: 200 nm. The boxed area is shown at higher magnification in F and G. (F) Magnified image of T-tubule and SR. The green arrow indicates RyR2 (6-nm gold particle); the red arrow indicates β1D integrin (10-nm gold particle); and blue arrows indicate colocalized particles in the T-tubule. Scale bar: 100 nm. (G) Diagram of F. Blue arrows indicate colocalization of β1D integrin (red dots) and RyR2 (green dots). Scale bar: 100 nm.
We also examined the expression of total and phosphorylated RyR2. While total RyR2 expression did not vary among groups (Figure 8A), RyR2 Ser2808 phosphorylation, which can increase RyR2 open probability and potentially sarcoplasmic reticulum (SR) Ca2+ release (29), was reduced in α7β1D-infected cells (Figure 8, A and B). No change was detected in phosphorylation of RyR2 at Ser2814. In addition, RyR2 Ser2808 phosphorylation was found to be decreased in heart tissue from α7β1D OE mice as compared with that from α5β1D OE mice. In contrast, RyR2 Ser2808 phosphorylation was increased when β1 integrin expression was reduced specifically in CMs (Supplemental Figure 8, A and B).
Figure 8. Integrins modulated RyR2 Ser2808 phosphorylation.

(A) β1D and α7β1D integrin reduce phosphorylation of RyR2 protein at Ser2808 but not Ser2814, following H/R (n = 6 per group). (B) Densitometry of Western blot in A. *P < 0.05 vs. LacZ, **P < 0.05 α7β1D vs. B1D (1-way ANOVA).
To confirm the β1 integrin–RyR2 interaction and determine the precise location within RyR2 that binds to β1D integrin, a full-length RyR2 cDNA or 7 different truncated and HA-tagged RyR2 fragments were transfected into human embryonic kidney (HEK) cells along with a full-length β1D integrin cDNA expression vector. First, we confirmed that β1D bound with full-length RyR2 in this system, as it did endogenously in CMs. Then, we evaluated which portion of RyR2 directed this binding. Using the truncated fragments shown in Figure 9A, we found that the N terminus of RyR2 (residues 1–530) directed this binding, while no interaction was detected with any of the other 6 fragments (Figure 9B).
Figure 9. Integrins interact with the RyR2 receptor.

(A) RyR2 fragments. Numbers represent amino acids spanned by the fragment. (B–D) Binding site of RyR2 and β1D integrin. The bottom blot in each panel is a Western blot of RyR2 fragment expression in HEK293 whole cell lysates. (B) Seven unique HA-tagged RyR2 fragments from A were transfected into HEK293 cells with full-length β1D integrin cDNA. IPs performed 48 hours after transfection using β1D integrin and anti-HA (for RyR2) antibodies showed that β1D integrin only bound the N-terminal domain of RyR2 (1–530) (arrow indicates the unique band only detected in the bound fragments). (C) Additional IPs using Flag-tagged RyR2 fragments 1–217, 205–395, and 392–532. Only the RyR2 1–217 fragment bound β1D integrin (arrow indicates the unique band only detected in the bound fragments). (D) IPs using Flag-tagged RyR2 fragments 89–395, 110–395, and 142–395. All fragments bound to β1D integrin, suggesting the binding site was within RyR2 residues 89–204. (E) RyR2 fragments examined using a peptide array. RyR2 amino acids 165–175 bound to β1D integrin. (F) IPs performed using RyR2 (fragment 1–217) WT or mutant (amino acids 169 [Arg {R} to Gln {Q}]) fragments. Binding between the WT RyR2 (fragment 1–217) domain and β1D integrin was destabilized by the mutation (arrow indicates the unique band only detected in the bound WT fragment, absent in the 169RQ mutant). Light chain indicates the immunoglobulin light chain detected as a nonspecific band in all samples. The bottom blot is a Western blot of WT and mutant RyR2 fragments in whole cell lysates.
Next, we performed studies to precisely determine the interaction site between the RyR2 N-terminal domain and β1D integrin. A recent report detailed the structure of the RyR2 N-terminal domain (30). This work indicated that the RyR2 N terminus had 3 major domains: (a) residues 1–205, which show structural homology with the Ca2+ release channel inositol-1,4,5-trisphosphate (IP3) receptors (31, 32); (b) the MIR domain (residues 206–394) that is named after the 3 proteins in which it occurs (mannosyltransferase, IP3 receptor, and RyR2) (33); and (c) the RIM domain (residues 395–532), which is formed by a bundle of 5 α-helices (30). Based on this work, we constructed 3 unique RyR2 N-terminal domain fragments and showed that β1D integrin interacted with the IP3 receptor domain (residues 1–217) (Figure 9, C and D). Then, using additional RyR2 fragments as well as a peptide array, we narrowed the binding region specifically to RyR2 amino acids 165–175 (Figure 9E). To definitively prove that this binding site was essential for the integrin-RyR2 interaction, we mutated Arg169 within the central portion of this binding region to Gln. This single amino acid mutation destabilized the binding between the N-terminal domain of RyR2 and β1D integrin (Figure 9F). Together, these results definitively show that β1D integrin binds to RyR2 IP3 receptor domain at amino acids 165–175.
