

Abstract
Acetylcholine (ACh) rapidly increases cardiac K+ currents (IKACh) by activating muscarinic K+ (KACh) channels followed by a gradual amplitude decrease within seconds. This phenomenon is called short-term desensitization and its precise mechanism and physiological role are still unclear. We constructed a mathematical model for IKACh to examine the conditions required to reconstitute short-term desensitization. Two conditions were crucial: two distinct muscarinic receptors (m2Rs) with different affinities for ACh, which conferred an IKACh response over a wide range of ACh concentrations, and two distinct KACh channels with different affinities for the G-protein βγ subunits, which contributed to reconstitution of the temporal behavior of IKACh. Under these conditions, the model quantitatively reproduced several unique properties of short-term desensitization observed in myocytes: 1), the peak and quasi-steady states with 0.01–100 μM [ACh]; 2), effects of ACh preperfusion; and 3), recovery from short-term desensitization. In the presence of 10 μM ACh, the IKACh model conferred recurring spontaneous firing after asystole of 8.9 s and 10.7 s for the Demir and Kurata sinoatrial node models, respectively. Therefore, two different populations of KACh channels and m2Rs may participate in short-term desensitization of IKACh in native myocytes, and may be responsible for vagal escape at nodal cells.
Introduction
Vagal nerve stimulation causes the release of acetylcholine (ACh) from axonal termini and then decelerates the heartbeat by increasing the amplitude of muscarinic K+ current (IKACh) in pace-making cells (1–4). IKACh activation is gradually decreased to a quasi-steady-state level despite the continuous presence of ACh (2). This phenomenon is classified into two distinct categories by timescale: short-term desensitization, which occurs within seconds immediately after exposure to ACh, and long-term desensitization, which is observed on a scale of minutes to hours. Although the latter appears to be attributable to sequestration of available receptors by modulations such as phosphorylation and internalization (5–9), the precise mechanism and physiological role of the former are still a matter of debate (2,10–16).
ACh binding to M2 muscarinic receptor (m2R) liberates the βγ subunits (Gβγ) from pertussis-toxin-sensitive G proteins (Gi/o) that activate IKACh. Interestingly, prestimulation of the A1-adenosine receptor, another Gi/o-coupled receptor, prevents ACh-induced short-term desensitization (2). This cross talk between receptors suggests that m2R is not involved in short-term desensitization. Constituents downstream of receptor activation, such as G protein and the muscarinic K+ (KACh) channel, have been proposed to cause this phenomenon (2,10–16). However, how these constituents could quantitatively account for the physiological IKACh response has not been fully examined.
Short-term desensitization of IKACh is a cellular response against overstimulation by ACh. At the organ level, excess vagal nerve stimulation eventually causes cardiac asystole, followed by resumption of the heartbeat. This desensitization is called vagal escape and has been explained by compensation from the sympathetic system (17–20). Although ACh causes short-term desensitization of IKACh and vagal escape, the functional relevance of these two different phenomena has not been previously examined.
A previously proposed mathematical model for IKACh activation consisted of only a single population of m2R, KACh channel, and G protein with the chemical cycle reactions (21). However, this linear system was insufficient to reproduce the temporal behavior of IKACh, suggesting the need for another hypothesis for the simulation of IKACh activation. At the macroscopic level, short-term desensitization is observed at higher ACh concentrations ([ACh]s) (2). The open probability of the KACh channel declines until a steady state is reached (22), and is modulated by the concentration of phosphatidylinositol 4,5-bisphosphate (PIP2) and the phosphorylation state (23,24). These observations allowed us to hypothesize an additional fraction of KACh channels that decrease their activity in a time-dependent manner. Because this fraction was expected to develop at higher [ACh]s, we designated them as KACh channels with low affinity for Gβγ (KACh/low channel). To differentiate between two populations of KACh channels, another fraction was presumed to have high affinity to Gβγ (KACh/high channel). This assumption contributed to reproduction of the apparent response of the peak and quasi-steady-state components in short-term desensitization, the effects of ACh preperfusion, and the recovery from short-term desensitization experimentally observed in atrial myocytes. We also implemented two populations of m2Rs with high and low affinities for ACh (25–29) to confer the IKACh response over a wide range of [ACh]s. Furthermore, integration of the these hypotheses reconstituted vagal escape when incorporated into mathematical models of action potential in the sinoatrial node (30–32). These results suggest that short-term desensitization may contribute to vagal escape originating in the sinus node.
Materials and Methods
KACh channel model
In this study, we used Monod’s allosteric model (33,34) to simulate the interaction between Gβγ and the KACh channels. Previously, we showed that Gβγ does not affect channel gating at steady state, but does increase the number of functionally active channels to enhance total channel activity (34). The allosteric model with four subunits could describe this regulation of functionally available channels; therefore, we used the allosteric model of a previous IKACh model study to reproduce the major features of steady-state IKACh and relaxation (21). To study the temporal behavior of IKACh, we adopted the experimental observation that high [ACh] modulates the gating kinetics of the KACh channel during short-term desensitization (22). To present the kinetically different populations in this phenomenon (22,35), we used two allosteric models with different affinities. Although the allosteric models of the KACh/low and KACh/high channels are structurally identical, they have different dissociation constants (Fig. 1, top). Similarly to the previous allosteric model (21,34), the KACh channel consists of four subunits. The KACh channel is a heterotetramer of Kir3.1 and Kir3.4. Because Kir3.1 homotetramers are not functional and the electrophysiological properties of functional Kir3.4 homotetramers are quite different from those of the KACh channel (36), we used four subunits with equal affinity and aimed at the reproduction of the experimental activation curve (Fig. S1 A in the Supporting Material). All of the subunits in a given channel are defined as being in the same state, either tense (T) or relaxed (R), and the subunits change state together (concerted transition). One Gβγ protein equilibrates with one subunit in either the tense or relaxed state with distinct dissociation constants defined as KT for tense and KR for relaxed. A tetramer without any Gβγ transitions between the tense and relaxed states according to the equilibrium constant L. The channels in the relaxed state are considered available to open with fast gating kinetics, and the channels in the tense state are considered unavailable. Gβγ generated by the G-protein cycle model was used in the allosteric model. By solving a system of equations (21,34), the channel availabilities for populations with high and low affinities (NPo,high and NPo,low, respectively) can be expressed by Eqs. 1 and 2:
| (1) |
| (2) |
The same parameters (L, the ratio between KT,i and KR,i) validated in the previous study were used so that the experimental densitometry profiles of membranes containing Kir3.4 preincubated with Gβγ (i.e., the binding relationship between KACh channel and Gβγ) could still be reproduced (21,37) (Table 1). The values of KT,high and KR,high successfully reproduced the experimental activation curve of the KACh channel by Gβγ (38) (Fig. S1 A). A simple sensitive analysis was conducted to see the effects of the KD of the KACh/low channel on the quantitative reproduction of the peak current (IP) and quasi-steady-state current (IS) (Fig. S2 A). Based on the analysis results the values of KT,low and KR,low were set to be increased threefold compared with those of KT,high and KR,high to represent low affinity of the KACh/low channel.
