

Summary
The CXCL12–CXCR4 chemokine signaling pathway is a well-established driver of cancer progression. One key process promoted by CXCR4 stimulation is tumor cell motility; however, the specific signaling pathways leading to migration remain poorly understood. Previously, we have shown that CXCL12 stimulation of migration depends on temporal regulation of RhoA. However, the specific RhoGEF that translates CXCR4 signaling into RhoA activity and cell motility is unknown. We screened the three regulator of G-protein signaling RhoGEFs (LSC, LARG and PRG) and found that PRG selectively regulated the migration and invasion of CXCR4-overexpressing breast tumor cells. Interestingly, we found that PDZ-RhoGEF (PRG) was required for spatial organization of F-actin structures in the center, but not periphery of the cells. The effects on the cytoskeleton were mirrored by the spatial effects on RhoA activity that were dependent upon PRG. Loss of PRG also enhanced adherens junctions in the epithelial-like MCF7-CXCR4 cell line, and inhibited directional persistence and polarity in the more mesenchymal MDA-MB-231 cell line. Thus, PRG is essential for CXCR4-driven tumor cell migration through spatial regulation of RhoA and the subsequent organization of the cytoskeletal structures that support motility. Furthermore, immunohistochemical analysis of human breast tumor tissues shows a significant increase of PRG expression in the invasive areas of the tumors, suggesting that this RhoGEF is associated with breast tumor invasion in vivo.
Key words: PDZ-RhoGEF, PRG, RhoA, Breast cancer, Invasion
Introduction
Multicellular organisms organize into tissues and organs in response to extracellular cues in the microenvironment (Bryant and Mostov, 2008). Alterations in cell morphology and motility occur during normal organ tissue remodeling, but deregulation of these processes contributes to pathological outcomes including tumorigenesis (Tervonen et al., 2011). The cytoskeleton is highly malleable and responds to extracellular signals by reorganizing its structure to effect changes in cell shape and drive migration (Gardel et al., 2010; Harris et al., 2009; Ridley et al., 2003). Chemokine ligand 12 (CXC-type; CXCL12) is a versatile and potent motomorphogen, promoting cell motility and morphological changes important for development and tissue remodeling (Kucia et al., 2005; Murdoch, 2000); and its receptor, chemokine receptor 4 (CXC-type; CXCR4) is frequently overexpressed in malignant tumors of the breast and other tissues (Balkwill, 2004; Müller et al., 2001; Teicher and Fricker, 2010; Zlotnik, 2006). Thus, CXCR4 is considered an important diagnostic and therapeutic target for human malignancies, and understanding the molecular mechanisms by which CXCL12 controls cytoskeletal organization and tumor cell motility may lead to future therapeutic modalities.
CXCR4-driven tumor cell migration depends on temporal regulation of RhoA signaling (Bartolomé et al., 2004; Molina-Ortiz et al., 2009; Struckhoff et al., 2010; Tan et al., 2006; Vicente-Manzanares et al., 2002), but the molecular mechanisms by which CXCR4 regulates RhoA activity to promote motility is currently unknown. As direct activators of RhoA, RhoGEFs (guanine nucleotide exchange factors) are good candidates to signals from CXCR4 into RhoA activation during tumor cell motility. The size and diversity of the Dbl RhoGEF family (Rossman et al., 2005) allows the cell to integrate signals from diverse extracellular stimuli to produce precise spatiotemporal regulation of RhoA activity and subsequent changes in downstream cytoskeletal dynamics (Birkenfeld et al., 2007; Heasman et al., 2010; Nalbant et al., 2009; Terry et al., 2011). Although RhoGEFs are best understood for their direct activation of Rho GTPases, additional regulatory mechanisms include influencing downstream signaling from Rho effectors (Bravo-Cordero et al., 2011; Mulinari et al., 2008; Mulinari and Häcker, 2010), restricting the spatial activation of Rho GTPases (Bravo-Cordero et al., 2011), and creating scaffolding platforms to recruit specific Rho effectors to produce differential signaling outputs (Buchsbaum et al., 2002).
Chemokine receptors, such as CXCR4, are G-protein-coupled receptors that couple to Gαi (Rubin, 2009; Tan et al., 2006; Teicher and Fricker, 2010), as well as Gαo (Maghazachi, 1997), Gαq (Maghazachi, 1997; Soede et al., 2000) and Gα12/13 G proteins (Kumar et al., 2011; Tan et al., 2006). Gα12/13 stimulates RhoA through direct activation of a subset of Dbl family RhoGEFs that contain regulator of G-protein signaling (RGS) domains (Fukuhara et al., 2001; Kelly et al., 2007; Siehler, 2009; Sternweis et al., 2007). Although it is known that RGS-RhoGEFs activate RhoA downstream of CXCR4 (Tan et al., 2006; Yagi et al., 2011), it is unknown which RGS-RhoGEF(s) is important for driving RhoA-dependent migration. The RGS-RhoGEF subfamily, comprising ARHGEF1 [p115 RhoGEF (LSC)], ARHGEF11 [PDZ-RhoGEF (PRG)] and ARHGEF12 (LARG) (Aittaleb et al., 2010; Sternweis et al., 2007), is relatively unique among the larger Dbl GEF superfamily in that each RGS-RhoGEF targets only RhoA, but not Rac or Cdc42, for activation. Furthermore, previous studies have shown that some Gα12/13-coupled receptors utilize a specific RGS-RhoGEF for the activation of RhoA and subsequent downstream biological effects (Wang et al., 2004). Thus, we reasoned that identifying the RGS-RhoGEF responsible for RhoA-mediated migration downstream from CXCR4, would provide a potential therapeutic target for CXCR4-expressing tumor cells.
We screened the three different RGS-RhoGEFs (LSC, LARG and PRG) and found that PRG regulated the migration and invasion of breast tumor cells in response to CXCL12. Interestingly, PRG was required for spatial organization of F-actin structures in the center, but not periphery of the cells. This was mirrored by the spatial effects on RhoA activity that were dependent upon PRG. Thus, the effects of PRG on tumor cell migration are tightly correlated with the spatial regulation of RhoA activity and its subsequent organization of the actin structures that support migration.
Results
Gα12/13 and RhoA are required for CXCR4-stimluated migration
The ability of the CXCR4–CXCL12 signaling axis to promote cancer metastasis is well established; however, considerably less is known about the downstream signaling molecules crucial for CXCR4 pro-migratory responses. To investigate the RhoA signaling pathways involved in CXCR4-driven breast cancer motility, we used an in vitro wound closure assay to assess changes in migration following CXCR4 stimulation. When MCF7-CXCR4 cells were treated with the CXCR4 ligand, CXCL12 (10 nM), there was a 60% increase in migration (Fig. 1A), verifying that activation of CXCR4 significantly stimulates breast cancer cell migration in our system. Pretreatment with the Rho inhibitor, C3-transferase, blocked CXCL12-stimulated cell migration, demonstrating Rho activity is required for cell migration, and that without Rho activity, CXCR4 cannot promote breast cancer migration.
Fig. 1.
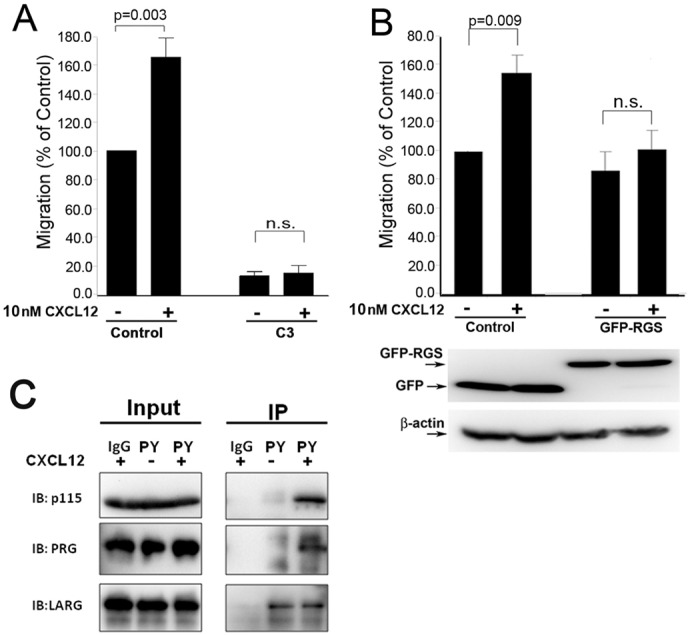
CXCR4-stimulated cell migration requires RhoA and Gα12/13, and results in tyrosine phosphorylation of RGS-RhoGEFs. (A) CXCL12 (10 nM) significantly stimulated migration (P = 0.003), whereas pretreatment of cells with C3-transferase (0.5 µg/ml) blocked basal and CXCL12-induced migration. Cells were imaged at 0 hours and 20 hours post-wounding, and the difference in wound area was determined using ImageJ software. (B) MCF7-CXCR4 cells were transfected with either control (GFP) vector or GFP-RGS 24 hours before wounding. Migration was calculated as change in wound area as described above. Representative immunoblots probed with anti-GFP confirm the expression of GFP (control cells) and GFP-RGS. Data from A and B are means ± s.e.m., n = 3. (C) MCF7-CXCR4 cells were transfected with LSC-, LARG- or PRG-specific siRNA. Serum-starved cells (16 hours) were treated with CXCL12 (10 nM, 10 minutes), lysed, and tyrosine-phosphorylated proteins were immunoprecipitated using a phosphotyrosine-specific antibody. Input represents 10% of IP sample.