The β1D cytoplasmic domain is specifically required for H/R protection.
The preceding data focused on the function of the β1D variant, since it is the dominant isoform found in adult CMs. To specifically evaluate whether the “D” cytoplasmic domain of β1 integrin was necessary for the beneficial effects noted when ARVMs were subjected to H/R, we compared the function of α7β1A with that of α7β1D. Increased expression of α7β1A integrin had no protective effect against H/R injury, in comparison with that of α7β1D (Figure 10, A and B). Further, α7β1A integrin had no effect on mitochondrial membrane potential or mitochondrial [Ca2+] in comparison with α7β1D (Figure 10, C and D). Finally, in contrast with β1D, β1A did not interact with RyR2 in IP assays (Figure 10, E and F). These results suggest that β1D integrin, which is expressed dominantly in muscle, plays an important protective role in H/R injury and functions uniquely as compared with the ubiquitously expressed β1A integrin isoform.
Figure 10. The β1D integrin cytoplasmic domain variant is necessary for protection from reoxygenation injury.

(A and B) Protection from reoxygenation injury occurs with the β1D but not the β1A integrin splice variant. (A) Survival rate of cells assayed by trypan blue staining after H/R injury. α7β1D integrin OE inhibited cell death compared with that in control cells. In contrast, α5β1D integrin had no beneficial effect against H/R. *P < 0.05 vs. LacZ (1-way ANOVA). (B) Cell survival evaluated by LDH release after H/R confirmed data in A. *P < 0.05 vs. LacZ following reoxygenation (1-way ANOVA). (C) Mitochondrial membrane potential was maintained by α7β1D but not α7β1A integrin following H/R injury. Mitochondrial membrane potential measured with the membrane potential–sensitive dye JC-1 after H/R. The excitation ratio (red/green) indicates the mitochondrial membrane potential. Cells in which α7β1D OE maintained the mitochondrial membrane potential during reoxygenation injury, in comparison with control virus (LacZ) or α7β1A integrin. *P < 0.05 vs. LacZ (1-way ANOVA). (D) Mitochondrial [Ca2+] measured using pericam excitation ratio during reoxygenation. In cells infected with α7β1D, mitochondrial [Ca2+] was maintained in comparison with the LacZ (control) or α7β1A-infected cells in which it increased. *P < 0.05 vs. LacZ (1-way ANOVA). (E and F) α7β1A did not interact with RyR2 in ARVM protein extracts. IPs were performed using IgG as a negative control and (E) anti-β1A integrin or (F) RyR2 antibodies.
The β1D cytoplasmic domain stabilizes the RyR2 protein.
The IP3 receptor domain is located in the N terminus of RyR2. The N-terminal and central regions of RyR2 are predicted to interact with one another and to stabilize this channel via interdomain interactions (34–36). To show that β1D could stabilize the RyR2 channel, we used a fluorescence resonance energy transfer (FRET) sensor, in which yellow fluorescent protein (YFP) was inserted into RyR2 after residue Ser437 of the N-terminal region, and a cyan fluorescent protein (CFP) was inserted after residue Ser2367 of the central region. This synthetic FRET sensor forms a dual YFP- and CFP-labeled RyR2 (RyR2S437-YFP/S2367-CFP) (36). The FRET sensor was transfected into HEK293 cells along with expression vectors for full-length α7, β1A, or β1D integrin subunits. Then, the FRET signal was evaluated in live cells (Figure 11, A and B).
Figure 11. The β1D cytoplasmic domain stabilizes RyR2; integrin heterodimers require ECM binding for RyR2 stabilization.

(A) FRET image of RyR2S437-YFP/S2367-CFP expressed alone or with α7, β1A, or β1D integrin (original magnification, ×63). (B) Graphic FRET ratio. Relative signal detected in the RyR2-transfected cells was set as 1. FRET signal detected in other samples is displayed relative to control. The RyR2 sensor FRET ratio was increased by dantrolene, an RyR2 stabilizer. β1D integrin cotransfected with the sensor increased the FRET ratio, indicating increased RyR2 stability. No change was seen with α7 or β1A integrin (*P < 0.05 vs. RyR2-transfected cells, n = 20 cells per group). (C) Western blots of control and β1D integrin stably transfected OE cells (OE). β1D integrin precursor (P) was substantially increased in OE cells, but the higher MW mature (M) form increased minimally. No β1D integrin was detected in untransfected cells. GAPDH was used as a loading control. (D) Graphic FRET ratio. FRET ratio in β1D OE cells transiently transfected with RyR2 alone and plated without ECM was set as 1. FRET detected in other samples is displayed relative to this amount. Cells expressing β1D integrin alone showed no change in FRET signal regardless of ECM coating, indicating that there was no stabilization of RyR2. With OE of the FN-binding integrin α5β1D and plating on FN matrix, a stronger FRET signal was found compared with cells plated on LN or no substrate. Cells expressing the LN-binding α7β1D integrin had the most robust FRET signal when plated on LN (*P < 0.05 vs. β1D OE cells transfected with RyR2 alone).
