

Background: Lipid peroxidation generates unsaturated aldehydes that form conjugates with histidyl dipeptides.
Results: Carnosine-aldehyde conjugates form covalent adducts with proteins and are reduced by aldose reductase.
Conclusion: Detoxification of carnosine-aldehyde by aldose reductase prevents protein carnosinylation.
Significance: Aldose reductase prevents tissue injury due to aldehyde-carnosine conjugates.
Keywords: Brain Metabolism, Cardiac Metabolism, Ischemia, Metabolism, Post Translational Modification, Protein Chemical Modification, Protein Cross-linking, Skeletal Muscle
Abstract
Oxidation of unsaturated lipids generates reactive aldehydes that accumulate in tissues during inflammation, ischemia, or aging. These aldehydes form covalent adducts with histidine-containing dipeptides such as carnosine and anserine, which are present in high concentration in skeletal muscle, heart, and brain. The metabolic pathways involved in the detoxification and elimination of these conjugates are, however, poorly defined, and their significance in regulating oxidative stress is unclear. Here we report that conjugates of carnosine with aldehydes such as acrolein are produced during normal metabolism and excreted in the urine of mice and adult human non-smokers as carnosine-propanols. Our studies show that the reduction of carnosine-propanals is catalyzed by the enzyme aldose reductase (AR). Carnosine-propanals were converted to carnosine-propanols in the lysates of heart, skeletal muscle, and brain tissue from wild-type (WT) but not AR-null mice. In comparison with WT mice, the urinary excretion of carnosine-propanols was decreased in AR-null mice. Carnosine-propanals formed covalent adducts with nucleophilic amino acids leading to the generation of carnosinylated proteins. Deletion of AR increased the abundance of proteins bound to carnosine in skeletal muscle, brain, and heart of aged mice and promoted the accumulation of carnosinylated proteins in hearts subjected to global ischemia ex vivo. Perfusion with carnosine promoted post-ischemic functional recovery in WT but not in AR-null mouse hearts. Collectively, these findings reveal a previously unknown metabolic pathway for the removal of carnosine-propanal conjugates and suggest a new role of AR as a critical regulator of protein carnosinylation and carnosine-mediated tissue protection.
Introduction
Short-chain aliphatic aldehydes are generated by several biochemical reactions such as the peroxidation of unsaturated fatty acids and phospholipids, glycation of proteins, and oxidation of amino acids (1, 2). Some of these aldehydes, e.g. 4-hydroxy-trans-2-nonenal (HNE),2 malondialdehyde, methylglyoxal, and acrolein are highly reactive (3) and cytotoxic (4). These aldehydes react readily with cellular nucleophiles such as glutathione, histidine, lysine, or arginine residues in proteins and guanosine bases in DNA. Previous studies have shown that aldehyde-modified proteins accumulate in diseased tissue during atherosclerosis (5, 6), myocardial ischemia (7, 8), arteritis (9), diabetes, and Alzheimer (10) and Parkinson (11, 12). Increased accumulation of aldehyde-modified DNA bases has also been associated with oxidative stress (13–15). Although the presence of aldehydes and aldehyde adducts is indicative of oxidative stress, it is unclear whether these aldehydes mediate and amplify the injurious effects of oxidative stress.
Because aldehydes are highly reactive, they are rapidly metabolized. In most cells aldehydes are either oxidized by aldehyde dehydrogenases or reduced by aldehyde reductases (16). In addition, unsaturated aldehydes such as acrolein and HNE form covalent conjugates with reduced glutathione (GSH). This conjugative reaction is further enhanced by glutathione S-transferases. Our previous studies have shown that the glutathione conjugates of aldehydes are further metabolized by a reductive transformation catalyzed by aldose reductase (AR) (17, 18) and that inhibition of this metabolic transformation increases aldehyde accumulation and exacerbates ischemic injury (19) and atherosclerotic lesion formation (6). These observations support the view that reduction diminishes the ability of aldehyde-glutathione conjugates to induce tissue toxicity.
In addition to glutathione, aldehydes also form stable conjugates with carnosine (20–22). Carnosine (histidyl-β-alanine) is an endogenous dipeptide. It is synthesized in tissues with high rates of glycolysis such as the anaerobic white skeletal muscle of birds, horses, and pigs. It is currently believed that during high anaerobic activity carnosine prevents tissue acidification by buffering changes in intracellular pH (23). Nevertheless, high levels of carnosine and related histidyl peptides (anserine, homocarnosine) are also present in aerobic tissues such as the heart and the brain, suggesting that carnosine might have other functions as well. Previous studies have shown that carnosine protects against ischemic injury (24–26), hyperglycemia (27), neurodegenerative diseases (28), and aging (29, 30). Collectively, these findings indicate that in addition to buffering intracellular pH, carnosine might have an antioxidant role in removing aldehydes (31). However, the metabolic fate of aldehyde-carnosine conjugates is unknown, and it is unclear whether, like glutathione-aldehyde conjugates, the carnosine-aldehyde conjugates are also enzymatically reduced and whether this metabolism regulates the antioxidant effects of carnosine. Accordingly, the current study was designed to elucidate the metabolism of carnosine-aldehyde conjugates. Our results show that carnosine-derived conjugates are major urinary products of unsaturated aldehydes such as acrolein and that these conjugates are reduced by AR before excretion in urine. Moreover, we found that enzymatic reduction by AR prevents these conjugates from modifying proteins and inducing tissue injury.
EXPERIMENTAL PROCEDURES
Materials
Carnosine, dl-glyceraldehyde, dithiothreitol (DTT), NADPH, EDTA, tyrosine-histidine, homocarnosine, anserine, hydralazine, pyridoxamine, aminoguanidine, and nonafluoropentanoic acid were obtained from Sigma. Anti-protein-acrolein antibody was purchased from U. S. Biological. The anti-protein-carnosine antibody was raised against carnosine conjugated to bovine serum albumin (BSA) that was a kind gift from Dr. Frank Margolis, University of Maryland. Synthetic model peptides Ac-RVAAKH, Ac-RVCAKH, and Ac-MCAAR and the chemical analogues of carnosine, octyl-d-carnosine, were purchased from AAPPTec, LLC (Louisville, KY). Enhanced chemiluminescence (ECL Plus) reagents and horseradish peroxidase-linked secondary anti-mouse and anti-rabbit antibodies were obtained from Amersham Biosciences. All other reagents were of analytical grade.
Animals
Adult male C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The AR-null mice were obtained from Dr. Stephen S. Chung, University of Hong Kong. Mice were treated in accordance with the Declaration of Helsinki and with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) as adopted and promulgated by the National Institutes of Health. Treatment protocols were approved by the University of Louisville Institutional Animal Care and Use Committee.
Preparation and Purification of Conjugates
For all the experiments, acrolein was freshly prepared by the acid hydrolysis of diethyl acetal (pH 3.0) for 1 h at room temperature. To synthesize conjugates, acrolein was incubated with an equimolar concentration of carnosine, anserine, homocarnosine, aminoguanidine, hydralazine, or pyridoxamine in 10 mm KH2PO4 (pH 7.0) at 37 °C for 3–24 h and purified by HPLC as described previously (32, 33). Briefly, the conjugates were separated using a C18 column (250 × 4.6 mm; particle size 5 μm) with a Waters (Milford, MA) 1525 Binary HPLC equipped with a Waters 2487 Dual wavelength detector. The conjugates were eluted using a gradient consisting of 100% water containing 5 mm nonafluoropentanoic acid (solvent A) and 100% acetonitrile (solvent B) at a flow rate of 1 ml/min. The gradient was established such that solvent B reached 15% in 10 min and was then held at this value for 50 min. The absorbance of the eluate was measured at 210 and 234 nm.
Mass Spectrometry
The conjugates of carnosine were analyzed using a Micromass ZMD mass spectrometer (Manchester UK) in the positive ion mode. The spectrometer was calibrated using NaCsI with the calibration routine included in the MassLynx 3.4 software. Samples were diluted in 70% water, 30% acetonitrile, and the solution was infused into the mass spectrometer using a glass syringe and a Harvard infusion pump at a rate of 10 μl/min. Tuning conditions were as follows: capillary 2.9 kV, cone 34 V, extractor 9 V, Rf lens 0.9 V, source temperature 100 °C, desolvation gas 200 °C, low mass resolution 15 relative setting, high mass resolution 15.2, ion energy 0.3 V, multiplier 650 relative setting. For accurate mass data, HPLC-purified fractions were minimally diluted with 70% water, 30% acetonitrile and analyzed using a Thermo Scientific LTQ XL mass spectrometer coupled with a LTQ Orbitrap XL mass spectrometer. Samples were analyzed by chip-based electrospray ionization using an Advion (Ithaca, NY) TriVersa NanoMate under the control of CHIP SOFT instrument control software. The ionization parameters were as follows: sample volume 6 μl, spray duration 3.5 min, N2 pressure 0.50 p.s.i., voltage 1.6 kV, and positive polarity.
Measurement of Urinary Mercapturic Acids
The 3-hydroxypropyl mercapturic acid (HPMA) conjugate in urine was measured as described previously (34). Briefly, the human urine was spiked with internal standard [13C3]HPMA and applied to solid-phase extraction cartridge. Fractions containing HPMA conjugates were separated on an HPLC column fitted with a UV detector. HPMA was detected after derivatization with bistrimethylsilytrifluoroacetamide for 1 h at 70 °C by GC/MS (Agilent Technologies). Ions of m/z 366 (endogenous) and 369 ([13C3]HPMA, internal standard) were monitored for measuring HPMA.
LC-MS/MS Analysis of Carnosinylated Proteins
Proteins immunoprecipitated by anti-carnosine antibody from ischemic hearts were separated on SDS-PAGE and identified by silver staining. Individual protein bands were excised and de-stained with 15 mm potassium ferricyanide, 50 mm sodium thiosulfate in water. The gel pieces were then washed twice with water and cut in half. One half of the band was digested with 60 ng of trypsin (Promega) as described previously (35); the other half of each band was digested with 60 ng of Arg-C (Sigma) in 50 mm triethylammonium bicarbonate. After digestion, peptides were sequentially extracted from the gel pieces using 5% v/v formic acid and acetonitrile (36). The extracts were dried, redissolved in 25 μl of 2% acetonitrile, 0.1% formic acid and filtered through 0.2-μm cellulose syringe filters.