Having established the importance of Rho in CXCR4 migration in our system, we set out to identify which signaling molecules are required. Previously it was shown that Gα12/13 activates RhoA downstream of CXCR4 in Jurkat cells (Tan et al., 2006) and therefore we anticipated that Gα12/13 would also be required for CXCR4-stimulated migration in our model. We transfected MCF7-CXCR4 cells with a GFP-tagged RGS domain and measured changes in cell migration following CXCL12 addition. Isolated RGS domains can act as dominant-negative inhibitors by blocking Gα12/13 stimulation of RhoA (Iguchi et al., 2008; Tan et al., 2006; Ziembicki et al., 2005). In control (GFP-transfected) MCF7-CXCR4 cells, CXCL12 significantly increased cell migration (58%, Fig. 1B), which is comparable to the increase in non-transfected MCF7-CXCR4 cells. Expression of GFP–RGS inhibited CXCL12-driven migration of MCF7-CXCR4 cells, confirming previous reports in other cell types showing a role for Gα12/13in CXCR4-driven RhoA activity (Tan et al., 2006; Yagi et al., 2011) and demonstrating Gα12/13 is important in CXCR4-driven migration in our system.
Stimulation of CXCR4 leads to tyrosine phosphorylation of LSC and PRG RhoGEFs
Tyrosine phosphorylation is a commonly used mechanism for the regulation of RhoGEF activity and increased tyrosine phosphorylation following receptor activation has been reported for the three RGS-RhoGEFs family members (Chikumi et al., 2002; Guilluy et al., 2010; Suzuki et al., 2009; Suzuki et al., 2003). We treated MCF7-CXCR4 cells with CXCL12 and used a phosphotyrosine (PY)-specific antibody to immunoprecipitate PY-proteins (Fig. 1C). Stimulation of CXCR4 sharply increased tyrosine phosphorylation of LSC and PRG, but not LARG. However, the basal level of PY-LARG was high, potentially masking a response to CXCL12. These data show that LSC and PRG are targets of CXCR4 signaling, suggesting that these two GEFs may be responsible for RhoA activation following activation of CXCR4.
Knockdown of PRG expression profoundly inhibits CXCR4-mediated migration
To determine whether LSC or PRG was required for cell migration following stimulation of CXCR4, we used siRNA to selectively target LSC, LARG or PRG expression and assessed the effect on cell migration (Suzuki et al., 2009; Tesmer, 2009). Each siRNA reduced expression of its target gene by 90% or greater, without altering the expression of other RGS-RhoGEF proteins (Fig. 2B). In control-siRNA-transfected cells, CXCL12 significantly increased cell migration (55%, Fig. 2A), which is similar to that observed in non-transfected MCF7-CXCR4 cells (Fig. 1A). We observed that LSC-deficient cells were still responsive to CXCR4 stimulation (P<0.05), suggesting that although CXCR4 stimulation results in tyrosine phosphorylation of LSC, LSC is not essential for CXCR4-driven migration (Fig. 2A). Knockdown of LARG attenuated migration following CXCR4 stimulation, albeit weakly (P = 0.041). The relatively small decrease in CXCR4-driven migration coupled with the lack of PY stimulation led us to conclude that although LARG contributes to CXCR4-driven migration it is not the primary regulator of CXCR4-driven migration. In contrast, PRG knockdown cells showed a dramatic decrease in both basal and CXC4-dependent migration. In wound-healing assays there was a slight increase in wound area during the assay in both non-stimulated and CXCL12-stimulated cells (Fig. 2A). Observing PRG knockdown cells during the assay confirmed PRG-deficient cells did not migrate into the wound and, in fact, contracted away from the empty space and increased their contact with neighboring cells. The slight increase in wound area was not due to cell death because PRG-deficient cells remained viable and attached for at least 2 days after the completion of the assay (data not shown). To confirm that effects on cell migration were due to loss of PRG and not siRNA off-target effects, we generated MCF7-CXCR4 cells that stably expressed mouse PRG (msPRG) cDNA, which is not targeted by the PRG siRNA (see Materials and Methods; Fig. 2D). Expression of msPRG restored the migratory response to CXCR4 stimulation (Fig. 2C). Although the migration response in the msPRG cells was not as high as in the controls, the restoration of the CXCR4-driven migration response was proportional to the expression of PRG (Fig. 2D), confirming that PRG expression correlates with the CXCR4-driven migration response. Because CXCL12-stimulated tyrosine phosphorylation of PRG, and PRG knockdown caused such a profound reduction in cell migration, we conclude that PRG is an essential mediator of MCF7-CXCR4 cell migration.
Fig. 2.

PRG is required for CXCR4-promoted migration. (A) CXCL12 (10 nM) significantly stimulated migration in control siRNA (P<0.001) and LSC-siRNA-transfected cells (P = 0.002) whereas LARG and PRG depletion prevented CXCL12-induced migration (P = 0.35). Values are means ± s.e.m., n = 6. (B) Representative immunoblot demonstrating the specificity and efficacy of RhoGEF siRNA in reducing target RhoGEF expression. (C) MCF7-CXCR4 cells stably expressing control vector or msPRG cDNA were used to assess changes in cell migration. In control cells, CXCL12 stimulated cell migration (P<0.001), which was inhibited by PRG siRNA (P = 0.41). In contrast, in cells stably expressing the siRNA-resistant msPRG, CXCL12 significantly stimulated cell migration in both control-siRNA- and PRG-transfected cells (P<0.001 and P<0.05, respectively). Values are means ± s.e.m., n = 3. (D) Representative immunoblot demonstrating that expression of siRNA-resistant msPRG is not affected by PRG siRNA. KD, knockdown.
RGS-RhoGEF knockdown results in defects in migration machinery components
Cell motility requires orchestrated changes in the cytoskeleton, thus, disruption of PRG-dependent cytoskeletal structures would impair cell migration. To specifically determine how cytoskeletal elements were disrupted in PRG knockdowns, we examined focal adhesion and F-actin structures in both sparsely plated cells (Fig. 3A) and migrating cells at the wound edge of a monolayer (Fig. 3C). Vinculin, a component of integrin adhesion complexes, was used to identify focal adhesions structures, and phalloidin was used to label F-actin. Using sparsely plated cells allowed us to image the structural components in greater detail, whereas examining cells at the wound edge allowed us to image cells in an environment that recreated that of the migration assays in Fig. 2. Non-stimulated MCF7-CXCR4 cells had modest focal adhesions at the cell periphery and cell protrusions, along with small stress fibers in the interior and at cell projections (Fig. 3A, top right panel). Stimulation with CXCL12 induced actin- and focal-adhesion-rich protrusions, increased focal adhesion number, and stimulated stress fiber formation (Fig. 3A, left panels). In contrast, PRG knockdown cells were large, symmetrical, and lacked obvious protrusive structures. Large, distinct focal adhesions were present but since these cells had no distinct protrusive areas, the focal contacts were uniformly spread about the periphery of the cell. No internal stress fibers were present and instead there were pronounced rings of cortical F-actin, and in cell clusters these rings defined the boundary of the cluster rather than defining individual cells within the cluster (Fig. 3A, bottom right panel). PRG-deficient cells exhibited few morphologic changes following CXCL12 treatment, with no increase in focal adhesions or protrusions. CXCL12-induced actin changes were restricted to the periphery of PRG-deficient cells, where it selectively induced formation of bundled actin (Fig. 3A, bottom panels).
Fig. 3.

PRG knockdown produces defects in focal adhesion and F-actin formation. (A) MCF7-CXCR4 cells were transfected with control or PRG-specific siRNA and plated on collagen-coated coverslips. Cells were serum-starved, treated with CXCL12 (10 nM, 20 minutes), fixed and probed with anti-vinculin and Alexa-Fluor-488–phalloidin. Overlay of vinculin (green) and phalloidin (red) labeling is shown in the ‘merged’ images. Arrows indicate areas of interest. The main image for PRG KD is of a cell cluster and the inset is that of a single PRG knockdown cell; both show the absence of internal actin and adhesion structures. (B) MCF7-CXCR4 cells stably expressing siRNA-resistant PRG (msPRG) were transfected with 5 µM PRG-specific siRNA and plated on collagen-coated coverslips. Cells were serum-starved, treated with CXCL12 (10 nM, 20 minutes), then processed as in A. Vinculin is shown in green, PRG in yellow, and Alexa-Fluor-488–phalloidin in red. White asterisks indicate cells with restored PRG expression, red arrows indicate actin- and adhesion-rich protrusions, and white arrows indicate cells with a typical PRG-deficient phenotype (large, symmetrical, with no protrusions and prominent cortical actin). (C) MCF7-CXCR4 cells were transfected with the indicated siRNAs and plated as a monolayer. 72 hours after transfection, monolayers were wounded, treated with CXCL12 (10 nM), and allowed to migrate overnight. The cells were then fixed and labeled as in A. Scale bars: 10 µm.