Figure 1.
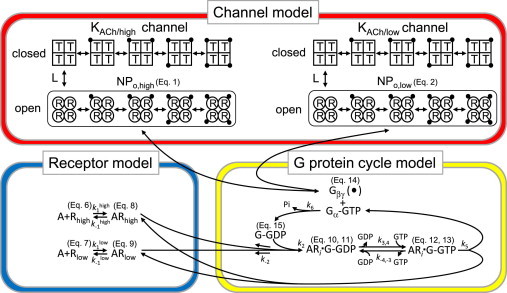
Schematic diagrams of the allosteric model for the KACh channel and the model for receptor-G-protein interaction. Top: Schematic representation of the allosteric model. In this scheme, each KACh channel with either high or low affinities to Gβγ (KACh/high or KACh/low, respectively) is assumed to be an oligomer composed of four identical subunits. Each subunit is in either the available (R, relaxed) or unavailable (T, tense) state, which is represented by a circle or a square, respectively. Each subunit in the R or T state binds to one dissociated G-protein βγ subunit (solid circles) independently of the other subunits, with the microscopic dissociation constants KR or KT, respectively. In this model, all subunits in the same oligomer must change their conformations simultaneously. R4 and T4 are in equilibrium with the allosteric constant L. Bottom: Models of receptor-G-protein interaction. The low-affinity state of m2Rs was incorporated into our previous model (21). A, ACh; Rhigh and Rlow, high-affinity and low-affinity m2Rs, respectively; G, G protein.
Table 1.
Parameters of the allosteric and G-protein cycle models
| Parameter | Value | Units |
|---|---|---|
| L | 1.41 × 103 | – |
| KR,high | 7.50 × 10−10 | M |
| KT,high | 1.36 × 10−8 | M |
| KR,low | 2.25 × 10−9 | M |
| KT,low | 4.09 × 10−8 | M |
| k1H | 2.50 × 106 | s−1·M−1 |
| k1L | 2.50 × 104 | s−1·M−1 |
| k-1H | 0.25 | s−1 |
| k-1L | 0.25 | s−1 |
| k2 | 5.40 × 10−1 | s−1·M−1 |
| k-2 | 1.0 × 103 | s−1 |
| k3,4 | 2.86 × 103 | s−1 M−1 |
| k-4,-3[GDP] | 0.68 | s−1·M−1 |
| k5 | 10 | s−1 |
| k6 | 0.03 + 2.4/(1 + e(−v−60)/−17) | s−1 |
| [Rhigh]total | 1.87 × 10−3 | M |
| [Rlow]total | 1.87 × 10−3 | M |
| [G]total | 5.60 × 10−2 | M |
The current-voltage relationship of IKACh with 5.4 mM [K+]o was obtained from a previous study (1) and fitted to the Boltzmann function: (v − EK) / (1 + exp[{v − EK − v1/2} / k]). The fitted equation reproduces the current-voltage relationship of IKACh (Fig. S1 B), which is used to calculate IKACh in Eq. 3. To present the kinetically different gating kinetics of the two populations (22,35), we calculated IKACh using Eq. 3:
| (3) |
where v is the membrane potential, gKACh is the maximum conductance for IKACh, f is the desensitization gate variable for the KACh/low channel, and EK is the equivalent potential for K+. The fast decrease in channel activity in short-term desensitization was associated with the gating kinetics of the KACh/low channel and characterized by exponential convergence to a steady state (22). Therefore, to represent this decrease, the kinetics of f were calculated using Hodgkin-Huxley-type equations:
| (4) |
| (5) |
where fss is the steady state of f, τf is the time constant for f, and [Gβγ] is the concentration of Gβγ in M.
G-protein cycle model
ACh causes a biphasic response in m2R over a wide range of [ACh]s (25–29). Therefore, we simulated Gβγ generation by the G-protein cycle model (21) using two m2Rs with either high or low affinity for ACh (Fig. 1, bottom). In the G-protein cycle model presented here, the reaction rates are represented by differential rate equations with rate constants and concentrations, as done in other studies (39–41). We calculated the values of 10 concentrations using the following ordinary differential equations (Eqs. 6–15):
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
where A is ACh; Rhigh and Rlow are m2Rs with high and low affinities for ACh, respectively; G is the Gi/o protein; and k is the reaction rate constant. The value of each rate constant was fixed, except for k6, which was defined as a function of membrane voltage (Table 1) to incorporate the regulation of G-protein signaling (RGS). RGS proteins regulate G-protein signaling by accelerating GTP hydrolysis in a voltage-dependent manner and are required to reconstitute short-term desensitization (42–44). On the basis of the voltage- and time-dependent characteristics of IKACh in atrial myocytes known as relaxation (44–46), k6 is defined as a function of membrane voltage so that depolarization of the membrane potential can accelerate GTPase activity. This relaxation of IKACh reflects an increasing suppression of channel open probability during depolarization (i.e., strong accelerated GTP hydrolysis at depolarization) and a gradual recovery during hyperpolarization (i.e., less accelerated GTP hydrolysis at hyperpolarization). The voltage-dependent Ca2+ influx and Ca2+/CaM modulation of RGS protein activity have been suggested as one of the underlying mechanisms (44). We did not incorporate the possible voltage dependence of m2R into our model because its electrophysiological function and properties have not yet been fully elucidated. The parameters in the G-protein cycle were mainly taken from the previous model (21) with minor modifications to incorporate the low-affinity interaction of m2R with ACh. On the basis of experimental results (25–29), we introduced m2R with low affinity to ACh into our model. The values of and were selected so that low affinity and high affinity differed by a factor of 100 (47). By a simple, sensitive analysis, we verified that this value could reproduce the unique characteristics of short-term desensitization in a wide range of [ACh]s, whereas factors of 10 and 1000 could not reproduce the constant increase in IP in the wide range of [ACh]s (Fig. S2 B). The values of k2 and k-2 were increased from those in the previous model so that the calculated Gβγ concentration would be sufficient to activate the allosteric model with a realistic activation curve. The Gβγ concentration generated by the G-protein cycle model was added to the allosteric model, and then channel availability and IKACh were calculated.
Incorporation of the IKACh model into the sinoatrial node models
We examined the effect of short-term desensitization on the action potential by incorporating the IKACh model into two mathematical models of action potential in the rabbit sinoatrial node (30–32). The original IKACh model in the sinoatrial node model was replaced with the IKACh model constructed in this study. The membrane potentials were calculated at 0.1 μM and 10 μM ACh. ACh activates Gi proteins that reduce adenylyl cyclase activity and therefore inhibits the hyperpolarization-activated current (If) and the L-type Ca2+ current (ICa,L) in sinoatrial nodes. On the basis of the experimental dose-dependent effects of ACh for If and ICa,L in single cells isolated from rabbit sinoatrial nodes (48), the effects of ACh on If and ICa,L were modeled and incorporated into the sinoatrial models. In the simulation, 0.1 μM ACh inhibits only If by a negative shift of its activation curve by 7.0 mV, whereas 10 μM ACh inhibits If by a 9.9 mV shift, and inhibits ICa,L by reducing its maximum conductance by 12.5%.
