

Abstract
Patients with hypogammaglobulinemia who do not fulfill all the classical diagnostic criteria for common variable immunodeficiency (reduction of two immunoglobulin isotypes and a reduced response to vaccination) constitute a diagnostic and therapeutic dilemma, because information concerning the clinical and immunological characteristics of these patients with idiopathic primary hypogammaglobulinemia is not available. In 44 common variable immunodeficiency and 21 idiopathic primary hypogammaglobulinemia patients we determined the clinical phenotypes and performed flow cytometric immunophenotyping to assess the pathophysiological B-cell patterns and memory B-cell subset counts. Age-matched B-cell subset reference values of 130 healthy donors were generated. Severe pneumonia and bronchiectasis occurred at similar frequencies in idiopathic primary hypogammaglobulinemia and common variable immunodeficiency. Although IgG levels were only moderately reduced compared to common variable immunodeficiency, 12 of 21 idiopathic primary hypogammaglobulinemia patients required immunoglobulin replacement. Non-infectious disease-related clinical phenotypes (autoimmune cytopenia, polyclonal lymphocytic proliferation and persistent unexplained enteropathy) were exclusively observed in common variable immunodeficiency and were associated with early peripheral B-cell maturation defects or B-cell survival defects. T-cell dependent memory B-cell formation was more severely affected in common variable immunodeficiency. Furthermore, 14 of 21 idiopathic primary hypogammaglobulinemia patients showed normal peripheral B-cell subset counts, suggestive for a plasma cell defect. In conclusion, idiopathic primary hypogammaglobulinemia patients who do not fulfill all diagnostic criteria of common variable immunodeficiency have moderately decreased immunoglobulin levels and often a normal peripheral B-cell subset distribution, but still suffer from serious infectious complications.
Introduction
Common variable immunodeficiency (CVID) is the most prevalent form of symptomatic primary antibody deficiency.1 It is defined by: 1) a marked decrease in serum IgG and IgA or IgM of at least 2 standard deviations (SD) below the mean for age; 2) absent isohemagglutinins and/or poor response to vaccines; 3) onset of immune deficiency after two years of age; and 4) exclusion of other defined causes of hypogammaglobulinemia.1 Patients with CVID suffer from recurrent infections and non-infectious complications (autoimmune cytopenia, polyclonal lymphocytic proliferation and persistent unexplained enteropathy), of which the latter are associated with increased mortality.2,3 By definition, CVID excludes a group of patients with idiopathic primary hypogammaglobulinemia, who suffer from hypogammaglobulinemia but do not fulfill CVID diagnostic criteria with respect to a reduction of two immunoglobulin isotypes and/or a reduced response to vaccination. According to the CVID diagnostic classification of the European Society for Immunodeficiency Diseases,4 some of these patients with idiopathic primary hypogammaglobulinemia (further referred to as IPH) can be classified as “possible” CVID, and in the ESID database of primary immunodeficiencies they are classified as “other hypogammaglobulinemias”. Remarkably, according to the primary immunodeficiency classification of the International Union of Immunological Societies (IUIS) these patients cannot be sufficiently classified within any of the subcategories of “Predominantly Antibody Deficiency”.5 In comparison, the International Classification of Diseases v10 (ICD10),6 classifies IPH as “hypogammaglobulinemia not otherwise specified” with the same ICD10 code as CVID. Patients with IPH are regularly encountered in clinical practice, but information concerning the prevalence and the clinical and immunological characteristics is not available. It is important to obtain insight into the frequency and severity of the clinical complications of IPH, to clarify whether IPH is a clinically relevant antibody deficiency, and to develop appropriate treatment strategies. In addition, analysis of immunological parameters will enable the comparison of pathophysiological aspects of IPH and CVID.