To verify the above observations were due to loss of PRG, we examined the cytoskeletal organization of cells in a rescue experiment. Fig. 3B demonstrates that expression of a siRNA-resistant PRG (msPRG) restored the actin and focal adhesion phenotype of cells transfected with PRG siRNA. A cell cluster with both high (white asterisks) and low levels of PRG shows cells with high PRG expression have obvious actin- and adhesion-rich protrusions following treatment with CXCl12 (red arrows). In contrast, adjacent cells with low PRG expression show the phenotype typical of PRG knockdown. These cells are large and symmetrical, with no protrusions and prominent cortical actin (white arrows). These data demonstrate that the cellular phenotype in PRG-depleted cells is due to lack of PRG and not due to off-target effects of siRNA transfection.
To determine whether the cytoskeletal alterations in PRG-deficient cells could account for the migration defects, we analyzed the actin and adhesion structures in cells during the wound closure assay. Control cells at the wound front protruded into the wound space with adhesions and actin stress fibers aligned in the direction of migration (Fig. 3C). Consistent with observations in sparsely plated cells, PRG-deficient cells do not protrude into the wound space and the actin fibers form a continuous cable along the wound front with focal adhesions distributed along the actin cable. The observed changes in the actin and adhesion characteristics provide insight into how loss of PRG prevents CXCR4-driven cell migration. The atypical cortical actin structure in PRG knockdowns appears to form a barrier to CXCR4-driven protrusion formation, while the lack of interior actin stress fibers prevents cells from generating the traction forces needed for motility.
The tightly clustered morphology observed in PRG-deficient cells suggested there were also alterations in cell–cell adhesion characteristics. E-cadherin is a key molecule involved in formation of adherens junctions, and E-cadherin expression is lost during the epithelial-mesenchymal transition that often accompanies breast cancer progression. In our previous study, we found that MCF7 cells expressing an activated mutant of CXCR4 showed a loss of E-cadherin expression, a more mesenchymal phenotype, and increased tumor cell motility and metastasis (Rhodes et al., 2011). We speculated PRG may regulate E-cadherin downstream from CXCR4, which may enhance tumor cell motility. To test this hypothesis we looked at expression of E-cadherin and β-catenin in MCF7-CXCR4 cells following PRG knockdown. Contrary to our expectations, we found PRG knockdown did not alter either E-cadherin or β-catenin expression (Fig. 4C). We then used immunofluorescence to investigate whether PRG influenced the localization of either protein. We found that localization of E-cadherin and β-catenin to cell–cell junctions was enhanced in PRG-deficient cells (Fig. 4B). Thus, loss of PRG did not increase E-cadherin expression, but clearly strengthened the presence of E-cadherin at cell junctions, which could act as a brake on CXCR4-promoted cell migration.
Fig. 4.
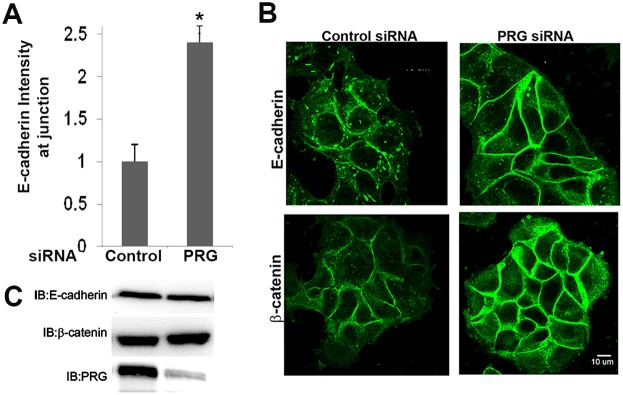
PRG knockdown enhances adherens junctions. (A) Quantification of E-cadherin labeling intensity at cell–cell junctions (*P = 0.03; values are means ± s.e.m.; 60 cells, n = 3). (B) Immunofluorescence of these same cells plated on collagen-coated coverslips and probed with anti-E-cadherin or anti-β-catenin antibody revealed enhanced localization of these proteins to cell–cell junctions in PRG-deficient cells. (C) Representative immunoblots for E-cadherin or β-catenin protein from cell lysates of control- and PRG-siRNA-transfected cells.
PRG is required for full activation of RhoA in response to CXCL12
Because PRG is Rho-specific GEF and its direct biochemical function is to activate RhoA, we investigated how changes in cell migration following PRG depletion correlated with RhoA activity, using an ELISA-based activity assay that detects GTP-loaded RhoA. CXCL12 increased GTP loading of RhoA in control siRNA-transfected cells (63% increase), verifying that RhoA is activated downstream of CXCR4 (Fig. 5A). Knockdown of PRG expression significantly (P<0.001) blocked stimulation of RhoA activity by CXCL12 (only 9% increase), establishing PRG in the signaling pathway between CXCR4 and activation of RhoA.
Fig. 5.

PRG activates RhoA downstream from CXCR4 and causes cell clustering. (A) Treatment with CXCL12 increased RhoA activity in control siRNA (P<0.001), whereas knockdown of PRG suppressed CXCL12-induced RhoA activation. C3-transferase (0.5 µg/ml, 4 hours) was used as a control for RhoA inhibition. RhoA activity in cellular lysates was determined by an ELISA-based protocol. Values are means ± s.e.m. of duplicate samples, n = 4. (B) Immunoblot analysis confirmed equivalent expression of RhoA in cell lysates and that PRG siRNA selectively reduced PRG expression. (C) MCF7-CXCR4 cells were transfected with control or PRG siRNA for 72 hours, or treated with C3-transferase (0.5 µg/ml, 4 hours). Phase-contrast images of cell morphology shows PRG knockdown induces cell clustering. Scale bars: 100 µm. (D) MCF7-CXCR4 cells stably expressing the Rho FRET biosensor were transfected with control or PRG siRNA and plated as a monolayer. Monolayers were scratched, treated with CXCL12 (10 nM), and allowed to migrate for 4 hours. Cells at the edge of the migrating monolayer were imaged for CFP and FRET and the FRET/CFP ratios, representative of RhoA activity, were calculated. Warmer colors correspond to high RhoA activity. High RhoA activity is present in the cell body and in membrane protrusions in control-siRNA cells, whereas RhoA activity is confined to the periphery of PRG-deficient monolayers. FRET emission ratios were plotted as a function of distance along lines drawn from cell edge to edge. A representative analysis line is shown in the picture and corresponding intensity plot from that line is shown in the inset. Averaged intensity plots are shown on the right (n = 24, 2 lines/cell, 3 cells/condition, n = 4). The dashed rectangle highlights area of greatest change in FRET emission (RhoA activity). Scale bar: 10 µm.
The inhibition of RhoA activity following PRG knockdown corresponds to distinct morphological changes, with PRG knockdown generating large, symmetrical, non-protrusive cells that formed tight clusters with neighboring cells (Fig. 5C; Fig. 3A). This morphology is very different from the morphological changes that resulted from treatment of cell with C3-transferase (Fig. 5C), which is known to produce a global inhibition of RhoA activity (Fig. 5A) (Kakudo et al., 2011). These observations suggested to us that PRG regulates the activity of a specific intracellular pool of RhoA and we thought that the observed changes in cell migration and morphology in PRG-deficient cells are due to dysregulation of RhoA activity at these sites.
RhoA FRET biosensor reveals that PRG knockdown profoundly alters the localization of RhoA activity
We directly tested the requirement of PRG for spatial regulation of RhoA using a well-characterized FRET biosensor to detect active RhoA (Machacek et al., 2009; Pertz and Hahn, 2004; Pertz et al., 2006). We created a wound in a monolayer of MCF7-CXCR4 cells stably transfected with the RhoA biosensor and measured RhoA activity only in cells migrating from the wound edge. In control siRNA-transfected cells, RhoA activity was high throughout the cell body, which is consistent with the role of RhoA in regulating the contractile machinery of the cell during cell migration (Fig. 5D). High RhoA activity was also seen in cellular protrusions that were extending into the open space, which is also consistent with reports showing high RhoA activity at the leading edge of the cell (El-Sibai et al., 2008; Pertz et al., 2006). As predicted, based on the differences in their morphology, the normal spatial regulation of RhoA activity required PRG. In cells lacking PRG, high RhoA activity was nearly absent from the cell body and cell junctions, and instead, was almost exclusively found at the periphery of the cell cluster. This pattern overlaps with that of the abnormal cortical actin structures previously observed in PRG-deficient cells (Fig. 3). The biosensor data show that PRG is required for RhoA activity in the center of cell and suggests that the specific spatial regulation of RhoA by PRG is an essential component of CXCR4-driven migration.
PRG is required for localization of RhoA activity in migrating MDA-MB-231 cells
Although the MCF7-CXCR4 cell line has enhanced migratory responses to CXCL12 (Rhodes et al., 2011; Struckhoff et al., 2010), it still retains most of the epithelial characteristics and relatively low migration rates of parental MCF7 cells. We speculated that PRG depletion may be a less potent inhibitor of migration of invasive, mesenchymally transformed breast cancer cells. MDA-MB-231 cells are a highly migratory and invasive breast cancer cell line, with high endogenous expression of CXCR4 (Lechertier et al., 2004; Kang et al., 2005; Zhao et al., 2008). They have undergone EMT and therefore do not express E-cadherin or form cell–cell junctions. We generated MDA-MB-231 cells stably expressing the RhoA biosensor to determine whether PRG depletion altered the spatial distribution of RhoA activity, and particularly the central pool of active RhoA, in these highly migratory cells. In 231 control cells, we observed an asymmetric cellular distribution of RhoA activity, with a wide region of high RhoA activity extending from the nucleus to the back of the cell (as highlighted by the FRET emission ratio line profile, blue box in Fig. 6A), and a sharp band of RhoA activity at the leading edge (Fig. 6B). In PRG-deficient cells, total RhoA activity was reduced and the cells lacked the asymmetric distribution of RhoA activity found in control cells. Rather than identifying a wide region of active RhoA in the rear of the cell and in a band at the protrusive front, PRG-depleted cells had only a thin band of RhoA activity around the periphery of the cell. These results are consistent with those observed in MCF7-CXCR4 cells, where loss of PRG resulted in a redistribution of active RhoA from the cell body and protrusive sites to the cell periphery, leading us to conclude that PRG is required for spatial regulation of RhoA in both cell types.