Results
Simulation of IKACh in response to ACh
First, induction of IKACh by various [ACh]s was simulated using the constructed model (Fig. 2) and quantitatively compared with experimental results (Fig. 3). The simulation conditions were the same as in the previous study (2), i.e., the membrane potential was held at −53 mV and EK was −87 mV. When various [ACh]s were applied, the outward IKACh responses exhibited the typical characteristics of short-term desensitization (Fig. 2 A, solid lines). As [ACh] increased, the maximum IKACh amplitude increased and the response was faster. At [ACh] > 0.1 μM, the IKACh gradually decreased after reaching a peak. The time constants for the simulated decreases were 12.9 s, 10.5 s, and 10.3 s for [ACh] = 1 μM, 10 μM, and 100 μM, which are comparable to values observed in previous experiments (11.8 s (2) and 7.3–12.7 s (16)). The IP during application of ACh was used to quantify the dependence of short-term desensitization on [ACh] (Fig. 3, row 1, upper solid line). The IP increased constantly over a wide range of [ACh]s, in accord with the values recorded experimentally (Fig. 3, row 1, upper circles) (2). In contrast to the IP, the IS that is measured at the end of ACh application exhibited saturation of current increase at [ACh]s > 1 μM (Fig. 3, row 1, lower solid line). This response of IS is consistent with that observed in isolated myocytes (Fig. 3, row 1, lower circles). The two KACh channel populations contributed to the responses of IP and IS: the IP is composed of the current response of the KACh/low and KACh/high channels (Fig. 3, rows 2 and 3), whereas the IS mainly consists of only the KACh/high channel (Fig. 2 A, dotted lines). When m2R with low affinity for ACh was removed from the model, the increase in the amplitude of IP was not observed at high [ACh]s (Fig. 2 B, solid line; Fig. 3, row 1, dashed lines). Further, removal of the KACh/low channels resulted in complete loss of short-term desensitization (Fig. 2 B, dotted lines). Previous mathematical models that dealt with the G-protein cycle (11) and were equipped with a single population of KACh channels (21) did not reproduce the responses of IP and IS over a wide range of [ACh]s. Therefore, mechanisms based on two different m2Rs and KACh channels quantitatively account for the short-term desensitization of IKACh.
Figure 2.
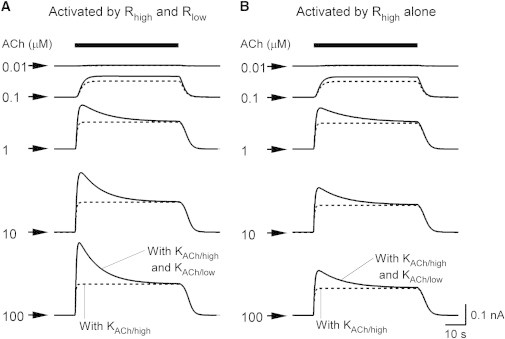
Simulated short-term desensitization. (A) Time course of IKACh activation by various [ACh]s. The simulated responses of IKACh at various [ACh]s (0.01–100 μM) are shown as solid lines. The value of gKACh was set to 0.16 to reproduce the IKACh amplitude in a quasi-steady state at −53 mV and 5.4 mM Ko (1). IKACh in the absence of the KACh/low channel is shown as dotted lines. The bar above the current traces represents the period of ACh perfusion. The [ACh]s are indicated in μM at each current trace. Arrows indicate the zero current. (B) IKACh in the absence of m2R with low affinity for ACh. Simulated IKACh without low-affinity m2R to ACh is shown in the same manner as in (A). IKACh in the absence of the KACh/low channel is shown as dotted lines.
Figure 3.
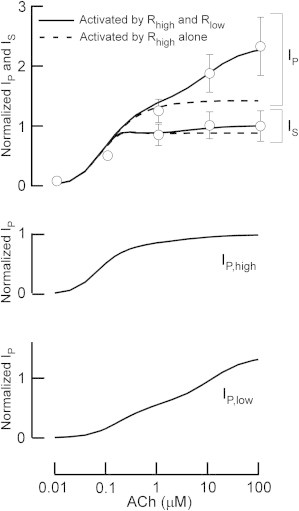
Quantitative analysis of simulated short-term desensitization. Dose-response curves for ACh-induced peak and quasi-steady IKACh. Top panel: The simulated peak current (IP) and quasi-steady-state current (IS) are shown as solid lines. Experimental data (2) for peak and quasi-steady IKACh are shown as circles with the standard deviation (SD). In the graph, the quasi-steady IKACh induced by 100 μM ACh is expressed as one. IKACh in the absence of m2R with low affinity for ACh is shown as dashed lines. Middle panel: Dose-response curve for IP through the KACh/high channel. The IP through KACh/high channel is shown as a solid line. The currents are normalized as in row 1. Bottom panel: Dose-response curve for IP through the KACh/low channel. The IP through the KACh/low channel is shown in the same manner as in the middle panel.
Characterization of short-term desensitization in the IKACh model
When atrial myocytes were serially perfused with bath solutions containing different [ACh]s, the amplitude of IP in the second ACh application was decreased by the high [ACh] of the first application (2). Therefore, the degree of experimental short-term desensitization is influenced by preperfusion with ACh. Using our model, we calculated IP during the second ACh application (I1, 10 μM) after applying various [ACh]s (0.01–1 μM) (Fig. 4 A), and then compared the amplitude of the I1 with that of the IP elicited by 10 μM ACh applied after a 5-min interval (I2) (Fig. 4 B). The I1 amplitude during preapplication of ACh at 0.1 μM did not differ significantly from the I2 amplitude (Fig. 4 A, top). However, when the [ACh] was increased to 1 μM, I1 decreased to approximately one-half of I2 (Fig. 4 A, bottom). The calculated ratio of I1 to I2 (Fig. 4 B, solid line) was comparable to that experimentally observed in atrial myocytes (Fig. 4 B, circles) (2). In contrast to the change in I1 amplitude, the [ACh] in the first application did not influence the fraction of IKACh through the KACh/high channel in I2 (Fig. 4 A, dotted lines). Therefore, our model reproduces the decreased IP amplitude after perfusion of high [ACh], suggesting that the desensitization of IKACh during preperfusion of ACh is attributable to desensitization of the KACh/low channel.
Figure 4.