An increase in the average FRET ratio measured from this sensor indicates a more stable interaction between the IP3 receptor and central RyR2 regions. The average FRET YFP/CFP ratio of cells expressing the sensor alone was increased significantly by 10 μM dantrolene, a substance known to stabilize the interdomain interactions of the RyR2 channel (37). Cotransfection of the sensor with α7 integrin, or β1A integrin alone, had no significant effect on the FRET ratio, while cotransfection with β1D caused a significant increase (Figure 11B). These results suggest that the β1D cytoplasmic domain stabilizes the RyR2 channel.
To further confirm the role of αβ integrin heterodimers in cardioprotection, particularly in contrast to single integrin subunits (e.g., β1D), we created a permanently transfected (stable) cell line that expressed β1D integrin (Figure 11C). We then cotransfected the RyR2 FRET construct into this stable cell line, along with vectors that expressed either α5 or α7 integrin subunits. In doing so, we created a FN-binding integrin (α5β1D) or a LN-binding integrin (α7β1D). Cells were examined for FRET signals when they were plated on ECM ligands that specifically bind to the integrin receptor that was produced, i.e., FN was used when α5β1D was tested, and LN was used when α7β1D was studied. As shown in Figure 11D, when cells only expressed β1D without a companion α integrin subunit, there was no change in FRET signal, regardless of the ECM coating, indicating that there was no stabilization of RyR2. Yet, when cells overexpressed the FN-binding integrin α5β1D and were plated on FN matrix, they elicited a stronger FRET signal compared with that when these cells were plated on either LN or no ECM substrate. Similarly, cells expressing the LN-binding integrin, α7β1D, had the most robust FRET signal when they were plated on LN. Given these data, we examined whether cells expressing α5β1D and plated on its suitable ECM ligand, FN, could also offer a beneficial effect against H/R injury, as was noted when α7β1D-expressing cells were plated on their appropriate ECM ligand, LN. For this purpose, ARVMs were infected with LacZ or α5β1D recombinant adenoviruses, and cells were plated on either LN matrix alone (which would bind to α7β1D but not α5β1D) or on a mixture of LN and FN, so that an appropriate matrix ligand for α5β1D was available. Subsequently, H/R injury was performed as above. Cell samples were then analyzed using trypan blue staining and LDH release as markers of cell damage. With the combined matrix coating, α5β1D integrin OE displayed a beneficial effect against H/R, while no protection was noted when the cells were plated on LN alone (Supplemental Figure 9). These data support the finding that integrin–ECM ligand engagement is necessary for protection against H/R injury.
Discussion
The current study shows that α7β1D integrin can protect CMs from I/R injury. Specifically, we showed that (a) combined expression of α and β integrin subunits is needed for expression of integrin receptors on the CM membrane, (b) α7β1D integrin OE protects against I/R injury in vivo and in cultured CMs, (c) loss of β1 integrins from the CM leads to deleterious effects following short-term I/R, (d) α7β1D OE reduces mPTP opening in the face of H/R and also limits injurious changes in mitochondrial Ca2+, and that β1D integrin (e) colocalizes with RyR2, (f) reduces phosphorylation of RyR2, (g) interacts with the N-terminal RyR2 domain at amino acids 165–175, and (h) stabilizes interdomain interactions of the RyR2 channel. ECM ligand binding of integrin was found to be necessary for its cardioprotective effects. Thus, α7β1D integrin OE offers a potentially novel means to protect myocytes and the intact heart from injury.
Previous work had shown that increased expression of integrins can protect against skeletal muscle sarcolemmal damage in mouse models of muscular dystrophy (38, 39). In Duchenne muscular dystrophy, caused by mutations in the dystrophin gene, the dystrophin-glycoprotein complex (DGC) is compromised and sarcolemmal integrity is reduced, but expression of integrins, including α7β1D, is increased, likely in compensation for loss of DGC components (40, 41). As in skeletal muscle studies that tested the effects of increased integrin expression in protection of dystrophic skeletal muscle, we found that combined expression of both integrin α and β subunits was required for appropriate expression of integrin receptors on the CM membrane. In contrast to the skeletal muscle studies, we found that β subunit expression did not provoke compensatory increases in α subunit expression but that dual expression of the α and β subunits was required (39) to increase integrin expression on the sarcolemma. Likewise, our data showed that coexpression of α5 and α7 integrin can increase both mature and precursor forms of β1D integrin and change its subcellular localization. These data are consistent with prior work in other cell types that showed that β1 subunits are generally synthesized in excess over α subunits (42). When expression of α subunits was increased, they assembled with β1 subunits, allowing for transport of the integrin heterodimer through the Golgi apparatus and then presentation of the αβ1 receptor on the cell surface (42).