Fig. 6.
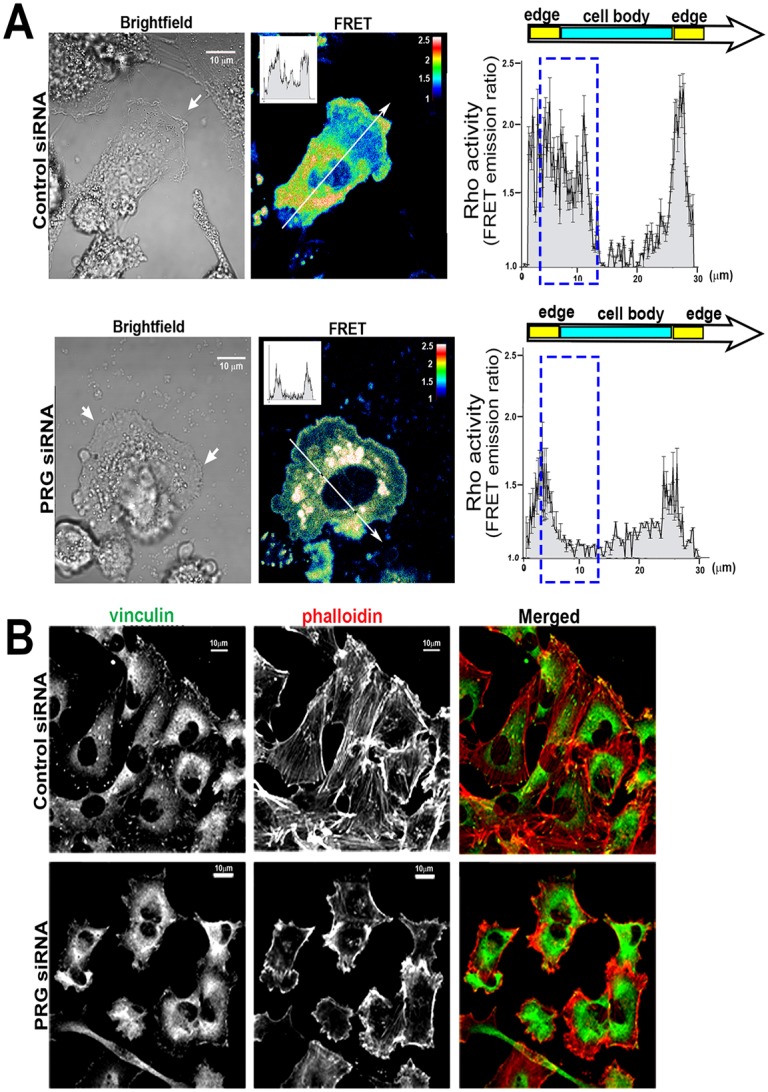
PRG knockdown causes redistribution of RhoA activity and cytoskeletal defects in the MDA-MB-231 cell line. (A) MDA-MB-231 cells stably expressing the RhoA FRET biosensor were transfected with control or PRG siRNA and imaged as described in Fig. 5. Control cells show asymmetric distribution of RhoA activity with high RhoA activity localized to the protrusive front, and the cell body from the nucleus to the rear of the cell. In PRG-depleted cells, total RhoA activity appears reduced and the polarized distribution is lost and is instead found in a thin band at the periphery and in regions of the cytoplasm. RhoA activity plotted along the length of control or PRG-deficient cells (n = 18, 2 lines/cell, 3 cells/condition, n = 3) is shown on the right. Dashed rectangles highlight areas of FRET emission (RhoA activity) change in the cell body. Inset graphs in the FRET images are intensity plots for the representative lines drawn in the main images. (B) Representative immunofluorescence images of PRG-deficient MDA-MB-231 cells plated on collagen-coated coverslips, fixed, and probed with anti-vinculin and Alexa-Fluor-488–phalloidin. An overlay of vinculin (green) and phalloidin (red) labeling is shown in the ‘merged’ image. Scale bars: 10 µm.
PRG is required for cytoskeletal organization in MDA-MD-231 cells
We then examined actin and adhesion structures by microscopy to determine whether changes in RhoA activity distribution corresponded to defects in MDA-MB-231 cytoskeletal organization, as observed in MCF7-CXCR4 cells. Control MDA-MB-231 cells were asymmetric with a broad protrusive front containing large focal adhesions that capped prominent actin stress fibers (Fig. 6B, top). In contrast, F-actin and focal adhesions were severely disrupted in PRG-depleted cells with few focal adhesions and diffuse vinculin localization (Fig. 6B, bottom). There were no internal actin stress fibers and instead we observed disorganized actin structures concentrated mainly at the cell cortex. These cytoskeletal changes correlate with the distribution changes of active Rho shown in Fig. 6A, and further confirm that PRG normally acts to localize active Rho to internal cellular sites and is needed for proper organization of cellular migration machinery.
PRG is required for CXCR4-driven migration and polarization
Fig. 6 demonstrates that PRG depletion disrupted the normal distribution of active RhoA and cytoskeletal machinery in MDA-MB-231 cells, similarly to the MCF7-CXCR4 cell line, making it probable that PRG was also required for the cell migration response in MDA-MB-231 cells. However, because of the high migration capacity and lack of cell–cell junctions in MDA-MB-231 cells, we hypothesized that the inhibition of cell migration would not be as profound as that in MCF7-CXCR4 cells. To test this hypothesis, we used a wound closure assay similar to that outlined in Fig. 2B to measure cell migration following loss of PRG. Since MDA-MB-231 cells migrate as individual cells, we modified our migration assay and tracked the movement of individual MDA-MB-231 cells. Quantification of distances traveled by control or PRG-deficient cells demonstrated that PRG depletion reduced basal cell migration and significantly inhibited the migration response to CXCL12 (58% decrease from control, Fig. 7A, left panel).
Fig. 7.

PRG is required for MDA-MB-231 migration and invasion and results in loss of migrational polarity. (A) MDA-MB-231 cells were transfected with control or PRG siRNA, then individual cell paths were tracked and total distance traveled was calculated. For each condition, cells in three separate locations on the wound were imaged and displacement values (µm) were averaged. CXCL12 (10 nM) stimulated migration in control siRNA-transfected cells (P = 0.002), whereas PRG knockdown prevented CXCL12-stimulated migration (P = 0.182) and decreased basal cell migration (P = 0.001). Directional persistence was calculated as described in Materials and Methods and is represented in a box-and-whisker plot (error bars show maximum and minimum values, box = 25% and 75% quartiles, line = mean persistence). Representative migration tracks are shown, and phase-contrast images show the cells at the end of the assay (20 hours), with the boundary of the wounds at the beginning of the assay indicated by superimposed white lines. Values are means ± s.e.m., 90–120 cells, n = 3. (B) MDA-MB-231 cells were transfected with control or PRG siRNA, plated as a monolayer, and scratch wounds were made. Six hours after wounding, the cells were fixed and probed with β-tubulin and anti-pericentrin (MTOC) antibodies. The percentage of cells with oriented MTOC was calculated as described in Materials and Methods. PRG knockdown significantly reduced the number of cells with polarized MTOC. Values are means ± s.e.m., 54–80 cells, n = 3. (C) PRG knockdown prevents MDA-MB-231 invasion into a 3D collagen matrix. Control- and PRG-siRNA-transfected MDA-MB-231 cells were plated in glass-bottomed dishes, overlayed with collagen (2 mg/ml), and cultured for 48 hours in SFM with 10 nM CXCL12. Cells were fixed, stained with Alexa-Fluor-488–phalloidin, and confocal images were taken at 5 µm intervals through 150 µm of the collagen matrix. 3D projections (top panel) and X-Z stacks (middle panel) of MDA-MB-231 in 3D collagen matrix revealed a nearly complete inhibition of invasion following PRG knockdown. The fluorescence intensity distributions 0–30 µm and 30–150 µm from the origin were calculated (bottom panel).Values are means ± s.e.m., n = 3.
Time-lapse movies from 231 cell migration assays (supplementary material Movies 1, 2) revealed that PRG-deficient cells appeared unable to sustain prolonged directional migration and instead traveled in random, meandering paths, suggesting PRG depletion led to defects in directional migration persistence. We calculated directional persistence in control and PRG-deficient MDA-MB-231 cells and found there was a downward shift in directional persistence following PRG depletion in MDA-MB-231 cell population and a 30% decrease in mean directional persistence. The reduced cytoskeletal asymmetry (Fig. 6B) and the decrease in directional persistence (Fig. 7A, center) suggested that PRG depletion led to loss of cell polarity, which would contribute to reduced cell migration. To test this, we immunolabeled cells with anti-β-tubulin and anti-pericentrin [a marker of the microtubule-orienting center (MTOC)] in migrating cells. Cells with MTOCs oriented in front of the nucleus towards the empty space were scored as positive for polarization. The majority of control cells (66.2%) had aligned MTOCs and the microtubule cytoskeleton projecting in the direction of migration (Fig. 7B). In contrast, PRG knockdown prevented polarization of the MTOC, with less than 30% of cells polarized to the wound edge.