The proteolytic digests were separated by a linear 30-min gradient (2–60% acetonitrile) using a Proxeon Easy nLC-1000 UPLC fitted with an Acclaim PepMap 100 (75 μm × 2 cm, nanoViper; C18, 3 μm, 100 Å) trap and Acclaim PepMap RSLC (50 μm × 15 cm, nanoViper; C18, 2 μm, 100 Å) separating column before MS data collection using a Thermo Scientific LTQ-Orbitrap ELITE hybrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) interfaced with a nanospray FLEX ion source operated in a data-dependent manner. MS data were collected on the Orbitrap ELITE with the MS scan from 300 to 2000 m/z in FTMS and a normal mass range with 120,000 resolution, full scan, positive polarity, and centroided data. The chromatography option was enabled on MS scans with an expected peak width at full width half mass (FWHM) of 10 s and a minimum signal threshold of 1000; the correlation option was used with a maximum area ratio to previous scan of one. Monoisotopic precursor selection was enabled. For MS2 data collection, dynamic exclusion was used with a repeat count of 1, 30-s repeat duration, exclusion list size of 50, and exclusion duration of 180 s; exclusion was +1.5 by mass. The “Predict ion injection time” and “Enable preview mode for FTMS master scans” options were enabled. Dependent MS2 scans were collected on the top three peaks of the MS scan with both high energy collision dissociation (HCD) and electron-transfer dissociation (ETD) fragmentation. The three HCD scans were acquired before the three ETD scans. The MS2 scans were acquired in FTMS with normal mass range, 60,000 resolution, and centroid data type. For collection of HCD data, the following instrument settings were used: normalized collision energy of 35, activation time of 0.1 ms, isolation width 2.0 m/z, default charge state of 2, minimum signal threshold for fragmentation of 500, and first mass in spectrum fixed at 100 m/z. For collection of ETD MS2 data, the activation time was 100 ms, isolation width 2.0 m/z, default charge state 2, minimum signal threshold for fragmentation 500. The first mass in spectrum was fixed at 100 m/z.
The MS2 data were analyzed by the Proteome Discoverer (Version 1.3.0.339). Spectra were filtered by mass range (350–5000 Da), total intensity threshold (500 ion counts), and minimum peak counts (3). Scan event filters were used to define HCD and ETD spectra. The Fourier transform-only spectra were filtered with a signal/noise ratio threshold of 4. Non-fragment filters were used to clip precursor spectra from 2.5 Da before and 5.5 Da after precursor masses. Non-fragment filters were used to remove charge reduced precursors and to remove FT-precursor overtones. All spectra were normalized, and a Top N Peaks filter of 10 within a 100-Da mass window was used to simplify data submitted for analysis. For Mascot analysis a SwissProt database selected for the Mus musculus taxanomy was used with Arg-C or trypsin enzyme designation, precursor mass tolerance of 10 ppm, fragment mass tolerance of 0.05 Da, and a peptide ion score cutoff of 10 was used to filter peptide assignments. Dynamic modifications to amino acids were selected for three adducts including C12H18N4O4 (+264.122 Da), C15H20N4O4 (+302.138Da), and C15H22N4O5 (+320.148 Da). All assigned peptide spectra were filtered for false discovery rates of 0.01 using Peptide Validator and again using a decoy database. For Sequest analysis, a mouse refseq database (Version 20120611.fasta) was downloaded from UniProt, and using either Arg-C or trypsin enzymes, 500 maximum peptides were considered with a maximum peptide output of 10. The absolute XCorr threshold was set to 0.4, and fragment ion cutoff at 0.1%. All other settings were similar to Mascot searches. Protein identification was accepted if it could be established at >95.0% probability and contained at least 1 or more identified peptides below the 1% false discovery rate. Data were aggregated and used to make lists of high probability/high confidence peptide/protein assignments.
Carnosinylation of Proteins and Peptides
To generate carnosinylated protein, purified carnosine-propanal conjugates were incubated with 1 mg of fat-free BSA at 37 °C for 24 h. After incubation, the samples were filtered through 3000 Mr cutoff filters, and the carnosinylated proteins were visualized on Western blots developed using anti-protein-carnosine and anti-propanal antibodies. For studying the reactivity of carnosine-propanals, the synthetic model peptide Ac-RVCAKH (10 nmol) was dissolved in 25 μl of 100 mm NH4HCO3 (pH 7.4) containing 1 mm DTT. The reaction mixture was incubated at 70 °C for 30 min. The samples were then cooled and incubated with 3 mm purified carnosine-propanals or carnosine-propanol for indicated times. The products were then identified by electrospray ionization/mass spectrometry (ESI+/MS).
Global Ischemia-Reperfusion ex Vivo
After anesthesia (sodium pentobarbital, 60 mg/kg body weight; heparin, 10 units/g body weight), the thorax of mice (8–12 weeks of age) was opened, and the hearts were removed and immediately placed in ice-cold modified Krebs-Henseleit buffer. The composition of the Krebs-Henseleit buffer was 118 mm NaCl, 4.7 mm KCl, 1.25 mm MgCl2, 2.5 mm CaCl2, 1.25 mm KH2PO4, 25 mm NaHCO3, and 11 mm glucose (pH 7.4). The buffer was heated to 37 °C and continuously gassed (95% O2, 5% CO2). Hearts were continuously perfused at a constant pressure of 80 mm Hg in the Langendorff mode and after 30 min of perfusion subjected to 30 min of ischemia and 45 min of reperfusion in the absence or presence of carnosine (1 mm). After the experimental protocol, the hearts were removed and snap-frozen in liquid N2. Functional recovery of the hearts was measured by placing a food-grade plastic wrap, fluid-filled balloon in the left ventricle through an incision in the left atrium and inflating the balloon to a pressure of 5–8 mm Hg. Left ventricular pressure was recorded using an APT300 pressure transducer (Harvard Apparatus, Holliston, MA) connected to a ML221 bridge amplifier, a PowerLab 16/30 A/D board, and a PC running LabChart Pro v7 (ADInstruments, Colorado Springs, CO). Perfusion flow rate was monitored using a 1PXN inline probe and TS410 flowmeter (Transonic Systems Inc., Ithaca, NY) and was typically between 1.5 and 2 ml/min. Left ventricle developed pressure was derived from the pressure trace as the difference between systolic and diastolic pressures. Perfusates were collected on ice, and the levels of creatinine kinase (CK) and lactate dehydrogenase were measured using kits purchased from Thermo-Electron. In a separate series of experiments, WT hearts were perfused 15 min and then perfused another 10 min without or with acrolein (10 μm). Hearts were removed and immediately snap-frozen.
Immunoprecipitation and Western Blot Analysis
Tissues were homogenized in 10 mm HEPES (pH 7.2) containing 0.5 mm EDTA, 0.5 mm EGTA, 10 mm NaF, 10 mm Na3VO4, 1% Nonidet P-40, 0.1% SDS containing a protease and phosphatase inhibitor mixture (1:100). The homogenates were centrifuged for 25 min at 13,000 × g, and the supernatants were incubated with the anti-protein-carnosine antibody overnight and then with agarose A beads for 3 h at 4 °C. Immunoabsorbent proteins were recovered by centrifugation at 1000 × g and washed with cell lysis buffer. Proteins were separated by SDS-PAGE and then transferred onto PVDF membranes. Western blots were developed using anti-protein-carnosine or anti-protein-propanal antibodies and the ECL Plus reagent (Amersham Biosciences) and detected with a Typhoon 9400 variable mode imager (Amersham Biosciences). Band intensity was quantified by using Image Quant TL software (Amersham Biosciences), and bands were normalized to actin or Amido Black staining as appropriate. For quantification, individual bands were selected, and the background intensity was subtracted from the band intensity using rolling-ball averaging. Western blots shown are representative of at least n = 3–4 samples.
Measurement of AR Activity
The human recombinant AR protein was generated as described previously (17). For measuring enzyme activity, the protein was reduced with 100 mm DTT for 1 h at 37 °C in 100 mm Tris-Cl (pH 8.0). The catalytic activity was measured in 150 mm potassium phosphate (pH 6.0) containing 0.15 mm NADPH and substrate (1 mm carnosine-aldehyde conjugates). To measure AR activity, hearts were rapidly snap-frozen using clamps precooled in liquid N2. Other tissues extracted were frozen in liquid N2. Tissue aliquots were thawed and homogenized in 3 volumes (30% v/v) of 150 mm KH2PO4 (pH 6.0) containing 1 mm EDTA, 1 mm DTT, and the protease inhibitor mix (1:100). The homogenates were centrifuged at 13,000 × g for 20 min, and the supernatant containing 1–2 mg of protein (0.1 ml tissue homogenate) was incubated with 1 mm carnosine-propanal conjugates. Protease inhibitors and NADPH (0.5–1.0 mm) were added to the mixture at regular time intervals, and the mixture was incubated at 37 °C in 150 mm potassium phosphate buffer (pH 6.0). Aliquots were withdrawn after 2, 6, and 24 h to measure conjugate reduction by ESI+/MS. Control samples contained all the components of the reaction mixture except NADPH.
Analysis of Urinary Metabolites
Adult (8–10 weeks old) WT and AR-null mice were housed in metabolic cages, and the urine was collected for 24 h. In some experiments, both the WT and AR-null mice were administered a single dose of octyl-d-carnosine (5 μmol) by oral gavage, and the urine collected was stored at −20 °C. During this time mice had free access to water and food. For Orbitrap analysis, urine samples were spiked with the internal standard tyrosine-histidine or histidine-[13C]propanal and deproteinized by the addition of 15 μl perchloric acid (70%) to 500 μl of the sample. After 15 min at 4 °C, the samples were centrifuged at 21,000 × g for 10 min. Urine samples collected from normal, healthy, non-smoking human volunteers were analyzed using the protocol described above. Cotinine concentrations in human urine samples were determined by a monoclonal antibody based ELISA kit (International Diagnostics). Urinary metabolites were measured by mass spectrometry as described above. The collection of urine from humans was approved by the University of Louisville, Institutional Review Board (IRB #10.0350).
Statistical Analysis
In all cases best fits to the data were chosen on the basis of the S.E. of the fitted parameters. All data are expressed as the mean ± S.E. Data were analyzed by one-way analysis of variance for multiple comparisons and by Student's t test for unpaired data. Statistical significance was accepted at p < 0.05.
RESULTS
Carnosine-Aldehyde Metabolites in Human Urine
Previous studies have shown that carnosine forms stable conjugates with unsaturated aldehydes such as HNE and acrolein (20–22). These aldehydes are generated upon oxidation of unsaturated lipids. In addition, acrolein is generated during the metabolism of biogenic amines (37) or by the oxidation of amino acids by myeloperoxidase (38). Acrolein is also present in a variety of food products and in cotton, wood, and tobacco smoke as well as automobile exhaust (39). To determine the role of carnosine in the systemic metabolism of these aldehydes, samples of urine from normal, healthy, non-smoking adult humans were analyzed. Exposure to tobacco smoke was estimated from measurements of urinary cotinine. Cotinine was not detected in any urine samples. Individual samples were separated by HPLC and analyzed by high precision mass spectrometry using methods optimized for the collection and identification of the aldehyde conjugates of carnosine. Because carnosine could be hydrolyzed to histidine and β-alanine, some of the aldehyde-carnosine conjugates could be excreted as histidyl conjugates (40). Therefore, histidine conjugates of HNE and acrolein were measured as well. On the basis of exact mass (<0.001 m/z difference from reagent standards), several ions in the urine could be assigned to carnosine or histidine conjugates of HNE and acrolein (Fig. 1A). In addition, corresponding reduced metabolites with m/z +2 were also detected. The most abundant reduced metabolites were carnosine-propanol (m/z 285), histidine propanal (m/z 212, His-propanal), and histidine-propanol (m/z 214, His-propanol) (Fig. 1B). These metabolites are likely to be derived from the hydrolysis of carnosine-propanal and/or enzymatic reduction of histidine conjugates. Reduced metabolites were present in high abundance (36% of the histidine and 75% of the carnosine conjugates of acrolein were in the reduced form), indicating that reduction is an important transformative step in the metabolism of these conjugates. Moreover, the average level of carnosine-acrolein adducts (carnosine propanal + carnosine propanol) was 266 pmol/mg of creatinine, which is between 0.2 and 5% of carnosine in human urine (5–125 nmol/mg of creatinine) (34), indicating that a significant proportion of carnosine in humans is utilized for acrolein metabolism.