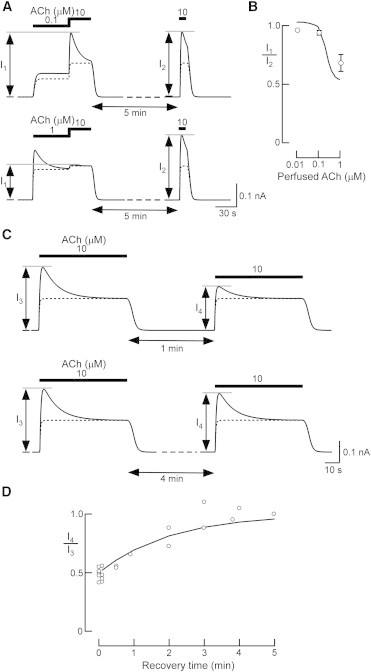
Analysis of simulated ACh preperfusion and recovery from short-term desensitization. (A) Traces of simulated IKACh with ACh preperfusion. The bars above the current traces represent perfusion with various [ACh]s (indicated in μM). (B) Simulated dose response after ACh preperfusion. IP induced with 0.01 to 1 μM ACh preperfusion (I1) was plotted with reference to the control IP value (I2). The experimental data (2) are shown as circles. (C) Traces of simulated recovery from short-term desensitization by ACh. ACh (10 μM) was applied during the two periods indicated by the bars below the current traces. (D) Time dependence of simulated recovery from short-term desensitization. The values of the control IP at 10 μM ACh before a washout period (I3) were set at one. The ratio of I4/I3 in (C) was plotted to determine the time dependence of recovery from short-term desensitization. The experimental data (2) are shown as circles.
As shown in Fig. 4 A, the response of IKACh to ACh is recovered from the desensitized state by perfusion with an ACh-free bath solution (2). Therefore, we determined the time course of recovery from desensitization. The IP amplitudes elicited by the first and second applications of 10 μM ACh (I3 and I4, respectively) were compared when the administration interval was changed (Fig. 4 C). As the washout time was prolonged, the ratio of I4 to I3 gradually increased and returned to one within 5 min (Fig. 4 D, solid line). This temporal response of simulated recovery is nearly identical to that measured experimentally (Fig. 4 D, circles) (2). The recovery rate was dependent on the time constant for Gβγ-dependent desensitization of the KACh/low channel. Since the current amplitude through the KACh/high channel was nearly constant regardless of the interval duration (Fig. 4 C, dotted lines), this recovery can be attributable to the recovery of the KACh/low channel from the desensitized state. Thus, the IP and IS in simulated short-term desensitization are comparable to those obtained in experiments. Short-term desensitization in experiments is usually evaluated quantitatively only by using the percentage of desensitization of IKACh at a certain, high [ACh] (11,14–16,49,50). We further validated the model by quantitatively comparing the simulated results of ACh preperfusion and recovery with the experimental results. Therefore, the major characteristics of short-term desensitization can be well captured by our IKACh model on the basis of the assumption that the current is due to two populations of KACh channels with different affinities for Gβγ.
Effects of nucleotide-bound states of G-protein on short-term desensitization
The transient change in the population of nucleotide-bound G proteins has been proposed to be responsible for the short-term desensitization of IKACh (11,14). Using our model, we examined the effects of nucleotides on the IKACh response (Fig. 5). In the presence of 0.1 μM ACh, application of high GTP (200 μM) over a short time period induced short-term desensitization (Fig. 5 A, solid line). The continuous presence of excess GDP (1 mM) in the bath solution (Fig. 5 B) and reduction of the m2R concentration by 50% (Fig. 5 C) effectively suppressed the GTP-induced short-term desensitization. These results are consistent with previously reported experimental results (11) and can be accounted for by the different degrees of the activation of the G-protein cycle (Fig. S3, B, D, and F). Next, we changed the rate parameters for nucleotide exchange on the G protein to reveal how its nucleotide-bound state affects short-term desensitization. When the rates for GDP/GTP exchange increased fivefold, ACh-induced short-term desensitization was strongly enhanced (Fig. 5 D, solid line). In contrast, when the rates were lowered to one-half, ACh-induced short-term desensitization was suppressed (Fig. 5 E). This is because fast GDP/GTP exchange enhances the formation of GTP-bound G protein (Fig. S3, H and J). Therefore, the nucleotide-bound state of G protein appears to be important in the development of short-term desensitization. However, when the KACh/low channel was omitted from the model, short-term desensitization was not produced under any condition (Fig. 5, dotted lines). Therefore, our theoretical analysis suggests that the KACh/low channel plays an essential role in reconstitution of short-term desensitization, and an additional role in modulating the nucleotide-bound state of G proteins.
Figure 5.
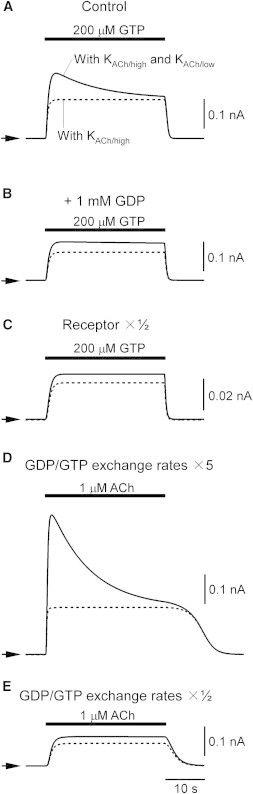
Effect of GDP/GTP exchange on short-term desensitization. (A) Traces of simulated IKACh elicited by 200 μM GTP. The bar above the current traces represents the period of GTP perfusion. To represent basal IKACh activity in the corresponding experimental condition (11), 0.1 μM ACh was applied during the simulation period. IKACh through the KACh/low channel only is shown as a dotted line. The arrowheads indicate the zero current level of each trace. The original values of initial [Rhigh] and [Rlow] were used to represent high receptor expression in the experiments (11). (B) Effect of 1 mM GDP on IKACh induced by 200 μM GTP. IKACh was elicited in the same way as in A except that 1 mM GDP was applied during the entire simulation period. (C) Traces of simulated IKACh elicited by 200 μM GTP with low receptor expression. IKACh was elicited in the same way as in A except that [Rhigh] and [Rlow] were lowered to 50% to represent low receptor expression in the experiments (11). (D) Effect of high GDP/GTP exchange activity on ACh-induced short-term desensitization. GDP/GTP exchange was enhanced by increasing the rate constants for GDP/GTP exchange (k3,4 and k-4,-3) by a factor of 5. The period during which ACh was perfused is shown as a bar above the current trace. (E) The effect of low GDP/GTP exchange activity on ACh-induced short-term desensitization. GDP/GTP exchange was lowered by reducing the rate constants to one-half.
Simulation of the effect of membrane potentials on short-term desensitization
M2R-dependent G protein signaling is enhanced at hyperpolarization (44,46). Because our IKACh model retains the voltage dependence incorporated into the previous model (21), we tested how membrane potential affects short-term desensitization of IKACh (Fig. 6). At 10 μM ACh, short-term desensitization occurred even at depolarized potential (0 mV), but it became more distinct at hyperpolarized potential (Fig. 6 A). Short-term desensitization was calculated in the presence of various [ACh]s at −80 mV, −50 mV, and 0 mV (Fig. 6 B). At hyperpolarization (−80 mV), IKACh started to exhibit short-term desensitization even at 0.1 μM ACh. In contrast, at depolarization (0 mV), this transient current response developed at [ACh] > 1 μM. These simulations demonstrate that hyperpolarization shifts the [ACh] threshold for short-term desensitization to lower concentrations.