Altering PRG expression in MDA-MB-231 cells had potent effects on the distribution of active RhoA, cytoskeletal organization, and polarity, but the inhibition of migration shown in MDA-MB-231 cells (Fig. 7A) was more modest than in MCF7-CXCR4 cells (only 50–60%) suggesting MDA-MB-231 cells retained substantial invasive and metastatic capacity. Therefore we measured the effect of PRG knockdown on the invasiveness of MDA-MD-231 cells in three-dimensional (3D) collagen gels. 3D environments more closely mimic the in vivo environment found in tissues and often reveal aspects of migration not identifiable in a two-dimensional system. We used confocal microscopy to determine the ability of MDA-MB-231 cells to invade into a 3D collagen matrix. We set 30 µm as the cutoff for invasion distance because cells that failed to invade remained below this distance. Using this method we observed that over 40% of the intensity of actin labeling was detected above the threshold distance in control siRNA cells (Fig. 7). 3D images revealed that many control cells migrated considerably further than the 30 µm threshold point, as we detected cells throughout the entire height of the gel with some cells migrating distances of up to 150 µm (Fig. 7C, top panels). In contrast, PRG knockdown prevented cell invasion, with only 8% of the actin intensity detected above the threshold distance. The difference between the control and PRG knockdown 3D projection images was particularly striking. Control cells were observed at all distances in the matrix, whereas we only rarely observed PRG knockdown cells in the higher regions of the collagen matrix. Figs 6 and 7 demonstrate that PRG is required for normal polarized orientation of migration machinery including the asymmetric spatial distribution of the active RhoA, F-actin, focal adhesions, and microtubules as the normal organization of each of these cytoskeletal elements in MDA-MB-231 cells is missing PRG-depleted cells.
Furthermore, these results demonstrate that PRG is required by MDA-MB-231 for invasion and that PRG is an essential component of cell motility in multiple breast cancer cell types.
PRG expression is associated with breast tumor invasion in vivo
To test whether PRG expression is associated with human breast cancer, we measured its expression by immunohistochemistry in a panel of 20 archival human breast tumor samples. PRG expression was measured in multiple areas of each tumor, categorized as in situ, invasive or lymphatic emboli. Fig. 8A shows images of PRG staining in several areas of a single tumor sample, in which PRG expression increases in the invasive areas. For each case, the level of PRG expression was scored on a scale of 0–4 and demonstrates a significant increase in the invasive areas and lymphatic emboli (Fig. 8B). Analysis of the difference in PRG expression between areas within the same tumor sample show significant increase in invasive areas over in situ (solid tumors that have invaded the surrounding stroma or individual cells that have spread to stromal and adipose tissue), and in lymphatic emboli over invasive areas (Fig. 8C). Thus, we find that consistent with our in vitro data, high PRG expression is correlated with an invasive phenotype in human breast cancer.
Fig. 8.

Expression of PRG in human ductal breast carcinomas. (A) Immunohistochemistry for PRG. In the in situ component (first panel) of a ductal breast carcinoma there is very weak labeling in in situ carcinoma cells (IS), and none in the stroma (S). Cells in the central tumor areas (solid tumor areas, second panel) demonstrate moderate expression of PRG. In contrast, less differentiated areas of invasive tumor infiltrating adipose (A) tissue show robust cytoplasmic PRG (third panel). Finally, neoplastic cells in tumor emboli inside lymphatic vessels show robust membrane-associated and cytoplasmic expression of PRG (fourth panel). Original magnification was 200×; scale bars: 10 µm. (B) PRG expression was scored on a scale of 0–4 from three different areas within the breast tumor sections (in situ, invasive or lymphatic emboli) and means ± s.e.m. were plotted for each area. (C) The change in PRG expression between areas within the same tumor was calculated and the means ± s.e.m. were plotted.
Discussion
CXCL12 signaling was first associated with tumor progression and metastasis 10 years ago, and studies since then have demonstrated that the involvement of CXCR4 in cancer is multi-faceted and includes the regulation of growth, motility and metastatic homing of primary tumor cells, as well as modification of the tumor microenvironment (Balkwill, 2004; Teicher and Fricker, 2010; Zlotnik, 2006). Our study is focused on CXCR4 signaling events that activate RhoA to regulate tumor cell motility. We screened the three RGS-RhoGEFs (LSC, LARG, and PRG) that were good candidates linking CXCR4 stimulation to RhoA activation, and discovered that PRG was essential for tumor cell motility (Figs 2, 7) and its depletion prevented CXCR4-driven tumor cell migration and invasion. Previous studies with fibroblasts and lymphocytes yielded conflicting results regarding the role of PRG in migration, but here we have clearly determined that PRG is essential for basal and CXCR4-driven breast tumor cell motility by using breast tumor cell lines that overexpress CXCR4: the weakly invasive MCF7-CXCR4 cells and the highly invasive MDA-MB-231 cells. Significantly, these findings correlate with our study of human breast tumor tissue samples where higher levels of PRG are observed in highly invasive neoplastic cells invading surrounding adipose tissue and inside lymphatic vessels, in contrast to in situ areas where PRG expression is weak, suggesting a direct correlation between PRG expression and an invasive phenotype.
To determine the cell biological mechanism responsible for PRG-dependent motility, we examined the cytoskeletal organization of the breast tumor cell lines by microscopy. In MCF7-CXCR4 cells, loss of PRG resulted in robust cell clustering (Fig. 3), and enhanced E-cadherin accumulation at cell junctions (Fig. 4). These data are consistent with reports demonstrating a role for Rho signaling in negatively regulating adhesion junction formation (Sahai and Marshall, 2002; Samarin et al., 2007), and with two recent papers demonstrating a role for PRG in the regulation of apical constriction and tight junctions formation in polarized epithelial tissues (Itoh et al., 2012; Nishimura et al., 2012). Similar to these studies, our FRET biosensor experiments showed an absence of RhoA activity (Fig. 5) and phosphorylated MLC (data not shown) at cell junctions in the absence of PRG, suggesting that PRG also modulates cellular junctions and tension in our system. However, our experimental system revealed another role for PRG at the base of the cell where we examined cytoskeletal structures involved in migration and invasion.
Our analysis of the actin cytoskeleton and integrin adhesion complexes revealed that organized actin and adhesion complexes in the center of the cell were completely absent in PRG knockdowns, but actin and adhesion complexes on the cell periphery were enhanced (Fig. 3). The strong band of actin and adhesions along the front of the wound edge, along with the increased E-cadherin adherens junctions, provides a cell biological mechanism to explain the lack of protrusions and migration observed in the absence of PRG in MCF7-CXCR4 cells. The mesenchymally transformed MDA-MB-231 cells do not express E-cadherin and therefore enhanced junction formation cannot contribute to the reduced motility in the absence of PRG. However, similar to the MCF7-CXCR4 cell line, PRG knockdown results in a loss of central actin and adhesion structures while retaining strong cortical actin (Fig. 6B). This was accompanied by a loss of a polarized F-actin front and a strong loss of polarity in the migration assay (Fig. 7A). This is consistent with a previous study in neuronal cells that implicated PRG in the acquisition of neuronal polarity (Longhurst et al., 2006). Together our data show that PRG regulates the motility of breast tumor cells of diverse phenotypes by controlling the organization of cytoskeletal structures in the center of the cell.
Spatial regulation of RhoA activity is known to be important during cell migration in many different systems (Ernkvist et al., 2009; Heasman et al., 2010; Machacek et al., 2009; Nalbant et al., 2009; Struckhoff et al., 2011; Timpson et al., 2011). Thus, to further investigate the molecular mechanisms underlying the role of PRG in cell motility and cytoskeletal regulation we tested its effects on the localization of RhoA activity using a FRET-based biosensor. This assay revealed a very distinctive change in the localization pattern of RhoA activity in PRG knockdown cells (Fig. 5D). Specifically, loss of PRG removed RhoA activity from the cell interior, while leaving significant RhoA activity in the periphery of the cell. This pattern of RhoA activity mirrors the spatially restricted effects on the cytoskeleton, in which internal actin and adhesion structures are lost, but peripheral structures are retained or even enhanced. Thus, in addition to acting as a RhoGEF to catalyze the activation of RhoA, PRG is required for normal spatial regulation of RhoA activity that controls cytoskeletal organization during breast tumor cell motility.