FIGURE 1.

Histidine and carnosine conjugates in human urine. A, Orbitrap mass spectra of human urine samples collected from healthy, non-smoking adults. The urine samples were de-proteinized, and the carnosine and histidine conjugates were separated by HPLC as described under “Experimental Procedures.” The spectra show prominent ions that were assigned to carnosine-propanal and carnosine-propanol (m/z = 283.139 and 285.155) (i), histidine propanal and histidine-propanol (m/z = 212.103 and 214.118) (ii), and histidine-HNE and histidine-DHN (m/z = 312.191 and 314.207), respectively (iii). B, the concentrations of urinary conjugates calculated using tyrosine-histidine (m/z 319) as an internal standard. Data are the mean ± S.E. (n = 7).
In addition to carnosine, unsaturated aldehydes are also metabolized via the glutathione-linked pathway (41, 42). Glutathione conjugates of aldehydes are excreted as reduced or non-reduced mercapturic acids. In case of acrolein, the most abundant glutathione-linked metabolite is the reduced HPMA (42). Therefore, to compare the relative contribution of carnosine and glutathione to acrolein metabolism, urinary HPMA levels were measured. These measurements indicate that the average concentration of HPMA in urine was 695 pmol/mg of creatine (Fig. 1B), which is much higher than the concentration of the carnosine conjugates (266 pmol/mg creatinine). Thus the carnosine conjugates represent 27% of the total conjugates (HPMA + carnosine conjugates) measured in the urine. Because HPMA represents only 60% of the total acrolein metabolism (43, 44), these measurements suggest that ∼15% of acrolein is metabolized via the carnosine-linked pathway. In comparison with acrolein conjugates, HNE conjugates were present at a much lower concentration. On average, the concentration of His-propanal in human urine was 60–75-fold higher than His-HNE. Unhydrolyzed conjugates of carnosine with HNE (e.g. carnosine-HNE and carnosine-DHN) were also detected but only at trace levels. Taken together, these results indicate that in humans, aldehyde conjugates of carnosine and histidine, generated during normal metabolism, are excreted in urine. In the case of acrolein, conjugation with carnosine represents a significant route of its overall metabolism, and a high proportion of the conjugate is excreted as a reduced metabolite. The appearance of predominantly reduced metabolites in urine suggests that during metabolism carnosine and histidine conjugates of acrolein are enzymatically converted to their corresponding alcohols.
Enzymatic Reduction of Propanal Conjugates
To identify the metabolic pathway that reduces carnosine-aldehyde conjugates, we examined the role of AR, an enzyme that catalyzes the reduction of S-linked glutathione-aldehyde conjugates (45). Because carnosine-propanol was the most abundant conjugate of its class in human urine, the properties and the reductive transformation of acrolein-derived conjugates of carnosine were studied in detail. The ESI+/MS spectra of reagent carnosine-propanal showed five major ions with m/z values of 265, 283, 301, 321, and 339 (Fig. 2A). The ion with m/z 265 is likely due to the formation of an imine formed by the Michael addition of acrolein to the histidine residue of carnosine and a Schiff base between carbonyl and primary amine of glycine. The ion with m/z 301 was ascribed to a geminal diol [M+H2O]+ of the 283 m/z ion. To determine the structure of the other major ions with m/z 283, 321, and 339, their fragmentation pattern was obtained by collision-induced dissociation (CID). The ion with m/z 283 fragmented into daughter ions with m/z values of 166 [MH- 3-oxobutan-1-aminium + acetic acid]+, 212 [MH-3-oxobutan-1-aminium]+, or [MH-formidamide+acetic acid], and 266 [MH-NH3]+ (Fig. 2B). On the basis of this pattern, this ion was assigned to a Michael adduct between carnosine and acrolein either at the imidazole nitrogen of histidine (to form β-Ala-His-propanal) or the amino group of β-alanine (to generate propanal-β-Ala-His). The ion of m/z 321 fragmented into 3 major daughter ions with m/z values of 166 [MH-His]+, 204 [M-4-(2-amino-2-carboxyethyl)-1H-imidazol-3-ium], and 232 [MH-(Z)-N-(methylamino) methylene) methanamium + acetic acid] (Fig. 2C), indicating that it was FDP-carnosine as previously identified. This ion could also be formed by the addition of acrolein to the ion with m/z 265, giving rise to propanal cycloamine. With the ion of m/z 339, two fragments with m/z 321 [MH-H2O]+ and 283 [MH-Acr]+, were detected (Fig. 2D), indicating that it was a conjugate formed by Michael addition of 2 acrolein molecules either to β-alanine, giving rise to dipropanal- β-Ala-His, or by the addition of one acrolein to histidine and another to β-alanine (propanal- β-Ala-His-propanal). The structures of these adducts are shown in Fig. 2, and their exact chemical names are listed in Table 1. Similar adducts were generated upon incubation of acrolein with anserine or homocarnosine (data not shown). Collectively, these results indicate that carnosine and related histidyl peptides form several stable and structurally distinct conjugates with acrolein. Moreover, several of these conjugates contain a free formyl group at the N or the C terminus or both or as a dihydropipyridine substituent (FDP-carnosine).
FIGURE 2.
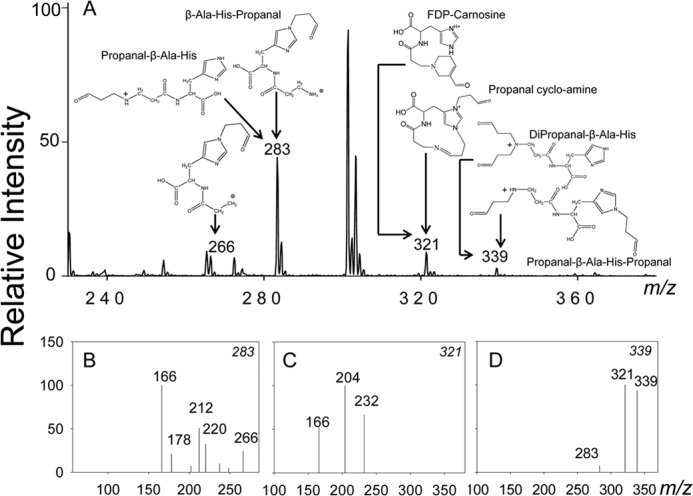
Identification of the carnosine conjugates of acrolein. A, ESI+/MS spectra of carnosine-propanal conjugates generated by incubating acrolein with carnosine as described under “Experimental Procedures.” Putative structures of the conjugates identified by mass spectrometry are shown in the figure for each m/z value. The fragmentation patterns of the major ions with m/z values of 283 (B), 321(C), and 339 (D) are shown.
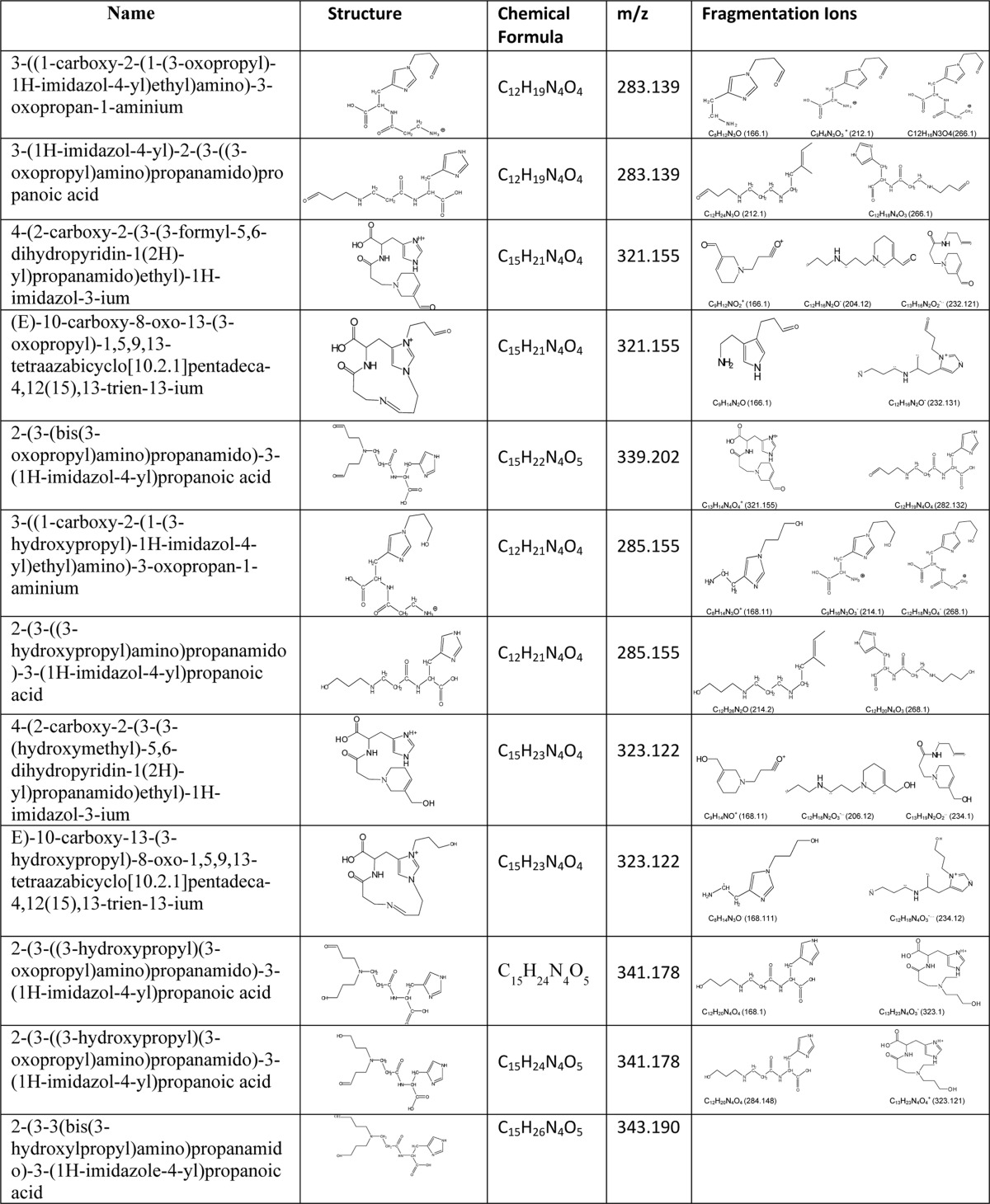
TABLE 1.