Figure 6.
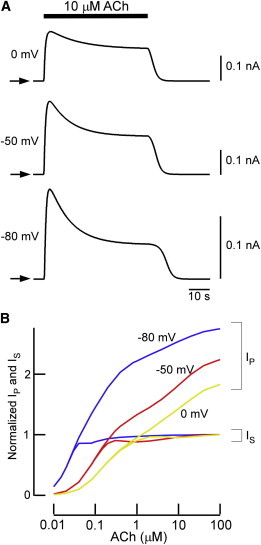
Voltage dependence in short-term desensitization. (A) The effect of 10 μM ACh on short-term desensitization at different membrane voltages. IKACh was activated by 10 μM ACh at potentials of 0 mV, −50 mV, and −80 mV. Arrows indicate the zero current levels of each trace. (B) Voltage-dependent dose-response curves for peak and quasi-steady IKACh. The peak current (IP) and quasi-steady-state current (IS) at clamped membrane potentials of −80 mV, −50 mV, and 0 mV are shown as blue, red, and yellow lines, respectively. IS induced by 100 μM ACh was set to one.
The role of short-term desensitization in action potential generation in the sinoatrial node
The two incorporated mechanisms quantitatively reconstituted short-term desensitization in the IKACh model. We next investigated how their characteristics were involved in the regulation of cardiac function. To address this question, we replaced the IKACh portion in the action potential models in the rabbit sinoatrial node with our IKACh model. Two models of cardiac pacemaker activity, the Demir model (30,31) and the Kurata model (32), were subjected to the simulation. The former is a classical Hodgkin-Huxley-type model, and the latter is a model with updated ion channels and intracellular Ca2+ models. Application of low [ACh] (0.1 μM) increased outward IKACh (Fig. 7 A, rows 2 and 4) and reduced the action potential frequency by 14.5% in the Demir model (Fig. 7 A, row 1) and by 12.4% in the Kurata model (Fig. 7 E, row 1). These values are in the range of the experimental responses to the applied [ACh]: 0.1 μM ACh decreased the action potential frequency by 0% (51) or 30% (30,52). The cycle length was prolonged by 46.0 ms and 43.0 ms, the maximum diastolic potential was reduced by 1.0 mV and 9.7 mV, and action potential amplitude was increased by 1.1 mV and 9.0 mV in the Demir model and Kurata model, respectively. In contrast, application of 10 μM ACh suspended the spontaneous generation of action potential in the two models (Fig. 7, B and F, row 1). This suspension was caused by the strong activation of IKACh (Fig. 7, B and F, row 2). However, action potential firing resumed in 8.9 s in the Demir model and in 10.7 s in the Kurata model. These time periods are comparable to those reported for the recovery from vagal escape observed in myocytes isolated from rabbit sinoatrial nodes (52). This phenomenon was observed even when a wide range of different values of gKACh were used (Fig. S4) or when ACh modulation on If and ICa,L was not incorporated. Resumption was mainly due to the gradual decrease in the outward current. Moreover, the hyperpolarization may facilitate short-term desensitization (see Fig. 6).
Figure 7.
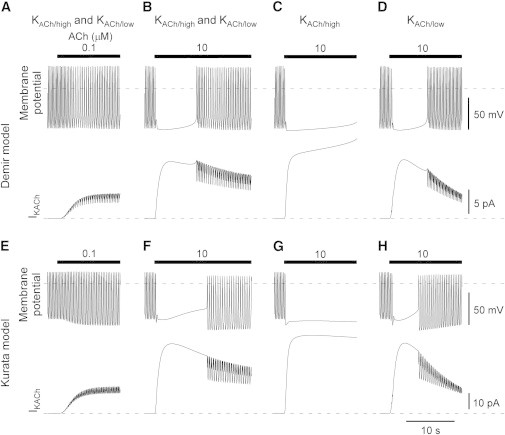
Role of short-term desensitization in spontaneous firing of the sinoatrial node. (A and E) Effects of 0.1 μM ACh on spontaneous firing in the Demir and Kurata sinoatrial node models. The current IKACh model was incorporated into the Demir (A) and Kurata (E) sinoatrial node models (30–32) and 0.1 μM ACh was applied during the period indicated by the bar. The dashed lines indicate the zero level of each trace. The value of gKACh was set at 1.1 (pS/pF) for the Demir model and at 6 (pS/pF) for the Kurata model. (B and F) Effects of 10 μM [ACh] on spontaneous firing. Parameters are the same as in A and E except that 10 μM ACh was applied. (C and G) Effects of 10 μM ACh on the KACh/high channel only. Parameters are the same as in B and F except that the KACh/low channel was replaced with the KACh/high channel. (D and H) Effects of 10 μM ACh on the KACh/low channel only. Parameters are the same as in B and F except that the KACh/high channel was replaced with the KACh/low channel.
To examine the effects of the two populations of KACh channels on the resumption, we replaced our IKACh model with an IKACh model that contained only KACh/high channels or only KACh/low channels. In both action potential models of the sinoatrial node, incorporation of the IKACh model with the KACh/high channel alone resulted in cardiac asystole with 10 μM ACh, and failed to resume during ACh application (Fig. 7, C and G). In contrast, the IKACh model with the KACh/low channel alone resulted in brief suspension of spontaneous firing (Fig. 7, D and H). These results suggest that the KACh/low channel is critical for the linkage between short-term desensitization and vagal escape.
Discussion
Short-term desensitization of IKACh is an adaptation mechanism for excessive vagal nerve stimulation in cardiac myocytes. In this study, we addressed the mechanisms underlying short-term desensitization of IKACh theoretically. On the basis of experimental evidence, we hypothesized the existence of two populations of m2R and KACh channels with different affinities to ACh and Gβγ, respectively. The introduction of m2Rs enabled the model to respond to a wide range of [ACh]s. The introduction of KACh channels resulted in quantitative reproduction of the temporal behavior of the IKACh current in short-term desensitization. Furthermore, the model conferred vagal escape on the mathematical action potential models of sinus node cells. These results allow us to propose that two functionally distinct populations of m2Rs and KACh channels underlie the physiological IKACh response of short-term desensitization.
To date, both G protein and the KACh channel have been proposed to be responsible for short-term desensitization. Experimental manipulations that lead to a rapid increase in free Gβγ induce short-term desensitization (11,14,16,42,43,53). However, neither previous models (11,21) nor our model without the KACh/low channel quantitatively reconstituted this phenomenon. This is simply because a linear model of the G-protein cycle cannot represent the transient response of IKACh. We hypothesized the KACh/low channel as the missing component. This fraction is likely a KACh channel with a different gating kinetic (35) or modulatory system, such as PIP2 (23,54) and phosphorylation (24), as discussed below. The K+ ion passing through the K+ channels has been proposed to account for short-term desensitization (15). This theory is based on the shift of EK by K+ flux in the microspace in close proximity to the plasma membrane. This explanation, however, cannot account for the constant IS after various amplitudes of IP under a wide range of [ACh]s. Although our mathematical model reproduced the effects of not only m2R stimulation (Figs. 2–4) but also G-protein-cycle modification (Fig. 5), there may be some discrepancy between the physiological phenomena and the model design and parameters. However, the theory we propose here is robust to various perturbations (Figs. 2–5; Fig. S4). Therefore, possible deviations in the model structure and parameters would not weaken our conclusion.