Therefore, we aimed to determine the position of IPH in the spectrum of idiopathic antibody deficiencies through clinical and immunological comparison with CVID. First, we recorded the patients with the clinical phenotypes as established by Chapel et al.2,7 In addition, we performed flow cytometric immunophenotyping in order to analyze T-cell dependent and independent memory B-cell subset counts8 and blood B-cell patterns, which are associated with differences in pathophysiological background.9
Methods
Patients
Peripheral blood samples and clinical data were collected for 44 CVID patients and 21 IPH patients. IPH was diagnosed if patients had a reduction of IgG at least 2 SD below the mean for age, an onset of the immunodeficiency after two years of age, exclusion of defined causes of hypogammaglobulinemia and if they did not fulfill the CVID diagnostic criteria with respect to a reduction of two immunoglobulin isotypes and/or a reduced response to vaccination. The group of CVID patients includes the 37 patients who were reported in our original description of the B-cell patterns.9 In addition, we collected blood from 130 healthy age-matched controls and 26 cord blood samples. This study was approved by the Medical Ethics Committee of the Erasmus MC.
Clinical phenotyping
Clinical data were collected from all IPH and CVID patients to record their clinical phenotypes as previously described by Chapel et al.2,5 These phenotypes are: 1) no disease-related complications (infections only); 2) autoimmune cytopenias; 3) polyclonal lymphoproliferation (granuloma/LIP/persistent unexplained lympadenopathy); and 4) unexplained persistent enteropathy. In addition, data were collected concerning the frequency and severity of infections and modes of treatment. Pneumococcal polysaccharide vaccination responses were interpreted according to Borgers et al.10 as an adequate response to half of the measured pneumococcal serotypes.
Flow cytometric analysis and assignment of B-cell patterns
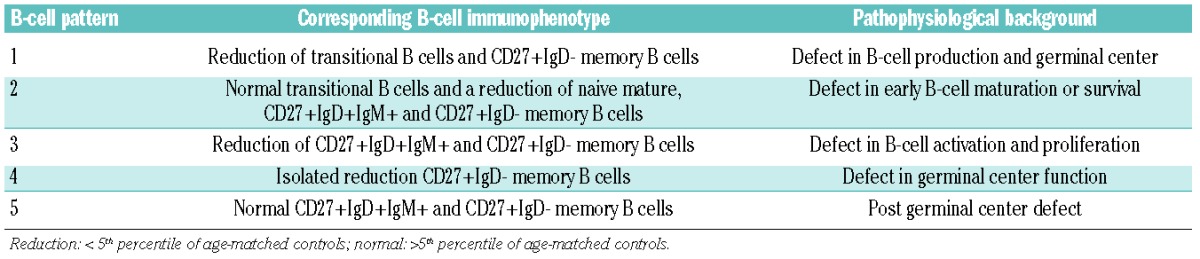
Six-color flow cytometric immunophenotyping of peripheral blood was performed on a CantoII (BD Biosiences) and data were analyzed using FACS Diva software (BD Biosiences). The following monoclonal antibodies were used: CD19-PerCP-Cy5.5, CD19-PE-Cy7, CD19-APC (all SJ25C1), CD5-APC ( L17F12 ), CD45-PerCP (2D1), CD19-APC (SJ25C1), CD38-PE, CD38-APC and CD38-PE-Cy7 ( HB7), CD27-APC (L128), CD3-PerCP-Cy5.5 (SK7) and CD8-APC-Cy7 (SK1) (all from BD Biosciences), polyclonal IgD-FITC, IgD-PE and IgM-PE (all from Southern Biotechnologies), polyclonal IgG-FITC (Kallestad), IgA-FITC and IgA-PE (IS11-8E10; Miltenyl Biotech), CD24-FITC ( gran-B-ly-1; Sanquin), CD21-PE ( LB21; Serotech), CD45RO-FITC ( UCHL1; DAKO), CD4-PC7 (SFCI12T4D11) and CD45-RA-RD1 (2H4; all from Beckman Coulter). The cell counts of the peripheral B-cell subsets (transitional B cells, naive mature B cells, and six memory B-cell subsets) were compared to age-matched healthy controls. A decrease or increase of a B-cell subset was defined as a value below the 5th or above the 95th percentile. B-cell patterns were determined as described previously9 and are summarized together with their pathophysiological background in Table 1.
Table 1.
B-cell patterns associated with different pathophysiological backgrounds in CVID.9
Statistical analysis
Statistical analysis was performed with Graphpad prism 5.0 software (Graphpad Software, San Diego, CA, USA). The Mann-Whitney test was used to compare continuous variables between two groups. The non-parametric Kruskal-Wallis rank sum test was used to compare multiple groups with continuous outcomes, followed by pair-wise Mann-Whitney tests if the former indicated significant differences. For categorical variables, the χ2 was used or Fisher’s exact test if required. Statistical significance was set at two-sided P<0.05.