Structures and fragmentation ions of carnosine-propanal and propanol conjugates
To determine whether these conjugates are reduced by AR, carnosine-propanal conjugates were incubated with recombinant human AR and NADPH. After incubation, the reaction was stopped, and the constituents of the mixture were separated by HPLC and analyzed by mass spectrometry. The ESI+/MS spectrum of HPLC-purified reaction products showed several ions with m/z values of 285, 323, 341, and 343 (Fig. 3A). These ions appear to be derived from the reduction of ions with m/z values of 283, 321, and 339 (as in Fig. 2A). The ion with m/z 285 was fragmented into ions of m/z 168 [MH-3-oxobutan-1-aminium+acetic acid]+, 214 [MH 3-oxobutan-1-aminium]+, and 268 [M-17]+ (Fig. 3B). This fragmentation pattern suggests that the ions could be due to the reduction of propanal-β-Ala-His or His-β-Ala-propanal. The ion with an m/z value of 323 generated a characteristic daughter ion of m/z 168 [MH-His]+, indicating that it was formed by the reduction of FDP-carnosine (Fig. 3C). Similarly, on the basis of an increased m/z value of 2, the ion with m/z 341 (Fig. 3D) was ascribed to carnosine semi-propanal generated from the reduction of one of the free formyl groups of propanal-β-Ala-His-propanal or dipropanal-β-Ala-His. A low-intensity ion with m/z 343 was also detected that could arise from the reduction of both the formyl groups of propanal-β-Ala-His-propanal or dipropanal-β-Ala-His; however, due to its low abundance, this ion could not be characterized further. No significant reduction of the carnosine-HNE conjugate by AR was observed, although trace level reduction of His-HNE to His-DHN was detected (data not shown). Taken together, these data indicate that AR catalyzes the reduction of several carnosine-propanal conjugates to their corresponding alcohols.
FIGURE 3.
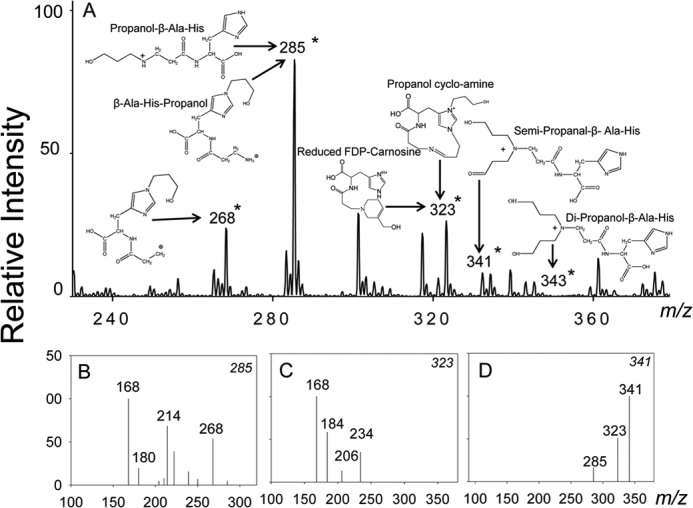
Aldose reductase catalyzes the reduction of carnosine-propanal conjugates. A, the ESI+/MS spectra of carnosine-propanal conjugates after incubation with AR. The carnosine-propanal conjugates were isolated by HPLC and incubated with recombinant AR and NADPH for 18–24 h. The ions generated in the presence of AR are indicated by an asterisk (*). Fragmentation patterns of the major ions with m/z values of 285 (B), 323 (C), and 341 (D) are shown.
To determine whether the conjugates of other histidyl peptides are also reduced by AR, propanal conjugates of anserine, homocarnosine, pyridoxamine, hydralazine, and aminoguanidine were synthesized. The identity of each of these conjugates was established by ESI+/MS (data not shown). HPLC-purified fractions containing individual conjugates were incubated with AR and NADPH. As shown in Table 2, appreciable reductive activity was observed with most of the conjugates tested. The highest activity was observed with carnosine-propanal (m/z 283), which was similar to that obtained with dl-glyceraldehyde. Progressively lower activity was observed with homocarnosine-propanal and pyridoxamine-propanal. Weak catalytic activity was also detected with other N-linked conjugates, and no activity was obtained with the FDP-homocarnosine conjugate (m/z 335) or with carnosine-HNE. These results indicate that AR catalyzes the reduction of several N-linked conjugates, albeit with variable efficiency.
TABLE 2.
Catalytic activity of aldose reductase with propanal conjugates
The enzyme activity was measured at the substrate concentration of 1 mm and is expressed as nmol of NADPH oxidized/min/mg of protein. Values are the mean ± S. E. NA: no activity.
| Substrate | m/z | Activity |
|---|---|---|
| Glyceraldehyde | 124 ± 6 | |
| Carnosine-propanal (β-Ala-His-propanal or propanal-β-Ala-His) | 283 | 113 ± 10 |
| FDP-carnosine or propanal-cycloimine | 321 | 20 ± 2 |
| Anserine-propanal (β-Ala-Me-His-propanal or propanal-β-Ala-Me-His) | 297 | 21 ± 1 |
| FDP-anserine or propanal-Me-cycloimine | 335 | 23 ± 4 |
| Homocarnosine-propanal (Me-β-Ala-His-propanal or propanal-Me-Ala-His) | 297 | 79 ± 7 |
| FDP-Homocarnosine or propanal-cyclo-methylimine | 335 (NA) | |
| Pyridoxamine propanal | 225 | 140 ± 9 |
| FDP-pyridoxamine | 263 | 35 ± 2 |
| Hydralazine-propanal | 273 | 13 ± 1 |
| FDP-hydralazine | 199 | 10 ± 1 |
| Aminoguanidine-propanal | 169 | 10 ± 2 |
| FDP-Aminoguanidine | 187 | 16 ± 1 |
Reduction of Carnosine-propanal Conjugates in Tissues
Because the data obtained so far indicate that AR catalyzes the reduction of carnosine-propanals and that carnosine-propanols were present in urine, additional experiments were designed to investigate whether these conjugates are generated endogenously by AR. For these experiments homogenates were prepared from the skeletal muscle of WT and AR-null mice and incubated with acrolein for 24 h without dialysis to prevent the loss of glutathione and carnosine. The conjugates generated in the homogenate were isolated and purified by HPLC and identified by ESI+/MS. Detectable quantities of carnosine-propanal conjugates were found in the skeletal muscle; however, the concentration of these conjugates was increased upon incubation of the homogenates with acrolein (Fig. 4). Notably, higher levels of carnosine-propanols were generated in WT than in the AR-null skeletal muscle lysates (Fig. 4B). These observations suggest that in tissue extracts, carnosine forms stable conjugates with acrolein, and that these conjugates are reduced by AR. To examine the time course of reduction, carnosine-propanal conjugates were incubated with WT and AR-null skeletal muscle lysates for 2, 6, and 24 h. Reduction of carnosine-propanal conjugates (m/z 283 and m/z 321) increased 2-fold after 6 h compared with 2 h of incubation (Fig. 5, C and D); however, no further increase in reductive activity was observed after 24 h compared with 2 h of incubation. No reduction of carnosine-propanal conjugates was observed in AR-null skeletal muscle lysates incubated with carnosine propanal conjugates (Fig. 5, C and D).
FIGURE 4.

Metabolic conversion of carnosine-propanal to carnosine-propanol via aldose reductase. A, ESI+/MS spectra of the skeletal muscle homogenates (2–4 mg of protein) from WT (solid line) and AR-null (dotted line) mice incubated with acrolein for 18–24 h. B, the relative abundance of reduced versus non-reduced carnosine conjugates calculated as intensity ratio (reduced/non-reduced) in lysates of WT and AR-null skeletal muscle. Values are the mean ± S.E.; n = 3; *, p < 0.05, WT versus AR-null.
FIGURE 5.

Reduction of carnosine-propanal conjugates in murine tissues. A, ESI+/MS spectra of tissue lysates prepared from mouse heart incubated with NADPH and carnosine-propanal conjugates. Tissue lysates (1–2 mg of protein) from WT (i and ii) AR-null (iii and iv) and FR-1-transgenic (FR-1-TG; v and vi) mice were incubated with HPLC-purified carnosine-propanal conjugates in the presence (solid line) and the absence (dotted line) of NADPH. B, Orbitrap analysis of WT (i and ii) and AR-null (iii and iv) heart homogenates. The ratio of ion intensities at m/z 285/283 and 323/321 are shown at different time intervals in lysates prepared from WT and AR-null skeletal muscle (C and D), WT AR-null and FR-1-transgenic hearts (E) and WT and AR-null brains (F). Data are the mean ± S.E.; n = 4; *, p < 0.05 versus WT + carnosine-propanal; #, p < 0.05 versus WT + carnosine-propanal + NADPH; $, p < 0.05 versus WT + carnosine-propanal + NADPH (2 h).
To determine whether carnosine conjugates are also reduced in tissues other than the skeletal muscle, carnosine-propanals were incubated with homogenates of mouse brain and heart. Although skeletal muscle contains the highest levels of carnosine (10–20 mm), significant levels of carnosine have been also been measured both in the heart and the brain (23). Therefore, in these tissues, carnosine-propanal conjugates could be generated and reduced by AR. To test this, lysates from the brain and heart of WT and AR-null mice were prepared and incubated with carnosine-propanals and NADPH. This led to an increase in peak intensity of the ions with m/z values of 285 and 323 (Fig. 5A, i and ii), which as before were assigned to carnosine-propanols. Significantly, the intensity of these ions was lower in homogenates of AR-null than in WT hearts (Fig 5A, iii and iv).
Because the levels of the conjugates were low and their mass spectra could be contaminated by noise or other co-eluting species, reduction of carnosine-propanals was examined using high precision Orbitrap mass spectrometry. This analysis revealed that although carnosine-propanols (m/z 285.1554 and 323.1112) were present in WT cardiac homogenates (Fig. 5, B, i and ii), they were not detected in AR-null homogenates (Fig. 5, B, iii and iv). Thus, the weak peak intensity in ESI+/MS analysis at m/z 285 in the AR-null group could not be due to carnosine-propanal. Similarly, even though a distinct molecular species of m/z 323.1334 was observed in AR-null heart (Fig. 5Biv), the m/z value was sufficiently different to preclude its identification as carnosine-propanol (m/z 323.1112). These analyses indicate that carnosine-propanols are not generated in AR-null hearts. Similarly, no reduction of carnosine-propanals was observed in AR-null heart (Fig. 5E) or brain (Fig. 5F) extracts.
In addition to AR, murine tissues also express the enzyme, FR-1 (46). FR-1 displays high sequence homology with AR, and like AR it catalyzes the reduction of unsaturated aldehydes and their glutathione conjugates (47). To determine whether FR-1 also catalyzes the reduction of carnosine-propanals, homogenates of hearts from WT and FR-1-transgenic mice were incubated with the carnosine-propanal adducts. As shown in Fig. 5, A and E, similar levels of carnosine-propanol adducts (m/z 285 and 323) were generated in the FR-1 transgenic and WT hearts, indicating that unlike AR, FR-1 does not catalyze the reduction of carnosine-propanals. These observations further reinforce the view that in the mouse heart AR is the major enzyme that catalyzes the reduction of carnosine-propanals to carnosine-propanols.