Although two different m2R populations with high and low affinities for ACh are well known, this has been ignored in previous models (25–29). Experimentally, cardiac m2R shows a biphasic response over a wide range of [ACh]s (to a factor of 104) that cannot be detected by a single receptor population with a single affinity. One cellular mechanism that could generate two distinct populations of KACh channel is different levels of PIP2. PIP2 is known as a prerequisite for KACh channel activation (23) and as a modulator for its gating kinetics (54). It has become apparent that PIP2 is enriched in membrane microdomains such as lipid rafts (55). Furthermore, the constituents of G-protein signaling and PIP2 metabolism are distributed differently within the plasma membrane, and after various stimuli, these constituents change their position with respect to these microdomains (49,56). In contrast, whereas phosphorylation of the KACh channel subunits (Kir3.1 and Kir3.4) by protein kinase A reportedly increases their open probability (24), phosphorylation of Kir3.1 by protein kinase Cδ has been shown to reduce PIP2 sensitivity (57). These reports suggest that phosphorylation state also has the potential to generate quantitatively different KACh channel populations. Therefore, although the precise mechanisms that contribute to the generation of two distinct KACh channel populations are not known, various cellular signals may account for this phenomenon.
This study also demonstrates the role of short-term desensitization in vagal escape. Vagal escape usually refers to the two responses of the heart to continuous vagal stimulation: the gradual return of the heart rate toward control level (58,59) and the development of pacemaker activity in multimodal origins (60). Compensation from the sympathetic system has been proposed to cause vagal escape (17–20). However, since this phenomenon could be observed even in isolated sinoatrial nodes in the presence of a β blocker (52), the cellular machinery associated with the postjunctional mechanism was expected to participate in vagal escape. The two assumptions that we proposed in this study are in agreement with this criterion. Therefore, the function of these two quantitatively different populations of m2Rs and KACh channels in short-term desensitization might be the missing cellular mechanisms in vagal escape from excessive parasympathetic nerve stimulation at the organ level. In general, the high-affinity m2R is most often measured and assumed to be physiologically relevant. However, it has been suggested that activation of the low-affinity m2R, rather than the high-affinity m2R, is responsible for atrial bradycardia in mice (27). The effects of RSG4 are another interesting link between short-term desensitization and vagal escape. In one study, the spontaneous generation of action potential firing in RGS4-null sinoatrial node myocytes was suspended by application of high concentrations of carbachol, whereas that of the wild-type was resumed (53). RGS proteins are also required to reconstitute short-term desensitization without affecting the amplitude of the IKACh (43). Because acceleration of the G-protein cycle enhances short-term desensitization (Fig. 5), RGS proteins are also important components to identify links between short-term desensitization and vagal escape in vivo.
In this study, we simulated short-term desensitization with a linear model of the G-protein cycle, which was improved on the basis of previous models (21). Although our model traced the IKACh recorded from electrophysiological experiments performed with isolated atrial myocytes under physiological conditions well (2) (Figs. 2–4), qualitative comparisons of the effects of nucleotides on the experiment and simulation remain to be done. Our model structure is different from that proposed by Chuang et al. (11), in which the nucleotide-free state of G proteins is responsible for short-term desensitization. In their qualitative reproduction of short-term desensitization, a large amount of G protein was required to populate the nucleotide-free state. The lack of consideration for such a drastic case might appear as a limitation of this study. However, when a model structure is linear, intracellular signaling cascades within the G-protein cycle do not appear to reproduce short-term desensitization quantitatively (Fig. 5). In other words, if a signal component is identified to transiently influence the receptor-dependent KACh channel activation, as we hypothesized in this study for the KACh/low channel, it could become another candidate for the mechanism of short-term desensitization. On the other hand, as shown in Fig. 6, our model possesses voltage dependence as a modulator of RGS protein activity (44–46). Ligand binding in m2R has been reported to show voltage dependence (61–63). How these voltage-dependent components should be considered with regard to the regulation of IKACh may need to be clarified in future studies. By incorporating new knowledge, the model presented here can be used to gain more insights into short-term desensitization as well as other phenomena associated with IKACh.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (HD Physiology) (22136001) to Y.K., a Grant-in-Aid for Scientific Research on Priority Areas (17079005) to Y.K., a Grant-in-Aid for Young Scientists (B) (20790206 and 22790251) to S.M., a Grant-in-Aid for Scientific Research (C) (25460331) to S.M., a Grant-in-Aid for Scientific Research (C) (235903011) to A.I., the Global COE Program “In Silico Medicine” at Osaka University, and a grant from the Vehicle Racing Commemorative Foundation.
Contributor Information
Shingo Murakami, Email: murakami@pharma2.med.osaka-u.ac.jp.
Yoshihisa Kurachi, Email: ykurachi@pharma2.med.osaka-u.ac.jp.