Results
Patients’ characteristics
This study involved 44 CVID patients (mean age 32 years, range 6–77 years) and 21 patients with IPH (mean age 27 years, range 4–74). Age and gender distribution was comparable between both groups. In the CVID group, 2 of 44 patients were from consanguineous families, and in 10 of 4 patients, the family history was positive for hypogammaglobulinemia and/or IgA deficiency, compared to one of 21 and 6 of 21 in IPH, respectively.
Details regarding immunoglobulin levels, response to immunization and infectious clinical complications of IPH patients are provided in Table 2. All IPH patients showed reduced serum IgG levels, but did not fulfill the diagnostic criteria for CVID due to: adequate response to immunization (n=7), normal levels of IgA and IgM (n=3) or both (n=7) (Table 2). Four patients had normal levels of both IgA and IgM (Table 2; patients 15–18), but vaccination data were not available. The median IgG levels at diagnosis were lower in patients with CVID (3.0 g/L, range 0–5.7) compared to IPH (4.9 g/L, range 3.6–6.6) (Figure 1A). Considering the IgG subclass levels, most patients had IgG1 levels just below the normal range, with IgG2, IgG3 and IgG4 levels in the (lower) normal range (Table 1). In 6 patients, IgG subclasses were in the (lower) normal range in the presence of a decreased total IgG. In addition, IgM and IgA levels were lower in CVID, which was an expected finding because of the clinical definition of CVID and IPH.
Table 2.
Clinical characteristics of patients with idiopathic primary hypogammaglobulinemia.
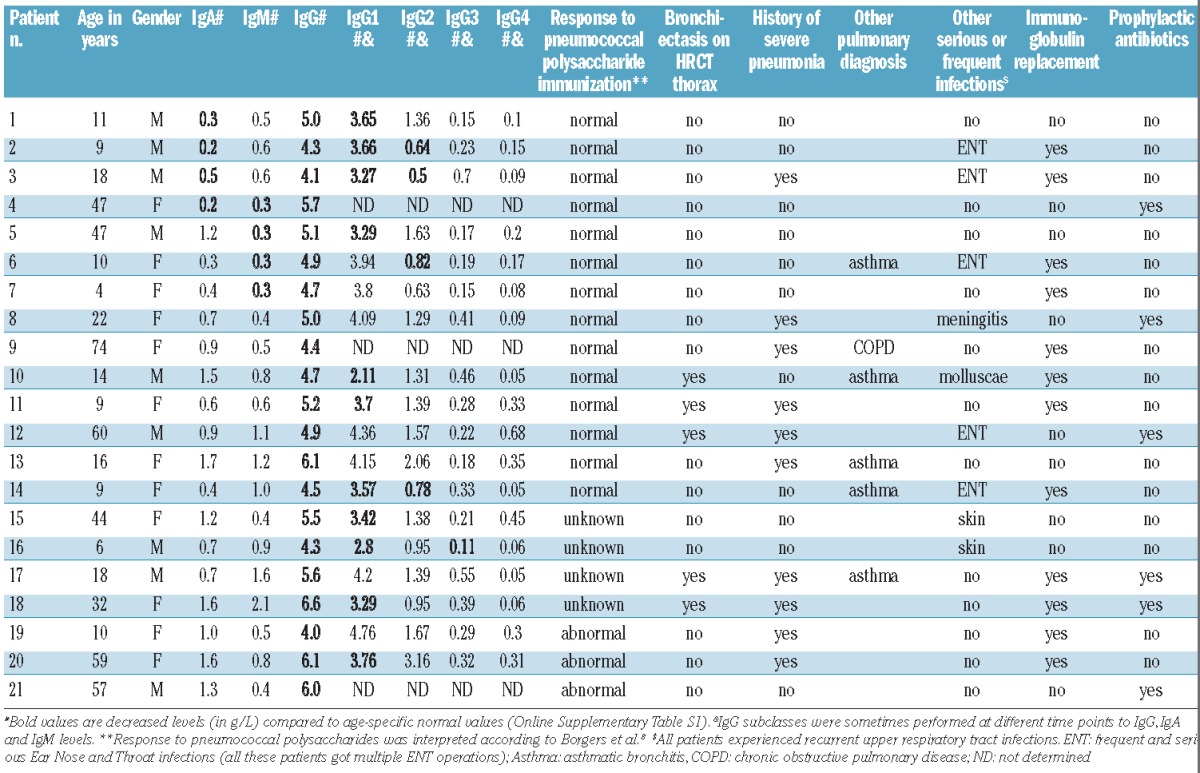
Figure 1.