Importantly, we found that integrin receptors are positioned at CM T-tubules. The T-tubule system is the dominant location for excitation-contraction coupling in ventricular myocytes (28). Integrin receptors require ECM binding to function as adhesive receptors, mechanotransducers, and direct intracellular signaling events. As noted in prior reports, ECM clearly exists at the T-tubule (43). The finding that integrins are critically positioned in the T-tubule is consistent with our results that integrin receptors participate in modifying proteins known to be essential for CM Ca2+ regulation and therefore can potentially protect against Ca2+-mediated myocyte injury. Our data indicated that the protective effect offered by α7β1D integrin to prevent Ca2+ overload does not appear to occur via an antiapoptotic mechanism but likely occurs by inhibition of cellular necrosis (44).
We also found that α7β1D integrin OE was inversely correlated with phosphorylation of RyR2 Ser2808. RyR2 phosphorylation is thought to be important in Ca2+ homeostasis, yet linking phosphorylation to specific changes in the single-channel function of RyR2 has remained quite controversial. Some prior work has shown that Ser2808 can be hyperphosphorylated by PKA (45) and that PKA-dependent phosphorylation of RyR2 can increase the open probability of RyR2 (45–47). In contrast, work by others has shown that dephosphorylation of RyR2 can also increase open probability, indicating that there must be an inhibitory component to phosphorylation (46, 48, 49). The controversy about the function of RyR2 phosphorylation in the myocardium is highlighted by recent work from Houser’s group (50), in which genetically manipulated mice with a RyR2 S2808A mutation were produced that could not undergo PKA-mediated hyperphosphorylation at the RyR2 S2808 site. Data from this mouse model strongly indicated that phosphorylation at this site was not linked to development of cardiac dysfunction after MI. A limitation of our study is that we cannot precisely link RyR2/integrin binding directly to RyR2 phosphorylation. This will require future studies that would ideally use a genetically manipulated knockin mouse model with specific mutation of RyR2 amino acid 169, which we showed to be required for RyR2/integrin binding.
OE of α7β1D integrin inhibited mPTP opening and also inhibited the increases in mitochondrial [Ca2+] that occur after myocyte H/R. mPTP opening can occur via 2 different mechanisms: oxidative stress or Ca2+ overload. In some reports, mPTP opening correlated to a greater extent with changes in oxidative stress than changes in [Ca2+] (25, 26). However, here we did not find integrins to cause changes in oxidative stress–related proteins. Ca2+ overload during I/R injury has also been shown to open the mPTP, resulting in collapse of the mitochondrial membrane potential, uncoupling of the respiratory chain, and efflux of cytochrome c and other factors, which can lead to cell death via apoptosis or necrosis (51). Many factors can change the mitochondrial [Ca2+] required to open the mPTP, and several of these may play a role in mediating mPTP opening during ischemia and reperfusion (24–26, 52, 53).
A primary means through which mitochondria uptake Ca2+ is via the mitochondrial calcium uniporter (54). Recent studies have demonstrated that mitochondrial Ca2+ uptake can be dependent on anatomical-functional microdomains established between the SR and mitochondria and that these microdomains play crucial roles separate from more global changes in cytosolic Ca2+ (55). While the precise mechanism whereby α7β1D protects CMs from “cell death via Ca2+ overload” is not fully understood, we would speculate that integrin OE and interaction with RyR2 reduces mitochondrial Ca2+ loading by altering Ca2+ availability from adjacent microdomains between SR and the mitochondrial membrane (56, 57). RyR activation by Ca2+ is very sensitive to the geometric arrangement of RyRs and LTCCs (58). Therefore, it is also possible that integrin binding to RyR2 alters critical interactions of RyR2 with other proteins such as LTCCs, leading to reduced Ca2+ influx. More detailed future experiments will be required to directly confirm these hypotheses.
This study is the first report showing that CM integrins, and specifically the β1D but not β1A cytoplasmic isoform variant, bind to RyR2. We detected this in human heart and then performed further studies in other species. Detailed analysis showed that β1D bound to the RyR2 N terminus domain at amino acids 165–175. For this, we used a series of IP studies, transfection experiments in HEK cells, and a peptide array. From these combined approaches it is clear that RyR2 and β1D integrin bind, but we cannot absolutely exclude that this interaction occurs through an intermediary protein.