Role of AR in the Systemic Metabolism of Carnosine-propanal Conjugates
Because reduction of carnosine-propanals was prevented in AR-null tissues, the role of AR in generating urinary carnosine-propanols was investigated using WT and AR-null mice. In urine collected from WT mice, a distinct molecular ion with m/z = 212.1032 attributable to histidine-propanal was detected (Fig. 6A). The abundance of this metabolite was significantly increased in the urine from AR-null mice (Fig. 6B), indicating that in the absence of AR the excretion of the non-reduced form of the histidine conjugate is increased.
FIGURE 6.

Identification of aldose reductase-generated carnosine metabolites in mouse urine. A, Orbitrap mass spectra of reagent histidine-propanal conjugate (i) and histidine [13C]propanal (ii) and the spectra of the m/z 210–218 region of urine collected from WT (iii) and AR-null (iv) mice. The urine samples were spiked with histidine-[13C]propanal (m/z = 215.1133) as indicated. B, the abundance of endogenous histidine-propanal conjugate was calculated as a ratio of its intensity to that of the internal standard in the urine of WT and AR-null mice. C, Orbitrap mass spectra of urine from WT (i) and AR-null (ii) mice fed octyl-d-carnosine. Mice were fed 50 μmol of octyl-d-carnosine by oral gavage, and urine samples were collected over 24 h, purified by HPLC, and analyzed by Orbitrap mass spectrometer. D, relative abundance of carnosine-propanol was calculated as a ratio of its intensity to that of the internal standard (Tyr-His, m/z 319). Data are the mean ± S.E. *, p < 0.05 versus WT (n = 4–5).
Although histidyl-conjugates are expected to be derived from the hydrolysis of carnosine conjugates, these conjugates could also be generated by the direct reaction of histidine with unsaturated aldehydes or from the hydrolysis of histidine-aldehyde adducts in proteins. Therefore, to determine whether AR participates in the systemic metabolism of carnosine-propanals, WT and AR-null mice were fed octyl-d-carnosine. A d configuration of the peptide bond was used to avoid hydrolysis by carnosinase. In the urine of mice fed octyl-d-carnosine, high levels of carnosine-propanol (m/z = 285.1554) were detected by Orbitrap mass spectrometry (Fig. 6C). The abundance of this adduct was significantly lower in the urine of AR-null than of WT mice (Fig. 6D), indicating that the absence of AR prevents the reduction of carnosine-propanals to carnosine-propanols. These data provide support to the view that during systemic metabolism, carnosine-propanals are enzymatically reduced by AR.
Reduction by AR Regulates Protein Carnosinylation
During enzymatic reduction, the reactive carbonyl function of carnosine-propanals is converted to an alcohol. Because alcohols do not form Schiff bases with amines, this conversion might decrease the chemical reactivity of the molecule. To determine if this may be the case, the relative reactivity of carnosine-propanals and carnosine-propanols with proteins was studied. When incubated with BSA, carnosine-propanals formed stable adducts with BSA. The adducted BSA was recognized by the anti-carnosine antibody, i.e. proteins bound to carnosine or “carnosinylated” proteins (Fig. 7A). The carnosine-propanal-bound protein was also recognized by the anti-protein-propanal antibody. Neither antibody recognized untreated BSA. These observations suggest that the entire carnosine-propanal conjugate was bound to the protein.
FIGURE 7.

Formation of protein-carnosine adducts. A, representative Western blots of albumin incubated with the carnosine-propanal. The blots were developed with either the anti-protein-carnosine or the anti-protein-propanal antibodies (Ab). B, ESI+/MS spectra of Ac-RVCAKH before (i) and after (ii) incubation with carnosine-propanal for 30 min or with carnosine-propanol for 60 min (iii) and percent modification of the model peptide by the carnosine-propanal conjugate (iv). C, Western blot of skeletal muscle lysates prepared from WT and AR-null mice incubated with 200–300 μm carnosine-propanal and 1 mm NADPH for 18 h developed using the anti-protein-carnosine antibody (i) and the relative intensity of bands of apparent molecular masses 130 (ii), 45 (iii), and 30 (iv) kDa normalized to total protein in the gel measured by Amido Black staining. Data are the mean ± S.E.; *, p < 0.01 versus WT + carnosine-propanal; #, p < 0.01 versus AR-null + carnosine propanal + NADPH (n = 3–4). D, accumulation of carnosinylated proteins in aged tissue from WT and AR-null mice. Total tissue lysates were prepared from the brain (i), heart (ii), and skeletal muscle (iii), and Western blots were developed using anti-protein-carnosine antibody. The relative intensity in specific immune-positive bands is shown in corresponding bar graphs (iv, v, and vi, respectively). vii, a representative Western blot developed using anti-propanal-protein antibody of skeletal muscle lysates immunoprecipitated (IP) with anti-protein-carnosine antibody. Data are expressed as the mean ± S.E. *, p < 0.01 versus WT tissues (n = 3–4). E, accumulation of carnosine-propanal-protein adducts in WT mouse hearts perfused with 10 μm acrolein (10 min). Blots were developed with anti-protein-carnosine antibody (inset). Data are expressed as mean ± S.E. *, p < 0.01 versus untreated hearts (n = 3–4).
To understand this reaction in greater detail, carnosine-propanals were incubated with a model peptide (Ac-RVCAKH), which contains two nucleophilic residues, cysteine and lysine, in close proximity. The ESI+/MS spectra of the peptide showed a single ion with m/z = 755 (Fig. 7Bi); however, incubation with carnosine-propanals led to the appearance of two new ions with m/z values of 1058 and 1076, respectively (Fig. 7Bii). Increases in m/z values by 303 and 321 suggest that propanal cycloamine or FDP-carnosine (m/z 321) and propanal-β-Ala-His-propanal or dipropanal-β-Ala-His (m/z 339) form dehydrated adducts (Schiff base) with the model peptide. When the reaction time was increased, the intensity of the ion with m/z 1058 or 1076 progressively increased, indicating that the two adducts are generated independent of each other. Approximately, 20–25% of the model peptide (m/z 755) was modified (Fig 7Biv). These results indicate that carnosinylated proteins could be generated by the reaction of the carbonyl groups of carnosine-propanals with nucleophilic residues in peptide side chains. Therefore, only carnosine-propanals, but not carnosine-propanols, should carnosinylate proteins. To test this notion, carnosine-propanals were incubated with AR. The carnosine-propanols generated by AR were isolated, purified, and incubated with Ac-RVCAAKH. As shown in Fig. 7Biii, no carnosinylated peptide was detected when the peptide was incubated with carnosine-propanols. Thus, it appears that only carnosine-propanals can generate carnosinylated protein, whereas carnosine-propanols cannot do so.
To examine the role of AR-catalyzed reduction in preventing protein carnosinylation, additional experiments were designed to determine whether AR prevents the formation of such adducts in tissues. Although Western blot analysis of skeletal muscle extract using the anti-protein-carnosine antibody revealed only faint immunoreactivity with a few proteins, several carnosinylated proteins were detected in the lysates of skeletal muscles that were incubated with carnosine-propanals (Fig. 7Ci). When lysates from WT mice were co-incubated with NADPH, the intensity of several carnosinylated protein bands (e.g. proteins of 30, 45, and 130 kDa) was significantly diminished (Fig. 7C, ii–iv). Notably, co-incubation with NADPH did not decrease protein carnosinylation in AR-null skeletal muscle lysates incubated with carnosine-propanals (Fig. 7C, ii–iv). These data reinforce the view that AR prevents protein-carnosinylation by reducing carnosine-propanals.
To determine whether AR also prevents endogenous carnosinylation of proteins, we measured the abundance of carnosinylated protein in aged tissue from WT and AR-null mice. Tissue aging is associated with an increase in oxidative stress, which could potentially lead to the accumulation of carnosinylated proteins. As shown in Fig. 7D, Western blot analyses of brain, heart, and skeletal muscle from 26-week old mice showed a high abundance of carnosinylated proteins in these tissues. The levels of carnosinylated proteins were higher in AR-null than WT tissues, indicating that AR prevents the endogenous formation of carnosinylated proteins.
Carnosinylated proteins can be generated by several mechanisms. Therefore, to determine whether protein adduction by carnosine might be due to the reaction of carnosine-propanals, carnosinylated proteins were immunoprecipitated from lysates of aged skeletal muscle using the anti-protein-carnosine antibody. The immunoprecipitated proteins were separated by SDS-PAGE, and Western blots were developed using the anti-protein-propanal antibody. As shown in Fig. 7Dvii several, but not all of the proteins immunoprecipitated by the anti-carnosine antibody showed positive immunoreactivity with the anti-protein-propanal antibody. Moreover, perfusion of isolated mouse hearts with acrolein resulted in the appearance of a 45-kDa band that was recognized by the anti-protein-carnosine antibody (Fig. 7E). These results indicate that at least some of the carnosinylated proteins are generated by the binding of carnosine-propanals to proteins or the binding of carnosine to protein-propanal adducts.
Role of AR in Regulating Protein Carnosinylation in the Ischemic Heart
To assess the pathophysiological significance of AR-catalyzed reduction of carnosine-propanal adducts, the effects of carnosine on ischemia-reperfusion injury in WT and AR-null hearts were studied. As shown in Fig. 8A, reperfusion of the ischemic heart led to <20% recovery of developed pressure, indicating significant post-ischemic dysfunction. However, when the WT hearts were perfused with 1 mm carnosine before ischemia, a more pronounced improvement in post-ischemic recovery was observed. Treatment with carnosine also decreased the release of myocardial enzymes (lactate dehydrogenase and CK) (Fig. 8B), indicating that carnosine prevents myocardial cell death precipitated by ischemia-reperfusion. In comparison with WT hearts, the AR-null heart showed slightly poorer recovery of function upon reperfusion. Moreover, pretreatment of AR-null hearts with carnosine neither improved post-ischemic recovery of function nor attenuated CK and lactate dehydrogenase release (Fig. 8). These observations suggest that consistent with previous work (24), carnosine attenuates myocardial ischemia-reperfusion injury but both AR and carnosine are required for the manifestation of the cardioprotective effects of carnosine.
FIGURE 8.

Aldose reductase-dependent protection of the ischemic heart by carnosine. A, changes in developed pressure in isolated mouse hearts subjected to 30 min of ischemia followed by 45 min of reperfusion. In the carnosine-treated group the hearts were continuously perfused with 1 mm carnosine before induction of ischemia and during reperfusion. Data are the mean ± S.E.; n = 4–7 hearts/group; *, p < 0.01 versus WT without carnosine or AR-null hearts with and without carnosine. B, levels of total released lactate dehydrogenase (LDH) (i) and CK (ii) in post-ischemic myocardial effluent collected from WT and AR-null hearts treated with and without carnosine. Data are expressed as the mean ± S.E.; n = 4–7 hearts/group. *, p < 0.01 versus WT without carnosine. C, Western blots from Langendorff-perfused hearts from WT and AR-null mice subjected to either 30 min of perfusion (P) or perfusion followed by 30 min of ischemia (i). At the end of the protocol, proteins from the heart homogenates were separated by SDS-PAGE and immunoblotted with anti-protein-carnosine antibody. Relative intensity of immuno-reactive bands of 45 (ii), 37 (iii), and 30 (iv) kDa was measured and normalized to total protein in the gel measured by Amido Black staining. Data are expressed as the mean ± S.E.; *, p < 0.01 versus perfused hearts; #, p < 0.01 versus WT ischemic hearts (n = 3–4 mice).