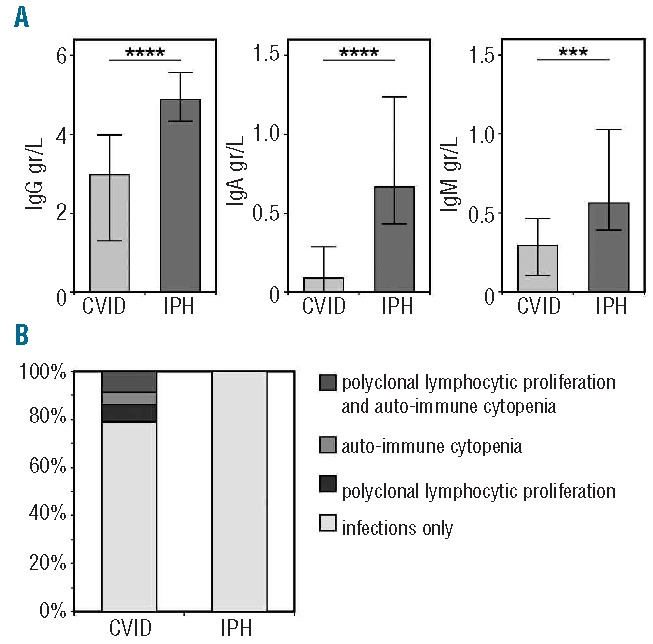
Immunoglobulin levels and clinical phenotypes in CVID and IPH. (A) Immunoglobulin levels at diagnosis in CVID and IPH. Groups are compared with the Mann Whitney test. Significant values are indicated. **** P<0.0001, *** P≤0.0005 (B) Clinical phenotypes according to Chapel et al.7,14
Clinical phenotypes in CVID and IPH
All patients with CVID and IPH suffered from recurrent upper respiratory tract infections. For IPH, the infectious complications are summarized in Table 2. Many IPH patients suffer from severe pneumonia (one or more episodes with hospital admission) and bronchiectasis. These complications were present in a similar frequency compared to CVID patients (50% vs. 48% and 24% vs. 27%, respectively).
Specific non-infectious disease-related clinical phenotypes (autoimmune cytopenia, polyclonal lymphocytic proliferation and persistent unexplained enteropathy) are associated with an increased mortality in patients with CVID.2,3 In our cohort, 79% of the CVID patients suffered from infections only, with the remaining 21% experiencing one or more disease-related non-infectious clinical complications (Figure 1B). In contrast, all IPH patients suffered only from infections. Therefore, non-infectious disease-related complications were observed exclusively in CVID (P=0.02).
Treatment
All CVID patients were treated with immunoglobulin replacement therapy. Of the IPH patients, 12 of 21 (57%) received immunoglobulin replacement (sometimes in combination with prophylactic antibiotics). Of the remaining 9 patients, 3 were treated with prophylactic antibiotics and 6 received antibiotics during infectious episodes as the only mode of treatment.
B-cell subsets in healthy controls
In order to detect abnormalities in peripheral B-cell subset distribution in patients, we generated age-related normal values of all B-cell subsets in a cohort of 130 healthy controls (Figure 2A–D, Table 3). Absolute numbers of transitional B cells and naive mature B cells directly increase after birth and decrease after the age of two years. The frequency of transitional B cells decreases over time, while the frequency of naive B cells remains stable (Figure 2B).
Figure 2.

Naive and memory B-cell subsets in normal controls. (A) Overview and definition of peripheral B-cell subsets. (B) Normal values of transitional and naive B cells. (C) Normal values of memory B-cell subsets. Gray bars represent absolute counts and open bars proportions of CD19+ cells. Bars indicate median with 25 and 75 percentiles. Whiskers represent 5th and 95th percentiles. (D) Relative distribution of memory B-cell subsets per age category.