The RyR2 N-terminal and central domains have been shown to physically interact with one another in 3D space, and changes in the strength of their interaction affect channel gating (34, 36). The interaction between these domains can serve as a regulatory switch for channel gating activity: a tight interaction or “zippering” of these domains stabilizes the channel in the closed state. Prior work has shown that mutations in either domain can weaken their interaction, thus increasing their tendency toward “unzipping,” causing activation and leakiness of the Ca2+ release channel (33, 59, 60). Interestingly, mutation of RyR2 amino acid 169, which is essential for integrin-RyR2 binding, has been previously linked to catecholaminergic polymorphic ventricular tachycardia, in which RyR2 dysfunction is caused by channel destabilization (30). Future studies will be required to see how integrin-RyR2 binding might be related to this phenotype. Further, it has been reported that dantrolene, which stabilizes RyR2, can attenuate I/R injury in various organs, including heart (61–63). Given this prior work, combined with our current findings, we suggest that β1D integrin contributes to RyR2 channel interdomain stabilization in the myocyte and, as above, in doing so can affect [Ca2+] microdomain availability within the myocyte.
The current study shows that ECM substrate binding to cell surface integrins stabilizes RyR2. It is interesting to note that cells in which we overexpressed the single subunit β1D integrin alone also showed evidence of RyR2 stabilization. We suggest that when β1D protein is increased, it heterodimerizes with endogenous α integrin subunits already present in these cells, allowing for expression of increased numbers of functional cell surface integrin receptors that can bind ECM ligand. It is for this reason that increased expression of β1D alone can still offer some degree of cardioprotection.
An important question is why β1D integrin has a preferential effect in allowing for cardioprotection and RyR2 binding, as compared with the β1A isoform. The β1D and β1A integrin isoforms are highly similar and vary only in their cytoplasmic domains. Like most β integrin subunits, the cytoplasmic domains of β1D and β1A are quite short (β1A = 47 amino acids, β1D = 50 amino acids) (10, 64). Both variants have the first 26 cytoplasmic N-terminal amino acids in common. Of the remaining sequence, β1D only differs from β1A by 13 amino acids. There is precedent for proteins showing exclusive binding to a specific β1 integrin cytoplasmic variant. For example, both integrin cytoplasmic domain–associated protein-1 (ICAP-1) and CD98 bind to the β1A but not the β1D integrin cytoplasmic domain and in doing so can modulate β1A integrin activation (65, 66). In addition, it has been shown that there are functional differences between the β1A and β1D isoforms, in that the β1D isoform binds more tightly to talin-2, which could allow for stronger ECM-cytoskeletal linkages required in myocytes (10). Here, we have shown that RyR2 binds preferentially to β1D but not β1A, the first protein to our knowledge to show this preference. Where RyR2 binds precisely in the β1D cytoplasmic domain, how this interaction leads to cardioprotection, and whether the RyR2-β1D interaction is the only means that offers the cardioprotection require further investigation.
In summary, our data clearly support an important physiological and new cardioprotective function of integrins in the CM. Basally, we would suggest the endogenous integrin/RyR2 interaction contributes to CM Ca2+ homeostasis by stabilizing the RyR2 channel. This is supported by our data showing that β1D integrin and RyR2 interact and our FRET studies indicating how integrin can stabilize the RyR2 channel and that β1 integrin–deficient hearts are more prone to ischemic damage than controls. In contrast, we would advocate that, in the face of pathophysiological stress produced by insults such as I/R, having an increased amount of α7β1D integrin expressed in CMs can protect the myocyte from damage. Perturbations in RyR2 function have been shown to be present in the face of I/R, and modulating their function has been proposed previously as a means toward cardioprotection (67). Integrin OE reduces mPTP opening and detrimental effects of elevated mitochondrial Ca2+ and stabilizes RyR2 interdomain interactions. While further studies are clearly required to prove this tenet, we would propose that the integrin OE may modify microdomain coupling between SR and mitochondria to ultimately protect the myocyte/myocardium (56, 57). While these results highlight the protective effect that occurs in part through α7β1D integrin interacting and colocalizing with RyR2, as above, it is possible that other integrin functions are also important for this therapeutic effect. Further data are necessary to elucidate all of the precise mechanism(s) that allow integrin OE to protect the heart from injury.
Methods
Further details regarding the materials and methods are available in the Supplemental Methods.
Animals, cells, and human heart tissue.
Tg mice with CM-specific OE of integrin α5, α7, or β1D were produced in a C57BL/6 background. Full-length integrin α5 (supplied by M. Valencik, University of Nevada, Reno, Nevada, USA), α7 (68), or β1D (69) cDNAs were cloned into a vector containing the α-myosin heavy chain promoter (70) and were used for microinjection in the UCSD Transgenic Core. Inducible and constitutively active CM-specific β1 integrin knockout mice were created as reported previously (19, 20).
CMs were isolated from adult mice or adult Sprague-Dawley rats as described previously (71).
Human ventricular tissue was obtained as either explanted samples at the time of heart transplant or from organs judged unsuitable for transplantation. Samples were either frozen rapidly in the operating room in liquid N2 or stored on ice for less than 4 hours at the time of acquisition. Then, all specimens were frozen at –80°C and thawed for processing.
H/R in isolated CMs.