PRG is known to bind actin stress fibers and to associate with microtubules where it is predicted to modulate Rho signaling at these specific sites to control cytoskeletal dynamics and, ultimately, cell migration. PRG localizes microtubules through its interaction between its PDZ domain and microtubule-associating protein-1 (MAP1) (Longhurst et al., 2006). Localization to the microtubules was required for cytoskeletal organization as drug-induced depolymerization of the microtubules or MAP1-binding-deficient PRG mutants had defects in neurite extension and polarity, as a consequence of dysregulated RhoA. Spatial regulation of RhoA at microtubules has been reported for the other two RGS-RhoGEF family members (Goulimari et al., 2008; Slattum et al., 2009), suggesting that modulation of RhoA at microtubules is a common feature of RGS-RhoGEF family members. The concept of RhoGEFs directing the spatial regulation of Rho to achieve specific biological outcomes is also well established for other RhoGEFs. GEF-H1 localizes to cell–cell junctions and cellular protrusions to direct regulate Rho-dependent cell biological events (Nalbant et al., 2009; Birkenfeld et al., 2007), and Ect2 modulates localized RhoA activity at the cleavage furrow of dividing cells during cytokinesis (Su et al., 2011), as well as cell–cell junctions (Ratheesh et al., 2012). Whether the localization of PRG to internal pools of actin or microtubules is responsible for the central activation of Rho and downstream effectors to regulate cytoskeleton organization, E-cadherin localization and polarity remains to be determined.
Materials and Methods
Reagents
Reagents were obtained as follows: recombinant CXCL12, R&D Systems (Minneapolis, MN); collagen-1, BD Biosciences (Bedford, MA) or Invitrogen (Carlsbad, CA); G-LISA RhoA activity assay and C3-transferase, Cytoskeleton (Boulder, CO); vinculin and β-tubulin antibodies, Sigma-Aldrich (St Louis, MO); anti-GFP, anti-LSC, anti-LARG, anti-PRG, Santa Cruz (Santa Cruz, CA); β-actin, Cell Signaling (Danvers, MA); anti-pericentrin, Covance (Princeton, NJ). Lipofectamine 2000, RNAiMAX, and Hoechst 33342 were purchased from Invitrogen. siRNA oligonucleotides (LSC: 5′-CATACCATCTCTACCGACG-3′, LARG: 5′-GAAACTCGTCGCATCTTCC-3′ and PRG: 5′-ACTGAAGTCTCGGCCAGCT-3′) (Zheng et al., 2006) were synthesized by Dharmacon.
pQCXIB-CMV/TO-DEST (w320) (created by Dr. Eric Campeau) and pBabe-puro-Rho biosensor (created by Dr. Klaus Hahn) were obtained from Addgene. The RGS domain of p115 RhoGEF (pENTR-RGS-p115), EGFP-DEST and mouse PRG (pENTR-msPRG) were gifts of Dr AdiDubash. Mouse PRG was subcloned into pQCXIB-CMV/TO-DEST to generate pQCXIB-msPRG by recombination (Invitrogen).
Cell culture
MCF7-CXCR4 cells stably overexpress the CXCR4 chemokine receptor in MCF7 cells (Mao et al., 2011; Rhodes et al., 2011; Struckhoff et al., 2010). MDA-MB-231 cells, obtained from ATCC (Manassas, VA), and Phoenix ecotropic retrovirus packaging cell lines (Garry Nolan via National Gene Vector Biorepository) were cultured in DMEM with 10% FBS and antibiotics.
Generation of RhoA FRET biosensor stable cell lines
Phoenix ecotropic packaging cells were transfected with pBABE-puro-RhoA FRET biosensor using calcium phosphate, and virus was collected at 36 hours and 48 hours after transfection. MCF7-CXCR4 or MDA-MB-231 cells were infected with viral supernatant plus Lipofectamine 2000 and selected with 2 µg/ml puromycin.
Generation of the siRNA-resistant PRG stable MCF7-CXCR4 cell line
The mouse PRG (msPRG) cDNA differs from human PRG by three nucleotides in the sequence targeted by the PRG siRNA, rendering it siRNA resistant. Invitrogen Gateway cloning was used to clone mouse full-length PRG (pENTR-PRG) into the retroviral vector pQCXIB-CMV/TO-DEST. MCF7-CXCR4 cells were infected with msPRG and stably expressing cells were selected with 8 µg/ml blasticidine.
Cell migration
To assess the role of Gα12/13 in CXCR4-regulated migration, cells were transfected with either GFP vector or GFP-RGS-p115 using Lipofectamine 2000. Migration experiments were begun 24 hours after transfection. For RGS-RhoGEF experiments, cells were transfected with RhoGEF siRNA (10 nM per well) using RNAiMAX (5 µl/10 nM).
Monolayers were scratched to form a wound and culture medium was replaced with serum-free DMEM or serum-free DMEM with 10 nM CXCL12 or 0.5 µg/ml C3-transferase (Rho inhibitor). Initial (t = 0 hour) and final (t = 20 hour) phase-contrast images were taken using an Olympus IX71 microscope with a 10× objective (NA = 0.87). Cells were maintained at 37°C, in 5% CO2 using a LiveCell Environmental Chamber (NEUE Group). The total area of the monolayer wound at 0 hours (t0) and 20 hours (t20) was determined using ImageJ software and cell migration was calculated as the change in wound area (t20−t0). Cell migration under various conditions was normalized to migration of non-stimulated, control cells (100%).
Single-cell analysis was used to measure migration in MDA-MB-231 cells. Migration assays were initiated as described for MCF7-CXCR4 cells, with images acquired every 30 minutes for 24 hours in order to create time-lapse movies. Individual cells (25–30 cells per field) were tracked and total displacement was calculated using Slidebook software. Cell migration was normalized to migration of non-stimulated, control siRNA-transfected cells (100%).
Directional persistence was calculated as a ratio of net migration distance versus total migration distance as described previously (Burdisso et al., 2013).
For all migration assays, Student's t-tests were used to determine significance of changes using GraphPad Prism software.
RhoA activity assays
To measure Rho GTP levels, cells were transfected with control or RGS-RhoGEF-specific siRNA as described above. After 72 hours, cells were serum-starved for 16 hours then treated with 10 nM CXCL12 for 10 minutes. Cells were lysed and RhoA activity was measured by G-LISA assay (Cytoskeleton). Levels of active RhoA obtained from the G-LISA assay were normalized by protein input levels. Additionally, equivalent amounts of cellular lysates were taken from assay lysates and analyzed by immunoblotting to confirm knockdown of RGS-RhoGEF expression. Student's t-tests were used to determine significance of differences using GraphPad Prism software.
FRET imaging of RhoA activity
MCF7-CXCR4 or MDA-MB-231 cells stably expressing the RhoA FRET biosensor were transfected with control or RGS-RhoGEF siRNA as described above. 6 hours after wounding cells at the wound edge were imaged in phenol-free DMEM. Confocal images were acquired with an Olympus BX51 laser scanning confocal microscope fitted with their Fluoview1000 system. Images were visualized through a 60× oil immersion objective. A 405 nm Ar/HeNe laser refined by a 405/473 dichroic mirror was used to excite CFP, and the CFP and FRET (CFP) emission signals were simultaneously recorded using BA465-495 nm (CFP) and BA535-565 nm (FRET) emission filters. The FRET emission signal was divided by the CFP signal to create a ratiometric image, which is reflective of the RhoA activity within the cell (Machacek et al., 2009; Pertz et al., 2006). A linear pseudocolor lookup table was applied to all ratio images with warm and cool colors representing high and low RhoA activity, respectively. Ratio values were normalized to the lower scale value, which was chosen to exclude the bottom 7% of the total histogram distribution to avoid areas of low signal-to-noise ratio (Machacek et al., 2009). Images were acquired and processed with Fluoview 1000 software, and exported as BMP files for further editing with Adobe Photoshop. For line profiles, FRET emission ratio images were rescaled so that all cells analyzed were approximately the same size. Emission ratios along a line drawn edge-to-edge through the long axis of the cells were recorded using Fluoview 1000 software. Compiled data for each condition were plotted as means ± s.d. using GraphPad Prism software.
Immunocytochemistry
MCF7-CXCR4 or MDA-MB-231 cells were transfected with control or RGS-RhoGEF siRNA as described above for cell migration assays. 48 hours after transfection, cells were plated onto collagen-1-coated coverslips (10 mg/ml) at high and low cell densities. Cells were fixed and labeled as previously described (Struckhoff et al., 2010). Images were obtained using the Olympus BX51 laser scanning confocal system, as described above. Quantification of E-cadherin intensity at cell–cell junctions was measured by extracting the integral intensities from a three-pixel-wide line around the perimeter of the cells using Fluoview software.
For analysis of MTOC polarity, MDA-MB-231 cells were transfected with control or PRG siRNA as described for cell migration assays. 48 hours after transfection, cells were plated onto collagen-1-coated coverslips. Scratches were made in confluent monolayers, and 6 hours later were fixed with ice-cold methanol/EGTA, stained with anti-β-tubulin and anti-pericentrin, and images were obtained and processed as described above. Cell polarity was scored positive if the MTOC fell within a 90° angle drawn relative to the wound front (Etienne-Manneville and Hall, 2003; Goulimari et al., 2008). Only cells along the wound edge were scored.
Immunoprecipitation and immunoblotting
Cells were serum-starved, treated with CXCL12 (10 nM, 10 minutes), washed with PBS, and lysed in TBS 1% Triton X-100 (TX) buffer with protease and phosphatase inhibitors. The lysates were precleared by incubation with protein-G–agarose beads, followed by incubation with a phosphotyrosine-specific antibody (clone 4G10) overnight at 4°C. The immunocomplexes were collected by protein-G-agarose beads, washed in ice-cold TBS 1% TX buffer, boiled in sample buffer, and resolved by SDS-PAGE. Immunoblots were performed as described previously (Struckhoff et al., 2010).