Table 3.
Reference values of peripheral B-cell subset absolute counts.
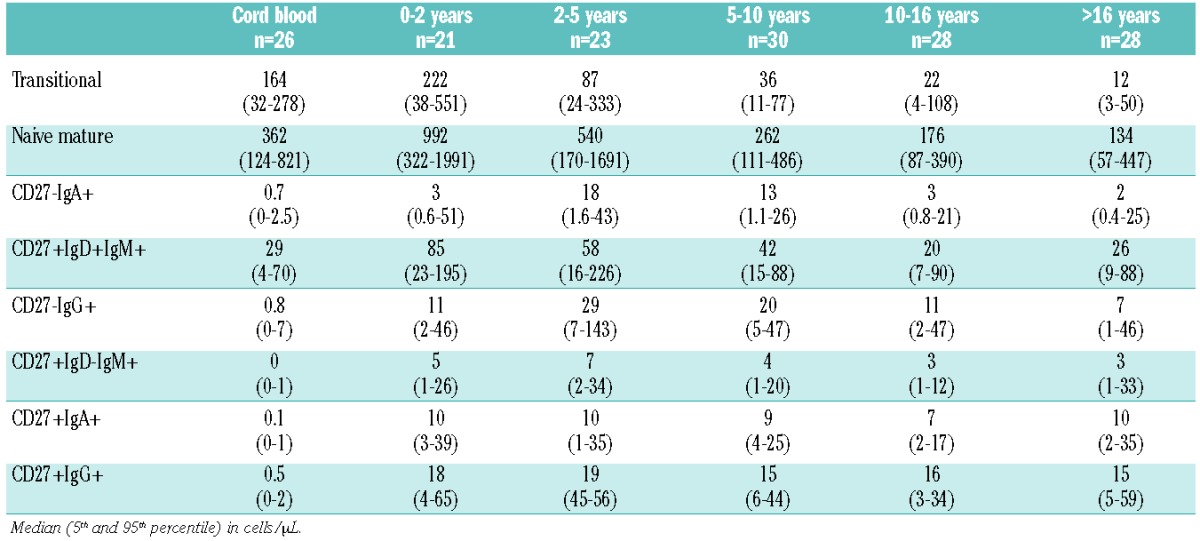
The human memory B-cell compartment consists of six subsets.8 CD27+IgD+IgM+ natural effector B cells are the only memory subset that constitutes a considerable part of the B-cell compartment in cord blood (Figure 2C). Absolute counts of natural effectors increase after birth to show a decline from 2–5 years. In contrast to absolute counts, the proportion of natural effector B cells gradually increases over time and form the largest proportion of memory B cells within the memory B-cell compartment. CD27− switched (IgA and IgG) memory B cells are present at birth in very small amounts and reach maximum values at 2–5 years, after which they decline to values just above those in cord blood (Figure 2C and D). CD27+IgM+IgD−(IgM-only) memory B cells and CD27+ switched (IgA+ and IgG+) memory B cells are hardly present in cord blood, but increase rapidly after birth to absolute counts that remain stable over all age groups until adulthood. The relative proportions of these CD27+ memory B-cell subsets show an impressive increase over time (Figure 2D), which mainly reflects the declining absolute number of transitional and naive mature B cells.
Peripheral B-cell subset patterns are different in CVID and IPH
CVID patients can display one of five distinct B-cell patterns.9 The patterns are based on the composition of the peripheral B-cell compartment and the replication history and somatic hypermutation frequency of the individual B-cell subsets and are indicative for the pathophysiological background of the antibody deficiency.9 The B-cell patterns can be easily determined by flow cytometry. Our earlier detailed molecular assays on sorted peripheral B-cell subsets were used to characterize the patterns, but are not used to define the pattern in individual patients.