CMs were cultured on plates coated with 10 μg/ml LN at 37°C. Hypoxia was induced by replacing the air content with a 95% N2 and 5% CO2 gas mixture at 2 l/min in a metabolic chamber (Columbus Instruments) and by replacing the media with a thin film of glucose-free media (glucose-free DMEM, Invitrogen), which covered the cells for 60 minutes. Subsequently, the culture was “reoxygenated” for 60 minutes by replacing the media with normal maintenance media and by incubating the cells in 21% O2 and 5% CO2. Cell death was quantified by trypan blue staining and by analyzing LDH release (Cytotoxicity Detection Kit; Roche) as a marker of cell damage.
Mitochondrial [Ca2+] measurements and measurement of mitochondrial membrane potential.
Adv-CMV-mito-pericam and its use have been described previously (22, 23). Mitochondrial membrane potential was measured using a JC-1 assay. After hypoxia treatment, as described above, JC-1 at a concentration of 10 μg/ml (Invitrogen) was added to the medium for 10 minutes. Cells were then washed twice in PBS. At 30 and 60 minutes following onset of reoxygenation, fluorescence measurements were performed at room temperature as described above with a Nikon Diaphot epifluorescent microscope. Fluorescence emission was filtered at 485 and 580 nm and directed to a photomultiplier tube (23).
In vivo I/R study.
Mice anesthetized with pentobarbital (80 mg/kg) were mechanically ventilated, and ischemia was produced by occluding the left coronary artery with a 7-0 silk suture on a tapered BV-1 needle (Ethicon, Johnson & Johnson) for 30 minutes. Following 30 minutes of occlusion, the ligature was released, and the heart was reperfused for 24 hours. The area at risk was determined by staining with 1% Evans blue (1.0 ml; Sigma-Aldrich). The heart was immediately excised and cut into 1-mm slices (McIlwain tissue chopper, Brinkmann Instruments). The LV was counterstained with 1% 2,3,5,-triphenyltetrazolium chloride (Sigma-Aldrich). Images were analyzed by Image-Pro Plus (Media Cybernetics), and infarct size was determined by planimetry.
RyR2 cDNA fragments.
PCR was used to produce 7X-HA–tagged RyR2 fragments and 6X-FLAG–tagged RyR2 fragments from the full-length RyR2 cDNA (supplied by S.R. Wayne Chen, University of Calgary, Calgary, Alberta, Canada). PCR primers used for this process are provided in Supplemental Table 1. Products were cloned into pcDNA3.1 using SalI or XhoI with BamHI or EcoRI restriction sites and sequenced to confirm fidelity. To produce the R169Q mutation located within the RyR2 IP3 receptor domain, the RyR2 cDNA was modified using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. All constructs were confirmed by sequencing.
Statistics.
Values are shown as mean ± SEM. Significance was evaluated with ANOVA, using GraphPad Prism software. Values of P < 0.05 were considered significant.
Study approval.
All animal use protocols were approved by the Veterans Administration San Diego Institutional Animal Care and Use Committee. These investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH. All investigations using human heart were approved by Stritch School of Medicine, Loyola University and Veterans Administration San Diego. Samples of LV tissue were obtained from Loyola University Medical Center’s Cardiovascular Institute Tissue Repository and from the Gift of Hope Organ and Tissue Donor Network. The investigation conformed to the principles outlined in the Declaration of Helsinki. A detailed protocol and informed consent document were reviewed by Loyola University Medical Center’s Institutional Review Board prior to tissue procurement. Explanted human LV tissue from patients undergoing heart transplantation for nonischemic dilated cardiomyopathy were obtained following informed consent. Donor heart tissue judged unsuitable for cardiac transplantation was similarly obtained following informed consent from organ donor family members by the Gift of Hope Organ and Tissue Donor Network.
Supplementary Material
Acknowledgments
The authors wish to thank Clayton Buck (University of Pennsylvania) and Lou Terracio (NYU) for the α5 integrin adenovirus, Maria Valencik (University of Nevada) for the α5 integrin antibody, Allen Samarel (Stritch School of Medicine, Loyola University Chicago) for assistance with procurement of human heart specimens, and Rui Zhang (Cedars-Sinai Heart Institute), Andrea Domenighetti, Britton W. Boras, Andrew McCulloch, Ruixia Li, and Tomoko Yajima (UCSD) for excellent advice and technical assistance. This work was supported by NIH PO1 HL 46345, RO1 HL088390, and RO1 HL103566 to R.S. Ross; Eli Lilly International Fellowship, Uehara Memorial Foundation Research Fellowship, and Kanae Foundation for The Promotion of Medical Science Research Fellowship to H. Okada; NIH RO1 HL070828 to J.I. Goldhaber; NIH R01-AG014632 and the Muscular Dystrophy Association to S.J. Kaufman; and NIH R01-HL095541 to Z. Liu.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(10):4294–4308. doi:10.1172/JCI64216.