Supporting Material
References
- 1.Kurachi Y., Nakajima T., Sugimoto T. On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins. Pflugers Arch. 1986;407:264–274. doi: 10.1007/BF00585301. [DOI] [PubMed] [Google Scholar]
- 2.Kurachi Y., Nakajima T., Sugimoto T. Short-term desensitization of muscarinic K+ channel current in isolated atrial myocytes and possible role of GTP-binding proteins. Pflugers Arch. 1987;410:227–233. doi: 10.1007/BF00580270. [DOI] [PubMed] [Google Scholar]
- 3.Logothetis D.E., Kurachi Y., Clapham D.E. The β γ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 4.Kurachi Y., Ito H., Ui M. Activation of atrial muscarinic K+ channels by low concentrations of β γ subunits of rat brain G protein. Pflugers Arch. 1989;413:325–327. doi: 10.1007/BF00583550. [DOI] [PubMed] [Google Scholar]
- 5.Shui Z., Boyett M.R., Zang W.J. ATP-dependent desensitization of the muscarinic K+ channel in rat atrial cells. J. Physiol. 1997;505:77–93. doi: 10.1111/j.1469-7793.1997.077bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bünemann M., Lee K.B., Hosey M.M. Desensitization of G-protein-coupled receptors in the cardiovascular system. Annu. Rev. Physiol. 1999;61:169–192. doi: 10.1146/annurev.physiol.61.1.169. [DOI] [PubMed] [Google Scholar]
- 7.Shui Z., Yamanushi T.T., Boyett M.R. Evidence of involvement of GIRK1/GIRK4 in long-term desensitization of cardiac muscarinic K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H2554–H2562. doi: 10.1152/ajpheart.2001.280.6.H2554. [DOI] [PubMed] [Google Scholar]
- 8.Shui Z., Khan I.A., Boyett M.R. Role of receptor kinase in long-term desensitization of the cardiac muscarinic receptor-K+ channel system. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H819–H828. doi: 10.1152/ajpheart.00515.2001. [DOI] [PubMed] [Google Scholar]
- 9.Yamanushi T.T., Shui Z., Boyett M.R. Role of internalization of M2 muscarinic receptor via clathrin-coated vesicles in desensitization of the muscarinic K+ current in heart. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1737–H1746. doi: 10.1152/ajpheart.01287.2005. [DOI] [PubMed] [Google Scholar]
- 10.Shui Z., Khan I.A., Boyett M.R. Role of receptor kinase in short-term desensitization of cardiac muscarinic K+ channels expressed in Chinese hamster ovary cells. J. Physiol. 1998;507:325–334. doi: 10.1111/j.1469-7793.1998.325bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang H.H., Yu M., Jan L.Y. Evidence that the nucleotide exchange and hydrolysis cycle of G proteins causes acute desensitization of G-protein gated inward rectifier K+ channels. Proc. Natl. Acad. Sci. USA. 1998;95:11727–11732. doi: 10.1073/pnas.95.20.11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D. Mechanism of rapid desensitization of muscarinic K+ current in adult rat and guinea pig atrial cells. Circ. Res. 1993;73:89–97. doi: 10.1161/01.res.73.1.89. [DOI] [PubMed] [Google Scholar]
- 13.Saitoh O., Masuho I., Kubo Y. Regulator of G protein signaling 8 (RGS8) requires its NH2 terminus for subcellular localization and acute desensitization of G protein-gated K+ channels. J. Biol. Chem. 2001;276:5052–5058. doi: 10.1074/jbc.M006917200. [DOI] [PubMed] [Google Scholar]
- 14.Leaney J.L., Benians A., Tinker A. Rapid desensitization of G protein-gated inwardly rectifying K(+) currents is determined by G protein cycle. Am. J. Physiol. Cell Physiol. 2004;287:C182–C191. doi: 10.1152/ajpcell.00540.2003. [DOI] [PubMed] [Google Scholar]
- 15.Bender K., Wellner-Kienitz M.C., Pott L. Acute desensitization of GIRK current in rat atrial myocytes is related to K+ current flow. J. Physiol. 2004;561:471–483. doi: 10.1113/jphysiol.2004.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sickmann T., Alzheimer C. Short-term desensitization of G-protein-activated, inwardly rectifying K+ (GIRK) currents in pyramidal neurons of rat neocortex. J. Neurophysiol. 2003;90:2494–2503. doi: 10.1152/jn.00112.2003. [DOI] [PubMed] [Google Scholar]
- 17.Raper C., Wale J. Sympathetic involvement in vagal escape and the effects of β-receptor blocking drugs. Eur. J. Pharmacol. 1969;8:47–57. doi: 10.1016/0014-2999(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 18.Campos H.A., Friedman A.H. The influence of acute sympathetic denervation, reserpine and choline Xylyl ether on vagal escape. J. Physiol. 1963;169:249–262. doi: 10.1113/jphysiol.1963.sp007254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts J., Stadter R.P. Effect of reserpine on ventricular escape. Science. 1960;132:1836–1837. doi: 10.1126/science.132.3442.1836. [DOI] [PubMed] [Google Scholar]
- 20.Obrink K.J., Essex H.E. Chronotropic effects of vagal stimulation and acetylcholine on certain mammalian hearts with special reference to the mechanism of vagal escape. Am. J. Physiol. 1953;174:321–330. doi: 10.1152/ajplegacy.1953.174.2.321. [DOI] [PubMed] [Google Scholar]
- 21.Murakami S., Suzuki S., Kurachi Y. Cellular modelling: experiments and simulation to develop a physiological model of G-protein control of muscarinic K+ channels in mammalian atrial cells. Philos. Trans. A Math. Phys. Eng. Sci. 2010;368:2983–3000. doi: 10.1098/rsta.2010.0093. [DOI] [PubMed] [Google Scholar]
- 22.Kim D. Modulation of acetylcholine-activated K+ channel function in rat atrial cells by phosphorylation. J. Physiol. 1991;437:133–155. doi: 10.1113/jphysiol.1991.sp018588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 24.Müllner C., Vorobiov D., Schreibmayer W. Heterologous facilitation of G protein-activated K+ channels by β-adrenergic stimulation via cAMP-dependent protein kinase. J. Gen. Physiol. 2000;115:547–558. doi: 10.1085/jgp.115.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christopoulos A., Grant M.K., El-Fakahany E.E. Transducer abstraction: a novel approach to the detection of partial agonist efficacy in radioligand binding studies. J. Pharmacol. Toxicol. Methods. 2000;43:55–67. doi: 10.1016/s1056-8719(00)00078-2. [DOI] [PubMed] [Google Scholar]
- 26.Mizushima A., Uchida S., Yoshida H. Cardiac M2 receptors consist of two different types, both regulated by GTP. Eur. J. Pharmacol. 1987;135:403–409. doi: 10.1016/0014-2999(87)90691-1. [DOI] [PubMed] [Google Scholar]
- 27.Stengel P.W., Cohen M.L. Low-affinity M2 receptor binding state mediates mouse atrial bradycardia: comparative effects of carbamylcholine and the M1 receptor agonists sabcomeline and xanomeline. J. Pharmacol. Exp. Ther. 2001;296:818–824. [PubMed] [Google Scholar]
- 28.Galper J.B., Dziekan L.C., Smith T.W. The biphasic response of muscarinic cholinergic receptors in cultured heart cells to agonists. Effects on receptor number and affinity in intact cells and homogenates. J. Biol. Chem. 1982;257:10344–10356. [PubMed] [Google Scholar]
- 29.Haga K., Haga T., Ichiyama A. Reconstitution of the muscarinic acetylcholine receptor. Guanine nucleotide-sensitive high affinity binding of agonists to purified muscarinic receptors reconstituted with GTP-binding proteins (Gi and Go) J. Biol. Chem. 1986;261:10133–10140. [PubMed] [Google Scholar]
- 30.Demir S.S., Clark J.W., Giles W.R. Parasympathetic modulation of sinoatrial node pacemaker activity in rabbit heart: a unifying model. Am. J. Physiol. 1999;276:H2221–H2244. doi: 10.1152/ajpheart.1999.276.6.H2221. [DOI] [PubMed] [Google Scholar]