In addition to functional responses to carnosine treatment, WT and AR-null heart also differed in the extent of protein carnosinylation. As shown in Fig. 8C, induction of global ischemia in isolated perfused mouse hearts led to an increase in the formation of carnosinylated proteins (e.g. 45 kDa; Fig. 8Ci). This observation indicated that the accumulation of carnosinylated proteins is increased in the ischemic heart. However, in comparison with WT hearts, there was a greater accumulation of carnosinylated proteins in the AR-null hearts, and higher levels of several carnosinylated proteins (e.g. at 30, 37, and 45 kDa) were detected (Fig. 8C, ii–iv). The greater accumulation of carnosinylated proteins in the AR-null versus WT hearts is consistent with the notion that AR prevents the accumulation of carnosinylated proteins in the ischemic heart.
Analysis of Carnosinylated Proteins by LC-MS/MS
To identify which proteins were carnosinylated, proteins from ischemic hearts were immunoprecipitated using the anti-carnosine antibody and analyzed by ThermoScientific LTQ-Orbitrap ELITE hybrid mass spectrometer. Because higher levels of carnosinylated proteins accumulated in the absence of AR, AR-null rather than WT hearts were used for the identification of carnosinylated proteins. SDS-PAGE analysis led to the identification of seven silver-stained bands in the immunoprecipitate. These bands were digested with trypsin and Arg-C proteases without reduction and alkylation. MS analysis identified 36 proteins in this digest (Table 3) that could be localized to the extracellular space, nucleus, mitochondria, and cytoplasm. These included functionally important myocardial proteins such as adenylate kinase, ATP synthase, glyceraldehyde-3-phosphate dehydrogenase, myosin, collagen, and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA), autophagy regulatory proteins, kinases, and peptidases. Collectively, these observations indicate extensive carnosinylation of myocardial proteins in the ischemic heart.
TABLE 3.
Proteins immuno-precipitated by the anti-carnosine antibody and identified by LC-LTQ-Oribitrap-ELITE hybrid mass spectrometer
| Sequence number | Accession number | Proteins | Location |
|---|---|---|---|
| 1 | Q62036 | 5-Azacytidine-induced protein 1 | Unknown |
| 2 | Q920P5 | Adenylate kinase isoenzyme 5 | Cytoplasm |
| 3 | 254910995 | α-N-Acetylglucosaminidase | Cytoplasm |
| 4 | Q03265 | ATP synthase subunit α, mitochondrial | Mitochondria |
| 5 | Q2MHE5 | Docking protein 6 | Cytoplasm |
| 6 | Q8BZ98 | Dynamin-3 | Cytoplasm |
| 7 | Q8R0W0 | Epiplakin | Cytoplasm |
| 8 | 46849812 | Fibronectin | Extracellular |
| 9 | P16858 | Glyceraldehyde-3-phosphate dehydrogenase | Cytoplasm |
| 10 | Q9WVD4 | H+/Cl(−) exchange transporter 5 | Plasma membrane |
| 11 | P02089 | Hemoglobin subunit β-2 | Unknown |
| 12 | Q922U2 | Keratin, type II cytoskeletal 5 | Unknown |
| 13 | Q8VED5 | Keratin, type II cytoskeletal 79 | Unknown |
| 14 | P17897 | Lysozyme C-1 | Unknown |
| 15 | Q3UIJ9 | Myocardial zonula adherens protein | Cytoplasm |
| 16 | Q02566 | Myosin-6 | Cytoplasm |
| 17 | Q8VDD5 | Myosin-9 | Cytoplasm |
| 18 | P70441 | Na+/H+ exchange regulatory cofactor NHE-RF1 | Cytoplasm |
| 19 | 309267203 | PREDICTED: keratin, type II cytoskeletal 78-like, partial | Unknown |
| 20 | Q9QYX7 | Protein piccolo | Cytoplasm |
| 21 | Q6ZQ06 | Protein QN1 homolog | Cytoplasm |
| 22 | 113866011 | Protocadherin β 9 | Plasma membrane |
| 23 | O55143 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | Cytoplasm |
| 24 | P07724 | Serum albumin | Secreted |
| 25 | Q80VP2 | Spermatogenesis-associated protein 7 homolog | Unknown |
| 26 | 269315863 | Collagen α-5 (VI) chain | Extracellular |
| 27 | 283135194 | Probable cation-transporting ATPase 13A1 | Unknown |
| 28 | 340745359 | Hemolytic complement-like isoform 3 | Unknown |
| 29 | 161484654 | Collagen α-1 (IV) chain | Unknown |
| 30 | 242397450 | Paired basic amino acid cleaving system 4 | Unknown |
| 31 | 241982771 | Phosphatidylinositol 4-kinase type 3 α | Cytoplasm |
| 32 | 262118187 | UPF0378 protein KIAA0100 | Unknown |
| 33 | 170671724 | UV radiation resistance associated gene | Nucleus |
| 34 | 113865905 | Zinc finger ZZ-type and EF-hand domain-containing protein 1 | Unknown |
| 35 | Q8VHK1 | Caskin 2 | Cytoplasm |
| 36 | 154240724 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 3 | Plasma membrane |
Further analysis of carnosinylated proteins in the ischemic heart led to direct identification of carnosine bound to 6 proteins. In these proteins, lysine, histidine, and cysteine residues were found to be bound to carnosine-propanal conjugates (m/z 283 and 339). As shown in Fig. 9, the carnosine-propanal conjugate (m/z, 283) was found to form a dehydrated adduct (m/z, 264 [3- [1-[1-[(3Z)-3-(5-amino-6-hydroxy-6-oxo-hexyl)iminopropyl]imidazol-4-yl]methyl]-2-hydroxy-2-oxo-ethyl]amino]-3-oxo-propyl]ammonium) with the lysine residue of paired basic amino acid cleaving system and zinc finger ZZ-type proteins (Table 4). A similar adduct was detected with a histidine residue of UV radiation resistance gene, a known regulator of autophagy (48, 49) and collagen α-1(IV) chain (Table 4). Similarly, dipropanal-β-Ala-His conjugates (m/z 339) were also found to form dehydrated adducts with the histidine residue of phosphatidylinositol kinase type 3α and the cysteine residue of UPF0378 proteins. Together, these data suggest that cardiac ischemia results in the carnosinylation of several cardiac proteins and that these adducts arise from the binding of the carnosine-propanal conjugates with the nucleophilic side chains of proteins.
FIGURE 9.

Mass spectrometric identification of carnosine-propanal-modified paired basic amino acid-cleaving system protein. Representative HCD fragmentation spectrum for +3-charged ion with a monoisotopic m/z 545.63898 Da was observed at +4.99 millimass unit (mmu)/9.14 ppm mass error using a 1D-LC-LTQ-Oribitrap-ELITE mass spectrometer. Analysis of the collected data by Sequest and Mascot (v1.20) identified a peptide (XCorr, 1.70; probability, 0.00) with the sequence WLQQQEVkRR, K8-C12H16N4O3 (264.1224 Da), and a MH+ of 1634.90238 Da. The fragmentation of the peptide yielded four c-ion and three z-ion series with sub 2.5 ppm mass accuracy. The inset shows the putative structures of the adducts that could be due to the formation of lysine-propanal-β-alanyl-histidine (i) or β-alanyl-histidine-propanal-lysine (ii) adducts.
TABLE 4.
Modification of cardiac proteins by carnosine-propanals
Site-specific modification of lysine (k), histidine (h), and cysteine (c) residues by carnosine propanal conjugates that are noted with the lowercase and bold letters.
| Sequence number | Accession number | Proteins | Modification | m/z | Peptide sequence |
|---|---|---|---|---|---|
| 1 | 161484654 | Collagen α-1(IV) chain | C12H16N4O3 | 264 | AhGQDLGTAGSCLR |
| 2 | 242397450 | Paired basic amino acid cleaving system 4 | C12H16N4O3 | 264 | WLQQQEVkRR |
| 3 | 241982771 | Phosphatidylinositol 4-kinase type 3α | C15H20N4O4 | 320 | GSQLhKYYMKR |
| 4 | 262118187 | UPF0378 protein KIAA0100 | C15H20N4O4 | 320 | LAGTEQSGQPcSR |
| 5 | 170671724 | UV radiation resistance associated gene | C12H16N4O3 | 264 | HhISNAIPVPKR |
| 6 | 113865905 | Zinc finger ZZ-type and EF-hand domain-containing protein 1 | C12H16N4O3 | 264 | AVIVDVKTRkR |
DISCUSSION
The results of this study show that endogenously generated unsaturated aldehydes such as HNE and acrolein are metabolized via carnosine conjugation and that carnosine-aldehyde conjugates are reduced before their elimination in urine. We found that this reductive transformation was catalyzed by AR and prevented in AR-null tissues and AR-null mice, indicating that AR plays an important role in the metabolism of carnosine-acrolein conjugates. Unlike carnosine-propanals, carnosine-propanols did not react with nucleophilic protein residues, suggesting that reduction by AR prevents protein modification by carnosine-propanals. Indeed there was greater accumulation of carnosinylated proteins in the heart, brain, and skeletal muscle of aged AR-null than WT mice. Moreover, even though carnosine attenuated ischemia-reperfusion injury in WT mice, its protective effects were not observed in AR-null hearts, and there was an increase in ischemia-induced accumulation of carnosinylated proteins in AR-null hearts. Taken together, these observations suggest that reduction of carnosine-propanals by AR prevents protein carnosinylation that could otherwise induce tissue dysfunction.
Previous studies have shown that carnosine forms stable conjugates with unsaturated aldehydes such as HNE and acrolein (32). In agreement with these data, we identified several structurally distinct carnosine-propanal (acrolein) conjugates. A variety of conjugates was generated by the formation of Michael adducts and Schiff bases between acrolein and carnosine as well as the addition of two acrolein molecules to a single carnosine dipeptide. We also found that acrolein also formed covalent conjugates with other related histidyl dipeptides such as homocarnosine and anserine, indicating that reaction with unsaturated aldehydes may be a general property of biological histidyl dipeptides.
Our measurements of urinary metabolites in both mice and humans revealed that the carnosine-aldehyde conjugates are mostly excreted as reduced conjugates, suggesting that reduction is an important metabolic transformation step in the elimination of these conjugates. Although the metabolic processes involved in the reduction of these conjugates in humans remain unknown, our studies in mice indicate that reduction of acrolein conjugates is catalyzed by AR. This conclusion is based on the observations that the reduction of carnosine-aldehyde adducts was catalyzed by purified AR protein in vitro and that genetic deletion of AR in mice attenuated the appearance of the reduced form of the conjugate in the urine. Collectively, these data reveal a previously unknown metabolic pathway involved in the detoxification of carnosine-aldehyde adducts and define a new role of AR in the metabolism of lipid peroxidation products.