References
- 1.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301(5):H1723–H1741. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- 2.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106(3):360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11–25. doi: 10.1016/0092-8674(92)90115-S. [DOI] [PubMed] [Google Scholar]
- 4.Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci. 2002;115(pt 19):3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 5.Ross RS. Molecular and mechanical synergy: cross-talk between integrins and growth factor receptors. Cardiovasc Res. 2004;63(3):381–390. doi: 10.1016/j.cardiores.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 6.Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88(11):1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 7.Altruda F, et al. A human integrin beta 1 subunit with a unique cytoplasmic domain generated by alternative mRNA processing. Gene. 1990;95(2):261–266. doi: 10.1016/0378-1119(90)90369-3. [DOI] [PubMed] [Google Scholar]
- 8.Languino LR, Ruoslahti E. An alternative form of the integrin beta 1 subunit with a variant cytoplasmic domain. J Biol Chem. 1992;267(10):7116–7120. [PubMed] [Google Scholar]
- 9.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70(3):422–433. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Anthis NJ, Wegener KL, Critchley DR, Campbell ID. Structural diversity in integrin/talin interactions. Structure. 2010;18(12):1654–1666. doi: 10.1016/j.str.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun M, et al. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107(7):1046–1052. doi: 10.1161/01.CIR.0000051363.86009.3C. [DOI] [PubMed] [Google Scholar]
- 12.Roman J, Little CW, McDonald JA. Potential role of RGD-binding integrins in mammalian lung branching morphogenesis. Development. 1991;112(2):551–558. doi: 10.1242/dev.112.2.551. [DOI] [PubMed] [Google Scholar]
- 13.Roman J, McDonald JA. Expression of fibronectin, the integrin alpha 5, and alpha-smooth muscle actin in heart and lung development. Am J Respir Cell Mol Biol. 1992;6(5):472–480. doi: 10.1165/ajrcmb/6.5.472. [DOI] [PubMed] [Google Scholar]
- 14.Nawata J, et al. Differential expression of alpha 1, alpha 3 and alpha 5 integrin subunits in acute and chronic stages of myocardial infarction in rats. Cardiovasc Res. 1999;43(2):371–381. doi: 10.1016/S0008-6363(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 15.Brancaccio M, et al. Differential onset of expression of alpha 7 and beta 1D integrins during mouse heart and skeletal muscle development. Cell Adhes Commun. 1998;5(3):193–205. doi: 10.3109/15419069809040291. [DOI] [PubMed] [Google Scholar]
- 16.van der Flier A, et al. Spatial and temporal expression of the beta1D integrin during mouse development. Dev Dyn. 1997;210(4):472–486. doi: 10.1002/(SICI)1097-0177(199712)210:4<472::AID-AJA10>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Wu RX, et al. Fibroblast migration after myocardial infarction is regulated by transient SPARC expression. J Mol Med (Berl). 2006;84(3):241–252. doi: 10.1007/s00109-005-0026-0. [DOI] [PubMed] [Google Scholar]
- 18.Krishnamurthy P, Subramanian V, Singh M, Singh K. Deficiency of beta1 integrins results in increased myocardial dysfunction after myocardial infarction. Heart. 2006;92(9):1309–1315. doi: 10.1136/hrt.2005.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li R, et al. beta1 integrin gene excision in the adult murine cardiac myocyte causes defective mechanical and signaling responses. Am J Pathol. 2012;180(3):952–962. doi: 10.1016/j.ajpath.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shai SY, et al. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90(4):458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 21.Sohal DS, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89(1):20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 22.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci U S A. 2001;98(6):3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belke DD, Swanson E, Suarez J, Scott BT, Stenbit AE, Dillmann WH. Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. Am J Physiol Heart Circ Physiol. 2007;292(4):H1755–H1763. doi: 10.1152/ajpheart.00884.2006. [DOI] [PubMed] [Google Scholar]
- 24.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787(11):1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Juhaszova M, et al. The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann N Y Acad Sci. 2008;1123:197–212. doi: 10.1196/annals.1420.023. [DOI] [PubMed] [Google Scholar]
- 26.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290(5):H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 27.Clark RB, Tremblay A, Melnyk P, Allen BG, Giles WR, Fiset C. T-tubule localization of the inward-rectifier K(+) channel in mouse ventricular myocytes: a role in K(+) accumulation. J Physiol. 2001;537(pt 3):979–992. doi: 10.1113/jphysiol.2001.012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei S, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107(4):520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 30.Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468(7323):585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 31.Bosanac I, et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420(6916):696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 32.Bosanac I, Yamazaki H, Matsu-Ura T, Michikawa T, Mikoshiba K, Ikura M. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol Cell. 2005;17(2):193–203. doi: 10.1016/j.molcel.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 33.Ponting CP. Novel repeats in ryanodine and IP3 receptors and protein O-mannosyltransferases. Trends Biochem Sci. 2000;25(2):48–50. doi: 10.1016/s0968-0004(99)01513-3. [DOI] [PubMed] [Google Scholar]
- 34.Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671–d683. doi: 10.2741/ikemoto. [DOI] [PubMed] [Google Scholar]