Three-dimensional invasion assay
MDA-MD-231 cells were transfected with control or PRG siRNA as described above. 48 hours after transfection, cells were plated on acid-etched glass coverslips (Mattek). The following day, siRNA-transfected cells were overlayed with collagen-1 gels (2 mg/ml) and cultured in complete medium for 48 hours (Hooper et al., 2006). The gels were fixed with 4% paraformaldehyde-PBS and stained with Alexa-Fluor-594–phalloidin. The collagen matrix was imaged in 5 µm optical sections to a distance of 150 µm from the origin. Three-dimensional images were generated using Slidebook software and projection images were generated using Fluoview software. The fluorescence intensity distribution above and below 30 µm (0–30 µm and 30–150 µm) was calculated and graphically represented as a percentage of the total signal intensity.
Immunohistochemistry
A total of 20 cases of formalin-fixed, paraffin-embedded biopsy samples of breast carcinomas were obtained from our pathology archives. Tumor classification and grading was done according to the WHO classification of breast tumors. Immunohistochemistry was performed using the avidin-biotin-peroxidase methodology according to the manufacturer's instructions (Vector Laboratories, Burlingame, California) as previously described (Knight and Parsons, 1991).
For statistical analysis of PRG expression, tumor samples were assigned a PRG expression value from 0 to 4 (0 = no expression and 4 = robust expression). The values for the three pathologically defined areas of the tumor sample (in situ, invasive, lymphatic emboli) were averaged to generate a mean expression value across all tumor samples. Additionally, the difference between each tumor area within the same sample was computed (in situ versus invasive, in situ versus emboli, and invasive versus emboli). The Wilcoxon singed ranks test was used to determine significance between patients' expression level for each pair of immunohistochemistry PRG categories.
Supplementary Material
Acknowledgments
We thank all of the members of the LSU Cell Migration Group for ongoing input and critique of this research.
Footnotes
Author contributions
A.P.S. and R.W. conceived experiments and wrote the manuscript; A.P.S., M.R., and S.K. performed experiments; J.H. performed statistical analysis, M.B. generated MCF7-CXCR4 cells; and L.D.V. performed histological analysis.
Funding
This work was supported by the National Institutes of Health [grant numbers NS055644 and P20RR021970 to L.D.V]; the National Center for Research Resources [grant number P20RR020160 to R.W.]; and the National Institute of Dental and Craniofacial Research [grant number R03DE19166 to R.W.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.132381/-/DC1
References
- Aittaleb M., Boguth C. A., Tesmer J. J. G. (2010). Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 10.1124/mol.109.061234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. (2004). The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 14, 171–179 10.1016/j.semcancer.2003.10.003 [DOI] [PubMed] [Google Scholar]
- Bartolomé R. A., Gálvez B. G., Longo N., Baleux F., Van Muijen G. N., Sánchez-Mateos P., Arroyo A. G., Teixidó J. (2004). Stromal cell-derived factor-1alpha promotes melanoma cell invasion across basement membranes involving stimulation of membrane-type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 64, 2534–2543 10.1158/0008-5472.CAN-03-3398 [DOI] [PubMed] [Google Scholar]
- Birkenfeld J., Nalbant P., Bohl B. P., Pertz O., Hahn K. M., Bokoch G. M. (2007). GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12, 699–712 10.1016/j.devcel.2007.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Oser M., Chen X., Eddy R., Hodgson L., Condeelis J. (2011). A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 21, 635–644 10.1016/j.cub.2011.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D. M., Mostov K. E. (2008). From cells to organs: building polarized tissue. Nat. Rev. Mol. Cell Biol. 9, 887–901 10.1038/nrm2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum R. J., Connolly B. A., Feig L. A. (2002). Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol. Cell. Biol. 22, 4073–4085 10.1128/MCB.22.12.4073-4085.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdisso J. E., González A., Arregui C. O. (2013). PTP1B promotes focal complex maturation, lamellar persistence and directional migration. J. Cell Sci. 126, 1820–1831 10.1242/jcs.118828 [DOI] [PubMed] [Google Scholar]
- Chikumi H., Fukuhara S., Gutkind J. S. (2002). Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF, and LARG by tyrosine phosphorylation: evidence of a role for focal adhesion kinase. J. Biol. Chem. 277, 12463–12473 10.1074/jbc.M108504200 [DOI] [PubMed] [Google Scholar]
- El-Sibai M., Pertz O., Pang H., Yip S. C., Lorenz M., Symons M., Condeelis J. S., Hahn K. M., Backer J. M. (2008). RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp. Cell Res. 314, 1540–1552 10.1016/j.yexcr.2008.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernkvist M., Luna Persson N., Audebert S., Lecine P., Sinha I., Liu M., Schlueter M., Horowitz A., Aase K., Weide T. et al. (2009). The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood 113, 244–253 10.1182/blood-2008-04-153874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S., Hall A. (2003). Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 421, 753–756 10.1038/nature01423 [DOI] [PubMed] [Google Scholar]
- Fukuhara S., Chikumi H., Gutkind J. S. (2001). RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene 20, 1661–1668 10.1038/sj.onc.1204182 [DOI] [PubMed] [Google Scholar]
- Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulimari P., Knieling H., Engel U., Grosse R. (2008). LARG and mDia1 link Galpha12/13 to cell polarity and microtubule dynamics. Mol. Biol. Cell 19, 30–40 10.1091/mbc.E06-11-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C., Brégeon J., Toumaniantz G., Rolli-Derkinderen M., Retailleau K., Loufrani L., Henrion D., Scalbert E., Bril A., Torres R. M. et al. (2010). The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 16, 183–190 10.1038/nm.2079 [DOI] [PubMed] [Google Scholar]
- Harris T. J. C., Sawyer J. K., Peifer M. (2009). How the cytoskeleton helps build the embryonic body plan: models of morphogenesis from Drosophila. Curr. Top. Dev. Biol. 89, 55–85 [DOI] [PubMed] [Google Scholar]
- Heasman S. J., Carlin L. M., Cox S., Ng T., Ridley A. J. (2010). Coordinated RhoA signaling at the leading edge and uropod is required for T cell transendothelial migration. J. Cell Biol. 190, 553–563 10.1083/jcb.201002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper S., Marshall J. F., Sahai E., William E. (2006). Tumor cell migration in three dimensions. Meth. Enzymol. 406, 625–643 10.1016/S0076-6879(06)06049-6 [DOI] [PubMed] [Google Scholar]
- Iguchi T., Sakata K., Yoshizaki K., Tago K., Mizuno N., Itoh H. (2008). Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J. Biol. Chem. 283, 14469–14478 10.1074/jbc.M708919200 [DOI] [PubMed] [Google Scholar]
- Itoh M., Tsukita S., Yamazaki Y., Sugimoto H. (2012). Rho GTP exchange factor ARHGEF11 regulates the integrity of epithelial junctions by connecting ZO-1 and RhoA-myosin II signaling. Proc. Natl. Acad. Sci. USA 109, 9905–9910 10.1073/pnas.1115063109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakudo N., Kushida S., Suzuki K., Matsumoto N., Kusumoto K. (2011). Effect of C3 transferase on human adipose-derived stem cells. Hum. Cell 24, 165–169 10.1007/s13577-011-0033-0 [DOI] [PubMed] [Google Scholar]
- Kang H., Mansel R. E., Jiang W. G. (2005). Genetic manipulation of stromal cell-derived factor-1 attests the pivotal role of the autocrine SDF-1-CXCR4 pathway in the aggressiveness of breast cancer cells. Int. J. Oncol. 26, 1429–1434 [PubMed] [Google Scholar]
- Kelly P., Casey P. J., Meigs T. E. (2007). Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry 46, 6677–6687 10.1021/bi700235f [DOI] [PubMed] [Google Scholar]
- Knight P., Parsons N. (1991). Modification of myofibrils by fluorophore-induced photo-oxidation. J. Muscle Res. Cell Motil. 12, 183–191 10.1007/BF01774037 [DOI] [PubMed] [Google Scholar]
- Kucia M., Reca R., Miekus K., Wanzeck J., Wojakowski W., Janowska-Wieczorek A., Ratajczak J., Ratajczak M. Z. (2005). Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells 23, 879–894 10.1634/stemcells.2004-0342 [DOI] [PubMed] [Google Scholar]
- Kumar A., Kremer K. N., Dominguez D., Tadi M., Hedin K. E. (2011). Gα13 and Rho mediate endosomal trafficking of CXCR4 into Rab11+ vesicles upon stromal cell-derived factor-1 stimulation. J. Immunol. 186, 951–958 10.4049/jimmunol.1002019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechertier T., Berard M., Vassy R., Herve M. A., Crepin M. (2004). Transendothelial migration of two metastatic breast carcinoma cells depend on the SDF-lalpha-CXCR4 complexes. Anticancer Res. 24, 4011–4017 [PubMed] [Google Scholar]
- Longhurst D. M., Watanabe M., Rothstein J. D., Jackson M. (2006). Interaction of PDZRhoGEF with microtubule-associated protein 1 light chains: link between microtubules, actin cytoskeleton, and neuronal polarity. J. Biol. Chem. 281, 12030–12040 10.1074/jbc.M513756200 [DOI] [PubMed] [Google Scholar]