AR is a multifunctional enzyme, and its kinetic and structural properties are well suited for the detoxification of peptidyl conjugates. Previous work from our laboratory has shown that the enzyme catalyzes the reduction of the glutathione conjugates of unsaturated aldehydes (17, 45). In agreement with these findings, current data showing that the catalytic efficiency of AR with carnosine-propanals is comparable with that of dl-glyceraldehyde reinforce the promiscuity of this enzyme and suggest that in addition to reducing S-linked aldehyde conjugates, the enzyme is also capable of reducing N-linked adducts generated by the stabilization of Michael adducts and Schiff bases.
In addition to carnosine-propanals, AR also catalyzed the reduction of conjugates containing other histidyl peptides (homocarnosine and anserine) as well as the conjugates of pyridoxamine, hydralazine, and aminoguanidine. Although most of these conjugates were reduced less efficiently than the carnosine conjugates, the catalytic activity of the enzyme with these substrates support the possibility that AR may be involved in the metabolism of several endogenous conjugates of acrolein and other related aldehydes. Surprisingly, only weak catalysis of the HNE conjugates was observed. We previously found that AR catalyzes the reduction of both GS-HNE and GS-propanal with similar efficiency; hence, we expected that both carnosine-HNE and carnosine-propanal will be reduced efficiently by AR. However, the poor activity of the enzyme with carnosine-HNE may be due to the proclivity of the HNE conjugates to cyclize, which could occlude the free aldehyde group required for recognition by AR. Nevertheless, detectable levels of histidine-DHN were found in human urine and trace levels of activity of the enzyme with histidine-HNE, suggesting that in vivo HNE conjugates might be reduced by AR.
Results obtained with AR-null mice suggest that the reduction by AR is a significant route for the detoxification of carnosine-propanal conjugates. These data show the reduction of carnosine-propanals was attenuated in AR-null tissues, and the levels of reduced conjugates was diminished in AR-null mice suggest that metabolism via AR is a unique, non-redundant pathway for the reductive transformation of carnosine-propanals. Nonetheless, on the basis of the current data alone, involvement of other reductases cannot be completely ruled out.
The levels of carnosine-acrolein conjugates in the urine were surprisingly higher than those of carnosine-HNE. HNE is one of the most abundant unsaturated aldehydes generated during lipid peroxidation, and in some conditions it represents >95% of the unsaturated aldehydes generated in oxidized lipids (50). However, recent studies show that in addition to reduction and oxidation, the carbon skeleton of HNE is broken down to 4-P-nonanoyl-Co A, 4-acyl-CoA esters, 4-hydroxynonaoate, 4-hydroxynonenoate, and 2-hydroxyheptanoate (51), and therefore, the levels of HNE available to form carnosine-HNE or carnosine-DHN may be decreased due to such metabolism. Additionally, it is likely that in comparison with acrolein, HNE is metabolized to a greater extent via glutathione-linked metabolism. It is also likely that the higher abundance of acrolein conjugates in the urine represent higher levels of acrolein production. Unlike HNE, which is generated mainly by lipid peroxidation, acrolein is also produced in cells from sources such as the oxidation of amino acids by myeloperoxidase (38) or the metabolism of biogenic amines (37), and therefore, its production may be higher than HNE. In addition to endogenous sources, acrolein is also present in several foods, tobacco smoke, and automobile exhaust (39). In the current study, urine from non-smoking adults only was analyzed; however, despite this selection, high levels of acrolein-derived carnosine conjugates were detected in the urine, indicating that acrolein is generated in appreciable quantities during the course of normal human metabolism.
The present study also led to the identification of a new mechanism of post-translational protein modification. Previous in vitro studies have shown that carnosine reacts with protein carbonyls generated by oxidation and glycation (52) and that carnosine could be adducted to proteins by the lipid peroxidation-derived ketoaldehyde, 4-oxo-2-nonenal (53). However, to the best of our knowledge, carnosinylated proteins have not yet been detected in biological tissues. Thus, data showing that carnosine-propanals bind to peptides and proteins and that perfusion of the heart with acrolein induces protein carnosinylation reveal a unique mechanism by which such proteins could arise in biological tissues. Moreover, these investigations show for the first time that the formation of carnosinylated proteins is increased under conditions of oxidative stress such as myocardial ischemia and aging. These observations suggest that protein carnosinylation may be a sensitive index of increased aldehyde generation in normal and diseased tissues.
The results of this study also provide assessments of the pathophysiological significance of protein carnosinylation. The observation that more carnosinylated proteins accumulate in aged and ischemic tissue from AR-null mice suggests that AR prevents protein carnosinylation by catalyzing the reduction of carnosine-propanal conjugates and that carnosine protects against tissue injury by forming stable conjugates with aldehydes generated by lipid oxidation. This view is consistent with the observation that perfusion with carnosine improved myocardial recovery after ischemia. The beneficial effects of carnosine against ischemic injury have been described before (24); however, the mechanisms by which carnosine exerts its anti-ischemic effects are not known. It has been speculated that carnosine could protect against ischemia by buffering changes in intracellular pH, by increasing the calcium sensitivity of the myofilaments, or by quenching singlet oxygen (23). The present data do not rule out any of these possibilities but instead suggest that carnosine protects against ischemia-reperfusion injury by removing aldehyde derived from lipid oxidation. Carnosine forms covalent conjugates with reactive aldehydes and thereby prevents them from reacting with proteins. However, conjugation with carnosine is not sufficient for detoxifaction because the aldehyde in the conjugate has to be reduced by AR for the conjugate to be fully inactivated. This is consistent with the observations that the cardio-protective effects of carnosine were abolished in AR-null hearts and that these hearts accumulated more carnosinylated proteins during ischemia than WT hearts. These results point toward an important role of AR in regulating the antioxidant effects of carnosine. However, this role of AR is not in agreement with the previous work showing that inhibition of AR prevents ischemia-reperfusion injury in rat hearts (54–56) and that hearts of AR-null mice are marginally protected from ischemia-reperfusion ex vivo (57). However, in contrast, previous studies from our laboratory have shown that inhibition of AR abolishes myocardial ischemic preconditioning (19) and increases infarct size in conscious rats, and our current data do not show attenuated injury in AR-null hearts (58). The reasons for such disparate results are unclear but may relate to different experimental conditions (buffer composition, extent of injury, animal age, etc). Clearly, further studies are required to identify the conditions in which inhibition of AR prevents ischemic injury and those under which AR is cardioprotective.
In summary, the results of this study reveal the presence of a new metabolic pathway for the removal and detoxification of carnosinylated acrolein. This pathway appears to involve reductive transformation of the conjugates by AR, and this process seems to be essential for the cardioprotective effects of carnosine. These findings indicate that carnosine and AR are important components of antioxidant protection against exogenous acrolein as well as acrolein generated during lipid peroxidation, oxidative metabolism, and inflammation. Carnosine is a natural component of the human diet, and it is also synthesized from β-alanine in food, and oral intake of carnosine or β-alanine has been shown to prevent hyperglycemic injury and age-dependent tissue degeneration and increases exercise capacity (23). Hence treatment with carnosine or carnosine-like drugs or peptides might be useful in treating several pathological conditions associated with oxidative stress such as cardiovascular, neurodegenerative, and skeleton-muscular diseases, especially under conditions in which the conjugates of carnosine are metabolized and detoxified by AR.
Acknowledgments
We are grateful to Dr. F. Margolis (University of Maryland) for providing the anti-protein-carnosine antibody. We thank L. Guo (University of Louisville) for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants HL55477, HL59378, HL89380, and RR024489.
- HNE
- 4-hydroxy-trans-2-nonenal (HNE)
- AR
- aldose reductase
- CK
- creatinine kinase
- HPMA
- 3-hydroxypropyl mercapturic acid
- FTMS
- Fourier transform mass spectrometry
- ESI
- electrospray ionization/mass spectrometry
- FDP
- dihydropipyridine
- HCD
- high energy collision dissociation
- ETD
- electron-transfer dissociation.