- 35.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108(7):871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, et al. Dynamic, inter-subunit interactions between the N-terminal and central mutation regions of cardiac ryanodine receptor. J Cell Sci. 2010;123(pt 10):1775–1784. doi: 10.1242/jcs.064071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene — a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59(4):364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Burkin DJ, Kaufman SJ. Increasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am J Physiol Cell Physiol. 2008;294(2):C627–C640. doi: 10.1152/ajpcell.00329.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Milner DJ, Boppart MD, Ross RS, Kaufman SJ. beta1D chain increases alpha7beta1 integrin and laminin and protects against sarcolemmal damage in mdx mice. Hum Mol Genet. 2012;21(7):1592–1603. doi: 10.1093/hmg/ddr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burkin DJ, Kaufman SJ. The α7β1 integrin in muscle development and disease. Cell Tissue Res. 1999;296(1):183–190. doi: 10.1007/s004410051279. [DOI] [PubMed] [Google Scholar]
- 41.Vachon PH, et al. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997;100(7):1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heino J, Ignotz RA, Hemler ME, Crouse C, Massague J. Regulation of cell adhesion receptors by transforming growth factor-beta. Concomitant regulation of integrins that share a common beta 1 subunit. J Biol Chem. 1989;264(1):380–388. [PubMed] [Google Scholar]
- 43.Kostin S, et al. The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 1998;294(3):449–460. doi: 10.1007/s004410051196. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117(9):2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 46.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98(12):1506–1513. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 47.Wehrens XH, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304(5668):292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 48.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552(pt 1):109–118. doi: 10.1113/jphysiol.2003.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487(pt 3):609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, et al. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110(6):831–840. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28(2):131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 52.Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245(3):915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. . Arch Biochem Biophys. 1979;195(2):460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 54.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem. 2000;275(20):15305–15313. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- 56.Dorn GW, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. 2013;55:42–49. doi: 10.1016/j.yjmcc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 57.Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res. 2013;98(2):259–268. doi: 10.1093/cvr/cvt032. [DOI] [PubMed] [Google Scholar]
- 58.Gomez AM, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276(5313):800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 59.Ikemoto N, Yamamoto T. Postulated role of inter-domain interaction within the ryanodine receptor in Ca(2+) channel regulation. Trends Cardiovasc Med. 2000;10(7):310–316. doi: 10.1016/S1050-1738(01)00067-6. [DOI] [PubMed] [Google Scholar]
- 60.Tateishi H, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. . Cardiovasc Res. 2009;81(3):536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boys JA, Toledo AH, Anaya-Prado R, Lopez-Neblina F, Toledo-Pereyra LH. Effects of dantrolene on ischemia-reperfusion injury in animal models: a review of outcomes in heart, brain, liver, and kidney. J Investig Med. 2010;58(7):875–882. doi: 10.231/JIM.0b013e3181e5d719. [DOI] [PubMed] [Google Scholar]
- 62.Kocogullari CU, et al. Can dantrolene protect spinal cord against ischemia/reperfusion injury? An experimental study. Thorac Cardiovasc Surg. 2008;56(7):406–411. doi: 10.1055/s-2008-1038731. [DOI] [PubMed] [Google Scholar]
- 63.Preckel B, Schlack W, Comfere T, Thamer V. Effect of dantrolene in an in vivo and in vitro model of myocardial reperfusion injury. Acta Anaesthesiol Scand. 2000;44(2):194–201. doi: 10.1034/j.1399-6576.2000.440211.x. [DOI] [PubMed] [Google Scholar]
- 64.van der Flier A, Kuikman I, Baudoin C, van der Neut R, Sonnenberg A. A novel β-1 integrin isoform produced by alternative splicing: unique expression in cardiac and skeletal muscle. FEBS Lett. 1995;369(2–3):340–344. doi: 10.1016/0014-5793(95)00814-P. [DOI] [PubMed] [Google Scholar]
- 65.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113(pt 20):3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 66.Millon-Frémillon A, et al. CaMKII-mediated intramolecular opening of integrin cytoplasmic domain associated protein-1 (ICAP-1α) negatively regulates β1 integrins. J Biol Chem. 2013;288(28):20248–20260. doi: 10.1074/jbc.M113.455956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zucchi R, Ronca F, Ronca-Testoni S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther. 2001;89(1):47–65. doi: 10.1016/S0163-7258(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 68.Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ. H36-alpha 7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J Cell Biol. 1992;117(3):643–657. doi: 10.1083/jcb.117.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pham CG, et al. Striated muscle-specific beta(1D)-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. Am J Physiol Heart Circ Physiol. 2000;279(6):H2916–H2926. doi: 10.1152/ajpheart.2000.279.6.H2916. [DOI] [PubMed] [Google Scholar]
- 70.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266(36):24613–24620. [PubMed] [Google Scholar]
- 71.Horikawa YT, et al. Caveolin-3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;44(1):123–130. doi: 10.1016/j.yjmcc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.