- Machacek M., Hodgson L., Welch C., Elliott H., Pertz O., Nalbant P., Abell A., Johnson G. L., Hahn K. M., Danuser G. (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103 10.1038/nature08242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghazachi A. A. (1997). Role of the heterotrimeric G proteins in stromal-derived factor-1alpha-induced natural killer cell chemotaxis and calcium mobilization. Biochem. Biophys. Res. Commun. 236, 270–274 10.1006/bbrc.1997.6937 [DOI] [PubMed] [Google Scholar]
- Mao L., Yuan L., Slakey L. M., Jones F. E., Burow M. E., Hill S. M. (2010). Inhibition of breast cancer cell invasion by melatonin is mediated through regulation of the p38 mitogen-activated protein kinase signaling pathway. Breast Cancer Res. 12, R107 10.1186/bcr2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Ortiz I., Bartolomé R. A., Hernández-Varas P., Colo G. P., Teixidó J. (2009). Overexpression of E-cadherin on melanoma cells inhibits chemokine-promoted invasion involving p190RhoGAP/p120ctn-dependent inactivation of RhoA. J. Biol. Chem. 284, 15147–15157 10.1074/jbc.M807834200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulinari S., Häcker U. (2010). Rho-guanine nucleotide exchange factors during development: Force is nothing without control. Small GTPases 1, 28–43 10.4161/sgtp.1.1.12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulinari S., Barmchi M. P., Häcker U. (2008). DRhoGEF2 and diaphanous regulate contractile force during segmental groove morphogenesis in the Drosophila embryo. Mol. Biol. Cell 19, 1883–1892 10.1091/mbc.E07-12-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N. et al. (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 10.1038/35065016 [DOI] [PubMed] [Google Scholar]
- Murdoch C. (2000). CXCR4: chemokine receptor extraordinaire. Immunol. Rev. 177, 175–184 10.1034/j.1600-065X.2000.17715.x [DOI] [PubMed] [Google Scholar]
- Nalbant P., Chang Y. C., Birkenfeld J., Chang Z. F., Bokoch G. M. (2009). Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 20, 4070–4082 10.1091/mbc.E09-01-0041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Honda H., Takeichi M. (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084–1097 10.1016/j.cell.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Pertz O., Hahn K. M. (2004). Designing biosensors for Rho family proteins – deciphering the dynamics of Rho family GTPase activation in living cells. J. Cell Sci. 117, 1313–1318 10.1242/jcs.01117 [DOI] [PubMed] [Google Scholar]
- Pertz O., Hodgson L., Klemke R. L., Hahn K. M. (2006). Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 440, 1069–1072 10.1038/nature04665 [DOI] [PubMed] [Google Scholar]
- Ratheesh A., Gomez G. A., Priya R., Verma S., Kovacs E. M., Jiang K., Brown N. H., Akhmanova A., Stehbens S. J., Yap A. S. (2012). Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nat. Cell Biol. 14, 818–828 10.1038/ncb2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes L. V., Short S. P., Neel N. F., Salvo V. A., Zhu Y., Elliott S., Wei Y., Yu D., Sun M., Muir S. E. et al. (2011). Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 71, 603–613 10.1158/0008-5472.CAN-10-3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704–1709 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J., Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- Rubin J. B. (2009). Chemokine signaling in cancer: one hump or two? Semin. Cancer Biol. 19, 116–122 10.1016/j.semcancer.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E., Marshall C. J. (2002). ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4, 408–415 10.1038/ncb796 [DOI] [PubMed] [Google Scholar]
- Samarin S. N., Ivanov A. I., Flatau G., Parkos C. A., Nusrat A. (2007). Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol. Biol. Cell 18, 3429–3439 10.1091/mbc.E07-04-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siehler S. (2009). Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41–49 10.1111/j.1476-5381.2009.00121.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattum G., McGee K. M., Rosenblatt J. (2009). P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J. Cell Biol. 186, 693–702 10.1083/jcb.200903079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soede R. D., Wijnands Y. M., Kamp M., van der Valk M. A., Roos E. (2000). Gi and Gq/11 proteins are involved in dissemination of myeloid leukemia cells to the liver and spleen, whereas bone marrow colonization involves Gq/11 but not Gi. Blood 96, 691–698 [PubMed] [Google Scholar]
- Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y. F., Singer W. D. (2007). Regulation of rho guanine nucleotide exchange factors by G proteins. Adv. Protein Chem. 74, 189–228 [DOI] [PubMed] [Google Scholar]
- Struckhoff A. P., Vitko J. R., Rana M. K., Davis C. T., Foderingham K. E., Liu C. H., Vanhoy-Rhodes L., Elliot S., Zhu Y., Burow M. et al. (2010). Dynamic regulation of ROCK in tumor cells controls CXCR4-driven adhesion events. J. Cell Sci. 123, 401–412 10.1242/jcs.052167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struckhoff A. P., Rana M. K., Worthylake R. A. (2011). RhoA can lead the way in tumor cell invasion and metastasis. Front. Biosci. 16, 1915–1926 10.2741/3830 [DOI] [PubMed] [Google Scholar]
- Su K. C., Takaki T., Petronczki M. (2011). Targeting of the RhoGEF Ect2 to the equatorial membrane controls cleavage furrow formation during cytokinesis. Dev. Cell 21, 1104–1115 10.1016/j.devcel.2011.11.003 [DOI] [PubMed] [Google Scholar]
- Suzuki N., Nakamura S., Mano H., Kozasa T. (2003). Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. USA 100, 733–738 10.1073/pnas.0234057100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N., Hajicek N., Kozasa T. (2009). Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 17, 55–70 10.1159/000186690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W., Martin D., Gutkind J. S. (2006). The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J. Biol. Chem. 281, 39542–39549 10.1074/jbc.M609062200 [DOI] [PubMed] [Google Scholar]
- Teicher B. A., Fricker S. P. (2010). CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 16, 2927–2931 10.1158/1078-0432.CCR-09-2329 [DOI] [PubMed] [Google Scholar]
- Terry S. J., Zihni C., Elbediwy A., Vitiello E., Leefa Chong San I. V., Balda M. S., Matter K. (2011). Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat. Cell Biol. 13, 159–166 10.1038/ncb2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervonen T. A., Partanen J. I., Saarikoski S. T., Myllynen M., Marques E., Paasonen K., Moilanen A., Wohlfahrt G., Kovanen P. E., Klefstrom J. (2011). Faulty epithelial polarity genes and cancer. Adv. Cancer Res. 111, 97–161 10.1016/B978-0-12-385524-4.00003-9 [DOI] [PubMed] [Google Scholar]
- Tesmer J. J. (2009). Structure and function of regulator of G protein signaling homology domains. Prog. Mol. Biol. Transl. Sci. 86, 75–113 10.1016/S1877-1173(09)86004-3 [DOI] [PubMed] [Google Scholar]
- Timpson P., McGhee E. J., Morton J. P., von Kriegsheim A., Schwarz J. P., Karim S. A., Doyle B., Quinn J. A., Carragher N. O., Edward M. et al. (2011). Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 71, 747–757 10.1158/0008-5472.CAN-10-2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Cabrero J. R., Rey M., Pérez-Martínez M., Ursa A., Itoh K., Sánchez-Madrid F. (2002). A role for the Rho-p160 Rho coiled-coil kinase axis in the chemokine stromal cell-derived factor-1alpha-induced lymphocyte actomyosin and microtubular organization and chemotaxis. J. Immunol. 168, 400–410 [DOI] [PubMed] [Google Scholar]
- Wang Q., Liu M., Kozasa T., Rothstein J. D., Sternweis P. C., Neubig R. R. (2004). Thrombin and lysophosphatidic acid receptors utilize distinct rhoGEFs in prostate cancer cells. J. Biol. Chem. 279, 28831–28834 10.1074/jbc.C400105200 [DOI] [PubMed] [Google Scholar]
- Yagi H., Tan W., Dillenburg-Pilla P., Armando S., Amornphimoltham P., Simaan M., Weigert R., Molinolo A. A., Bouvier M., Gutkind J. S. (2011). A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci. Signal. 4, ra60 10.1126/scisignal.2002221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Mueller B. M., DiScipio R. G., Schraufstatter I. U. (2008). Akt plays an important role in breast cancer cell chemotaxis to CXCL12. Breast Cancer Res Treat 110, 211–222 [DOI] [PubMed] [Google Scholar]
- Zheng R., Iwase A., Shen R., Goodman O. B., Jr, Sugimoto N., Takuwa Y., Lerner D. J., Nanus D. M. (2006). Neuropeptide-stimulated cell migration in prostate cancer cells is mediated by RhoA kinase signaling and inhibited by neutral endopeptidase. Oncogene 25, 5942–5952 10.1038/sj.onc.1209586 [DOI] [PubMed] [Google Scholar]
- Ziembicki J., Tandon R., Schelling J. R., Sedor J. R., Miller R. T., Huang C. (2005). Mechanical force-activated phospholipase D is mediated by Galpha12/13-Rho and calmodulin-dependent kinase in renal epithelial cells. Am. J. Physiol. 289, F826–F834 10.1152/ajprenal.00412.2004 [DOI] [PubMed] [Google Scholar]
- Zlotnik A. (2006). Chemokines and cancer. Int. J. Cancer 119, 2026–2029 10.1002/ijc.22024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.