The five B-cell patterns with their corresponding pathophysiological background are summarized in Table 1. The distribution of B-cell patterns was different between CVID and IPH (Figure 3A; P=0.003). B-cell patterns 1 and 2, which reflect B-cell production or early peripheral B-cell maturation/survival defects, were exclusively present in CVID (9 of 44 and 4 of 44, respectively) and not in IPH. B-cell patterns 3 and 4, which are associated with B-cell activation, proliferation and germinal center (GC) defects, were more common in CVID (12 of 44 and 10 of 44, respectively) as compared to IPH (3 of 21 and 4 of 21, respectively). In contrast, most IPH patients showed a normal peripheral B-cell subset distributions (B-cell pattern 5, 14 of 21), which is higher in frequency than CVID (9 of 44). The differences in B-cell patterns between CVID and IPH patients were reminiscent of the differences in clinical phenotypes: CVID patients with non-infectious complications predominantly displayed B-cell patterns 1 and 2 (Figure 3; P=0.003).
Figure 3.
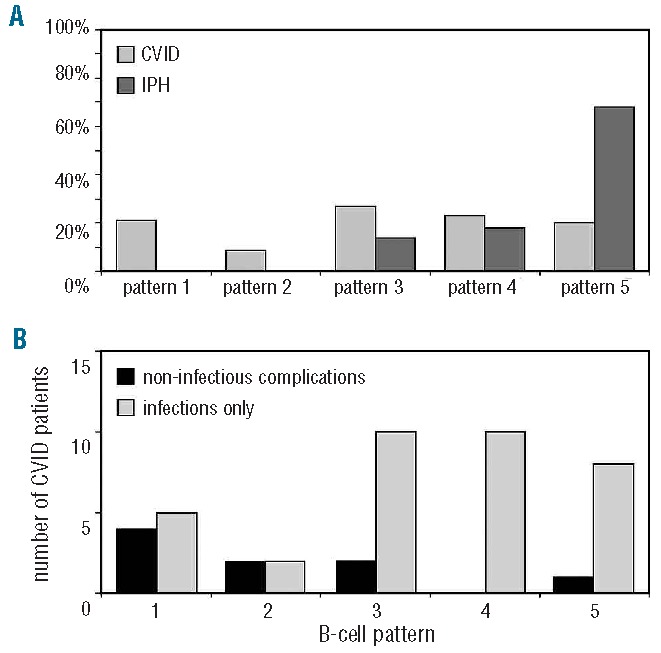
Pathophysiological B-cell patterns in CVID and IPH and relation to clinical phenotypes. (A) Comparison of B-cell patterns between CVID (n=44) and IPH (n=21). B-cell patterns 1 and 2 are exclusively observed in CVID (P =0.02). (B) B-cell patterns and clinical phenotypes in CVID (n=44). Non-infectious complications (auto-immunity and polyclonal lymphocytic proliferation) were more often observed in B-cell pattern 1–2 compared to B-cell pattern 3–5 (P =0.003).
Memory B-cell subsets are more severely affected in CVID than in IPH patients
Data of memory B-cell subsets of CVID and IPH patients were compared to age-matched controls and the proportion of patients with normal (>5th percentile of age-matched normal controls) and reduced (<5th percentile) memory B-cell subset size was determined (cut-off values are summarized in Table 3). The results are shown in Figure 4. The T-cell independent CD27−IgA+ memory B-cell subset was normal in the majority of CVID and IPH patients. In CVID, four memory B-cell subsets were more frequently reduced compared to IPH. T-cell dependent class switched CD27+IgA+ and CD27+IgG+ memory B cells showed the most significant difference between the groups. In line with these findings, 19 of 21 (90%) IPH patients could be classified as smB+ (CD27+IgD− B cells >2% of B cells) according to the EUROclass CVID classification11 and only 2 of 21 (10%) as smB-(CD27+IgD− B cells ≤ 2% of B cells), whereas in CVID, 25 of 44 (57%) patients were smB- and 19 of 44 smB+ (43%). In conclusion, memory B-cell formation was more severely affected in CVID compared to IPH, primarily affecting GC-dependent memory B-cell subsets.
Figure 4.