- 31.Demir S.S., Clark J.W., Giles W.R. A mathematical model of a rabbit sinoatrial node cell. Am. J. Physiol. 1994;266:C832–C852. doi: 10.1152/ajpcell.1994.266.3.C832. [DOI] [PubMed] [Google Scholar]
- 32.Kurata Y., Hisatome I., Shibamoto T. Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H2074–H2101. doi: 10.1152/ajpheart.00900.2001. [DOI] [PubMed] [Google Scholar]
- 33.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 34.Hosoya Y., Yamada M., Kurachi Y. A functional model for G protein activation of the muscarinic K+ channel in guinea pig atrial myocytes. Spectral analysis of the effect of GTP on single-channel kinetics. J. Gen. Physiol. 1996;108:485–495. doi: 10.1085/jgp.108.6.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D., Duff R. Activation of kinetically different populations of K channels by adenosine in rat atria. Biophys. J. 1990;57:312a. [Google Scholar]
- 36.Mintert E., Bösche L.I., Bender K. Generation of a constitutive Na+-dependent inward-rectifier current in rat adult atrial myocytes by overexpression of Kir3.4. J. Physiol. 2007;585:3–13. doi: 10.1113/jphysiol.2007.140772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corey S., Clapham D.E. The stoichiometry of Gβγ binding to G-protein-regulated inwardly rectifying K+ channels (GIRKs) J. Biol. Chem. 2001;276:11409–11413. doi: 10.1074/jbc.M100058200. [DOI] [PubMed] [Google Scholar]
- 38.Ito H., Tung R.T., Kurachi Y. On the mechanism of G protein βγ subunit activation of the muscarinic K+ channel in guinea pig atrial cell membrane. Comparison with the ATP-sensitive K+ channel. J. Gen. Physiol. 1992;99:961–983. doi: 10.1085/jgp.99.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi T.M., Kitano H., Simon M.I. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl. Acad. Sci. USA. 2003;100:10764–10769. doi: 10.1073/pnas.1834247100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saucerman J.J., Brunton L.L., McCulloch A.D. Modeling β-adrenergic control of cardiac myocyte contractility in silico. J. Biol. Chem. 2003;278:47997–48003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 41.Mosser V.A., Amana I.J., Schimerlik M.I. Kinetic analysis of M2 muscarinic receptor activation of Gi in Sf9 insect cell membranes. J. Biol. Chem. 2002;277:922–931. doi: 10.1074/jbc.M104210200. [DOI] [PubMed] [Google Scholar]
- 42.Doupnik C.A., Davidson N., Kofuji P. RGS proteins reconstitute the rapid gating kinetics of Gβγ-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mutneja M., Berton F., Slesinger P.A. Endogenous RGS proteins enhance acute desensitization of GABAB receptor-activated GIRK currents in HEK-293T cells. Pflugers Arch. 2005;450:61–73. doi: 10.1007/s00424-004-1367-1. [DOI] [PubMed] [Google Scholar]
- 44.Ishii M., Inanobe A., Kurachi Y. Ca2+ elevation evoked by membrane depolarization regulates G protein cycle via RGS proteins in the heart. Circ. Res. 2001;89:1045–1050. doi: 10.1161/hh2301.100815. [DOI] [PubMed] [Google Scholar]
- 45.Inanobe A., Fujita S., Kurachi Y. Interaction between the RGS domain of RGS4 with G protein α subunits mediates the voltage-dependent relaxation of the G protein-gated potassium channel. J. Physiol. 2001;535:133–143. doi: 10.1111/j.1469-7793.2001.t01-1-00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishii M., Inanobe A., Kurachi Y. PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G protein cycle in a cardiac K+ channel. Proc. Natl. Acad. Sci. USA. 2002;99:4325–4330. doi: 10.1073/pnas.072073399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ikegaya T., Nishiyama T., Yamazaki N. Interaction of atrial muscarinic receptors with three kinds of GTP-binding proteins. J. Mol. Cell. Cardiol. 1990;22:343–351. doi: 10.1016/0022-2828(90)91467-l. [DOI] [PubMed] [Google Scholar]
- 48.Zaza A., Robinson R.B., DiFrancesco D. Basal responses of the L-type Ca2+ and hyperpolarization-activated currents to autonomic agonists in the rabbit sino-atrial node. J. Physiol. 1996;491:347–355. doi: 10.1113/jphysiol.1996.sp021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho H., Hwang J.Y., Ho W.K. Acetylcholine-induced phosphatidylinositol 4,5-bisphosphate depletion does not cause short-term desensitization of G protein-gated inwardly rectifying K+ current in mouse atrial myocytes. J. Biol. Chem. 2002;277:27742–27747. doi: 10.1074/jbc.M203660200. [DOI] [PubMed] [Google Scholar]
- 50.Kobrinsky E., Mirshahi T., Logothetis D.E. Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat. Cell Biol. 2000;2:507–514. doi: 10.1038/35019544. [DOI] [PubMed] [Google Scholar]
- 51.Boyett M.R., Roberts A. The fade of the response to acetylcholine at the rabbit isolated sino-atrial node. J. Physiol. 1987;393:171–194. doi: 10.1113/jphysiol.1987.sp016818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DiFrancesco D. Current If and the neuronal modulation of heart rate. In: Zipes D., Jalife J., editors. Cardiac Electrophysiology. Saunders; Philadelphia, PA: 1990. pp. 28–35. [Google Scholar]
- 53.Cifelli C., Rose R.A., Heximer S.P. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 2008;103:527–535. doi: 10.1161/CIRCRESAHA.108.180984. [DOI] [PubMed] [Google Scholar]
- 54.Sui J.L., Petit-Jacques J., Logothetis D.E. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc. Natl. Acad. Sci. USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gamper N., Shapiro M.S. Target-specific PIP2 signalling: how might it work? J. Physiol. 2007;582:967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer T., Wellner-Kienitz M.C., Pott L. Depletion of phosphatidylinositol 4,5-bisphosphate by activation of phospholipase C-coupled receptors causes slow inhibition but not desensitization of G protein-gated inward rectifier K+ current in atrial myocytes. J. Biol. Chem. 2001;276:5650–5658. doi: 10.1074/jbc.M009179200. [DOI] [PubMed] [Google Scholar]
- 57.Brown S.G., Thomas A., Leaney J.L. PKC-delta sensitizes Kir3.1/3.2 channels to changes in membrane phospholipid levels after M3 receptor activation in HEK-293 cells. Am. J. Physiol. Cell Physiol. 2005;289:C543–C556. doi: 10.1152/ajpcell.00025.2005. [DOI] [PubMed] [Google Scholar]
- 58.Bain W.A. A method of demonstrating the humoral transmission of the effects of cardiac vagus stimulation in the frog. Exp. Physiol. 1932;22:269–274. [Google Scholar]
- 59.McDowall R.J. On the nature and significance of vagus escape. J. Physiol. 1926;61:131–140. doi: 10.1113/jphysiol.1926.sp002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wallace A.G., Daggett W.M. Pacemaker activity during vagal escape rhythms. Circ. Res. 1964;15:93–102. doi: 10.1161/01.res.15.2.93. [DOI] [PubMed] [Google Scholar]
- 61.Ben-Chaim Y., Chanda B., Parnas H. Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- 62.Ben-Chaim Y., Tour O., Parnas H. The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J. Biol. Chem. 2003;278:22482–22491. doi: 10.1074/jbc.M301146200. [DOI] [PubMed] [Google Scholar]
- 63.Moreno-Galindo E.G., Sánchez-Chapula J.A., Navarro-Polanco R.A. Relaxation gating of the acetylcholine-activated inward rectifier K+ current is mediated by intrinsic voltage sensitivity of the muscarinic receptor. J. Physiol. 2011;589:1755–1767. doi: 10.1113/jphysiol.2010.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.