REFERENCES
- 1. Esterbauer H., Schaur R. J., Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde, and related aldehydes. Free Radic. Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 2. Awasthi Y. C. (2007) Toxicology of Glutathione Transferases, Taylor & Francis, Galveston, TX [Google Scholar]
- 3. LoPachin R. M., Gavin T., Petersen D. R., Barber D. S. (2009) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity. Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 22, 1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riahi Y., Cohen G., Shamni O., Sasson S. (2010) Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 299, E879–E886 [DOI] [PubMed] [Google Scholar]
- 5. Steinberg D. (1997) Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 272, 20963–20966 [DOI] [PubMed] [Google Scholar]
- 6. Srivastava S., Vladykovskaya E., Barski O. A., Spite M., Kaiserova K., Petrash J. M., Chung S. S., Hunt G., Dawn B., Bhatnagar A. (2009) Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ. Res. 105, 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eaton P., Li J. M., Hearse D. J., Shattock M. J. (1999) Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am. J. Physiol. 276, H935–H943 [DOI] [PubMed] [Google Scholar]
- 8. Hill B. G., Awe S. O., Vladykovskaya E., Ahmed Y., Liu S. Q., Bhatnagar A., Srivastava S. (2009) Myocardial ischaemia inhibits mitochondrial metabolism of 4-hydroxy-trans-2-nonenal. Biochem. J. 417, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rittner H. L., Hafner V., Klimiuk P. A., Szweda L. I., Goronzy J. J., Weyand C. M. (1999) Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J. Clin. Invest. 103, 1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butterfield D. A., Bader Lange M. L., Sultana R. (2010) Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim. Biophys. Acta 1801, 924–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jenner P. (2003) Oxidative stress in Parkinson's disease. Ann. Neurol. 53, S26–S36; discussion S36–S28 [DOI] [PubMed] [Google Scholar]
- 12. Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E. R., Mizuno Y. (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 93, 2696–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung F. L., Pan J., Choudhury S., Roy R., Hu W., Tang M. S. (2003) Formation of trans-4-hydroxy-2-nonenal- and other enal-derived cyclic DNA adducts from omega-3 and omega-6 polyunsaturated fatty acids and their roles in DNA repair and human p53 gene mutation. Mutat. Res. 531, 25–36 [DOI] [PubMed] [Google Scholar]
- 14. Nair J., Barbin A., Velic I., Bartsch H. (1999) Etheno DNA-base adducts from endogenous reactive species. Mutat. Res. 424, 59–69 [DOI] [PubMed] [Google Scholar]
- 15. Minko I. G., Kozekov I. D., Harris T. M., Rizzo C. J., Lloyd R. S., Stone M. P. (2009) Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem. Res. Toxicol. 22, 759–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srivastava S., Chandra A., Wang L. F., Seifert W. E., Jr., DaGue B. B., Ansari N. H., Srivastava S. K., Bhatnagar A. (1998) Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 273, 10893–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srivastava S., Watowich S. J., Petrash J. M., Srivastava S. K., Bhatnagar A. (1999) Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry 38, 42–54 [DOI] [PubMed] [Google Scholar]
- 18. Dixit B. L., Balendiran G. K., Watowich S. J., Srivastava S., Ramana K. V., Petrash J. M., Bhatnagar A., Srivastava S. K. (2000) Kinetic and structural characterization of the glutathione-binding site of aldose reductase. J. Biol. Chem. 275, 21587–21595 [DOI] [PubMed] [Google Scholar]
- 19. Shinmura K., Bolli R., Liu S. Q., Tang X. L., Kodani E., Xuan Y. T., Srivastava S., Bhatnagar A. (2002) Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ. Res. 91, 240–246 [DOI] [PubMed] [Google Scholar]
- 20. Zhou S., Decker E. A. (1999) Ability of carnosine and other skeletal muscle components to quench unsaturated aldehydic lipid oxidation products. J. Agric. Food Chem. 47, 51–55 [DOI] [PubMed] [Google Scholar]
- 21. Aldini G., Carini M., Beretta G., Bradamante S., Facino R. M. (2002) Carnosine is a quencher of 4-hydroxy-nonenal. Through what mechanism of reaction? Biochem. Biophys. Res. Commun. 298, 699–706 [DOI] [PubMed] [Google Scholar]
- 22. Liu Y., Xu G., Sayre L. M. (2003) Carnosine inhibits (E)-4-hydroxy-2-nonenal-induced protein cross-linking. Structural characterization of carnosine-HNE adducts. Chem. Res. Toxicol. 16, 1589–1597 [DOI] [PubMed] [Google Scholar]
- 23. Hipkiss A. R. (2009) Carnosine and its possible roles in nutrition and health. Adv. Food Nutr. Res. 57, 87–154 [DOI] [PubMed] [Google Scholar]
- 24. Lee J. W., Miyawaki H., Bobst E. V., Hester J. D., Ashraf M., Bobst A. M. (1999) Improved functional recovery of ischemic rat hearts due to singlet oxygen scavengers histidine and carnosine. J. Mol. Cell. Cardiol. 31, 113–121 [DOI] [PubMed] [Google Scholar]
- 25. Dobrota D., Fedorova T., Stvolinsky S., Babusikova E., Likavcanova K., Drgova A., Strapkova A., Boldyrev A. (2005) Carnosine protects the brain of rats and Mongolian gerbils against ischemic injury. After-stroke-effect. Neurochem. Res. 30, 1283–1288 [DOI] [PubMed] [Google Scholar]
- 26. Kurata H., Fujii T., Tsutsui H., Katayama T., Ohkita M., Takaoka M., Tsuruoka N., Kiso Y., Ohno Y., Fujisawa Y., Shokoji T., Nishiyama A., Abe Y., Matsumura Y. (2006) Renoprotective effects of l-carnosine on ischemia/reperfusion-induced renal injury in rats. J. Pharmacol. Exp. Ther. 319, 640–647 [DOI] [PubMed] [Google Scholar]
- 27. Sauerhöfer S., Yuan G., Braun G. S., Deinzer M., Neumaier M., Gretz N., Floege J., Kriz W., van der Woude F., Moeller M. J. (2007) L-carnosine, a substrate of carnosinase-1, influences glucose metabolism. Diabetes 56, 2425–2432 [DOI] [PubMed] [Google Scholar]
- 28. Boldyrev A., Fedorova T., Stepanova M., Dobrotvorskaya I., Kozlova E., Boldanova N., Bagyeva G., Ivanova-Smolenskaya I., Illarioshkin S. (2008) Carnosine [corrected] increases efficiency of DOPA therapy of Parkinson's disease. A pilot study. Rejuvenation Res. 11, 821–827 [DOI] [PubMed] [Google Scholar]
- 29. McFarland G. A., Holliday R. (1994) Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp. Cell Res. 212, 167–175 [DOI] [PubMed] [Google Scholar]
- 30. Yuneva A. O., Kramarenko G. G., Vetreshchak T. V., Gallant S., Boldyrev A. A. (2002) Effect of carnosine on Drosophila melanogaster lifespan. Bull. Exp. Biol. Med. 133, 559–561 [DOI] [PubMed] [Google Scholar]
- 31. Carini M., Aldini G., Beretta G., Arlandini E., Facino R. M. (2003) Acrolein-sequestering ability of endogenous dipeptides. Characterization of carnosine and homocarnosine/acrolein adducts by electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 38, 996–1006 [DOI] [PubMed] [Google Scholar]
- 32. Orioli M., Aldini G., Benfatto M. C., Facino R. M., Carini M. (2007) HNE Michael adducts to histidine and histidine-containing peptides as biomarkers of lipid-derived carbonyl stress in urines. LC-MS/MS profiling in Zucker obese rats. Anal. Chem. 79, 9174–9184 [DOI] [PubMed] [Google Scholar]
- 33. Srivastava S., Dixit B. L., Cai J., Sharma S., Hurst H. E., Bhatnagar A., Srivastava S. K. (2000) Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes. Role of aldose reductase. Free Radic. Biol. Med. 29, 642–651 [DOI] [PubMed] [Google Scholar]
- 34. Tsuruta Y., Maruyama K., Inoue H., Kosha K., Date Y., Okamura N., Eto S., Kojima E. (2010) Sensitive determination of carnosine in urine by high performance liquid chromatography using 4-(5,6-dimethoxy-2-phthalimidinyl)-2-methoxyphenylsulfonyl chloride as a fluorescent labeling reagent. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 327–332 [DOI] [PubMed] [Google Scholar]
- 35. Vladykovskaya E., Sithu S. D., Haberzettl P., Wickramasinghe N. S., Merchant M. L., Hill B. G., McCracken J., Agarwal A., Dougherty S., Gordon S. A., Schuschke D. A., Barski O. A., O'Toole T., D'Souza S. E., Bhatnagar A., Srivastava S. (2012) Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. J. Biol. Chem. 287, 11398–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen O. N., Shevchenko A., Mann M. (1997) Protein Analysis by Mass Spectrometry, 2nd ed., Oxford University Press, Oxford [Google Scholar]
- 37. Toninello A., Pietrangeli P., De Marchi U., Salvi M., Mondovì B. (2006) Amine oxidases in apoptosis and cancer. Biochim. Biophys. Acta 1765, 1–13 [DOI] [PubMed] [Google Scholar]
- 38. Anderson M. M., Hazen S. L., Hsu F. F., Heinecke J. W. (1997) Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive α-hydroxy and α,β-unsaturated aldehydes by phagocytes at sites of inflammation. J. Clin. Invest. 99, 424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stevens J. F., Maier C. S. (2008) Acrolein. Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 52, 7–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aldini G., Orioli M., Carini M., Maffei Facino R. (2004) Profiling histidine-containing dipeptides in rat tissues by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 39, 1417–1428 [DOI] [PubMed] [Google Scholar]
- 41. Kuiper H. C., Miranda C. L., Sowell J. D., Stevens J. F. (2008) Mercapturic acid conjugates of 4-hydroxy-2-nonenal and 4-oxo-2-nonenal metabolites are in vivo markers of oxidative stress. J. Biol. Chem. 283, 17131–17138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carmella S. G., Chen M., Zhang Y., Zhang S., Hatsukami D. K., Hecht S. S. (2007) Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry. Effects of cigarette smoking. Chem. Res. Toxicol. 20, 986–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Linhart I., Frantík E., Vodicková L., Vosmanská M., Smejkal J., Mitera J. (1996) Biotransformation of acrolein in rat. Excretion of mercapturic acids after inhalation and intraperitoneal injection. Toxicol. Appl. Pharmacol. 136, 155–160 [DOI] [PubMed] [Google Scholar]
- 44. Parent R. A., Paust D. E., Schrimpf M. K., Talaat R. E., Doane R. A., Caravello H. E., Lee S. J., Sharp D. E. (1998) Metabolism and distribution of [2,3–14C]acrolein in Sprague-Dawley rats. II. Identification of urinary and fecal metabolites. Toxicol Sci. 43, 110–120 [DOI] [PubMed] [Google Scholar]
- 45. Ramana K. V., Dixit B. L., Srivastava S., Balendiran G. K., Srivastava S. K., Bhatnagar A. (2000) Selective recognition of glutathiolated aldehydes by aldose reductase. Biochemistry 39, 12172–12180 [DOI] [PubMed] [Google Scholar]
- 46. Hsu D. K., Guo Y., Peifley K. A., Winkles J. A. (1997) Differential control of murine aldose reductase and fibroblast growth factor (FGF)-regulated-1 gene expression in NIH 3T3 cells by FGF-1 treatment and hyperosmotic stress. Biochem. J. 328, 593–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Srivastava S., Harter T. M., Chandra A., Bhatnagar A., Srivastava S. K., Petrash J. M. (1998) Kinetic studies of FR-1, a growth factor-inducible aldo-keto reductase. Biochemistry 37, 12909–12917 [DOI] [PubMed] [Google Scholar]
- 48. Zhao Z., Ni D., Ghozalli I., Pirooz S. D., Ma B., Liang C. (2012) UVRAG. At the crossroad of autophagy and genomic stability. Autophagy 8, 1392–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Z., Oh S., Li D., Ni D., Pirooz S. D., Lee J. H., Yang S., Lee J. Y., Ghozalli I., Costanzo V., Stark J. M., Liang C. (2012) A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev. Cell 22, 1001–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benedetti A., Comporti M., Esterbauer H. (1980) Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 620, 281–296 [DOI] [PubMed] [Google Scholar]
- 51. Zhang G. F., Kombu R. S., Kasumov T., Han Y., Sadhukhan S., Zhang J., Sayre L. M., Ray D., Gibson K. M., Anderson V. A., Tochtrop G. P., Brunengraber H. (2009) Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J. Biol. Chem. 284, 33521–33534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brownson C., Hipkiss A. R. (2000) Carnosine reacts with a glycated protein. Free Radic. Biol. Med. 28, 1564–1570 [DOI] [PubMed] [Google Scholar]
- 53. Zhu X., Gallogly M. M., Mieyal J. J., Anderson V. E., Sayre L. M. (2009) Covalent cross-linking of glutathione and carnosine to proteins by 4-oxo-2-nonenal. Chem. Res. Toxicol. 22, 1050–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramasamy R., Trueblood N., Schaefer S. (1998) Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am. J. Physiol. 275, H195–H203 [DOI] [PubMed] [Google Scholar]
- 55. Hwang Y. C., Sato S., Tsai J. Y., Yan S., Bakr S., Zhang H., Oates P. J., Ramasamy R. (2002) Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J. 16, 243–245 [DOI] [PubMed] [Google Scholar]
- 56. Ananthakrishnan R., Kaneko M., Hwang Y. C., Quadri N., Gomez T., Li Q., Caspersen C., Ramasamy R. (2009) Aldose reductase mediates myocardial ischemia-reperfusion injury in part by opening mitochondrial permeability transition pore. Am. J. Physiol. Heart Circ. Physiol. 296, H333–H341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abdillahi M., Ananthakrishnan R., Vedantham S., Shang L., Zhu Z., Rosario R., Zirpoli H., Bohren K. M., Gabbay K. H., Ramasamy R. (2012) Aldose reductase modulates cardiac glycogen synthase kinase-3β phosphorylation during ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 303, H297–H308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaiserova K., Srivastava S., Hoetker J. D., Awe S. O., Tang X. L., Cai J., Bhatnagar A. (2006) Redox activation of aldose reductase in the ischemic heart. J. Biol. Chem. 281, 15110–15120 [DOI] [PubMed] [Google Scholar]