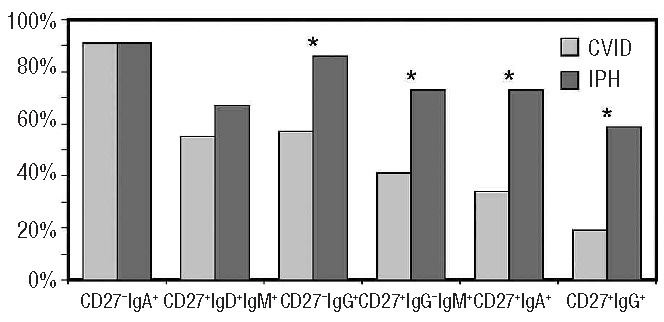
Memory B-cell subsets in CVID and IPH. The proportion of patients with normal memory B-cell subset size (>5th percentile of age-matched normal controls) is depicted. Data of CVID and IPH were compared with Fisher’s exact test. * P <0.05.
Discussion
This study describes the clinical and immunological characteristics of a group of patients with idiopathic primary hypogammaglobulinemia who do not fulfill the criteria for CVID with respect to a reduction of two immunoglobulin isotypes and/or an impaired response to vaccination. These IPH patients have not yet been well described in the literature and cannot be sufficiently classified within the current PID diagnostic classification system.4 It has been previously shown that some “CVID” patients have the ability to respond to vaccination,12,13 so these patients might show similarities to some of the IPH patients described in this present study. Our data raise the question as to whether an impaired response to vaccination should be used for the diagnosis of CVID, especially since solid criteria for the interpretation of vaccination responses are lacking.
Important differences were observed between IPH and CVID in clinical phenotypes, pathophysiological B-cell pattern and memory B-cell subset sizes, but similarities were also seen with respect to infectious complications. IPH patients have less severe hypogammaglobulinemia compared to CVID, but most patients with IPH still suffered from recurrent or serious infections. There was no significant difference in the occurrence of severe pneumonia and bronchiectasis to patients with CVID and more than half of the patients with IPH required immunoglobulin replacement. These data support the idea that IPH is a clinically relevant antibody deficiency. In contrast to CVID, several IPH patients were in good clinical condition without immunoglobulin replacement or antibiotic prophylaxis. Apparently, the specific antibody production in these patients was sufficient to prevent the occurrence of severe or frequent infections. Long-term follow-up studies are needed to examine which IPH patients require immunoglobulin replacement and/or antibiotic prophylaxis, and to monitor whether IPH can develop into a full CVID phenotype over time.
Over recent years, Chapel et al. have described the different clinical phenotypes of CVID patients2,14 and convincingly showed that non-infectious clinical complications (autoimmune cytopenia, polyclonal lymphocytic proliferation and persistent unexplained enteropathy) are associated with increased mortality compared to patients with infections only.2,14 We showed that non-infectious clinical complications were present in CVID but not in IPH, suggesting that the latter condition might have a better prognosis. Due to the relatively small cohort of IPH patients, we cannot exclude the possibility that some of them could present with or develop non-infectious clinical complications.
In line with the clinical differences, analysis of the earlier presented B-cell patterns9 revealed that defective B-cell production and early peripheral B-cell maturation or survival defect (B-cell patterns 1 and 2, respectively) were seen exclusively in CVID. In addition, we showed that these two B-cell patterns are associated with non-infectious disease-related clinical complications. Early defects in B-cell development apparently tend to result in the full CVID phenotype and more often give rise to immune deregulation. Further analysis of B-cell patterns showed that more than half of the CVID patients display B-cell patterns 1–3, indicative of defects in peripheral B-cell development before the GC stage. In contrast, the majority of IPH patients did not show abnormalities in peripheral B-cell distribution, suggesting a defect in post-GC (terminal) B-cell or a plasma-cell defect such as a differentiation, a survival and/or homing defect. Thus, the identification of B-cell patterns is useful to detect differences in pathophysiological background and has the potential to become clinically relevant in the follow up of CVID and IPH, because of the association with non-infectious clinical complications.
We analyzed memory B-cell subsets in CVID and IPH and compared these to 130 age-matched controls. We showed that absolute numbers of memory B-cell subsets in healthy individuals reach adult levels within two years of age and do not substantially increase afterwards. The observed relative increase in memory B cells mainly reflects a reduction of transitional and naive B cells over time. Our data are in line with previously published B-cell subset reference values.15–19 Memory B cells were reduced in the majority of CVID patients and mainly involved GC-dependent maturation pathways. IPH patients less frequently showed abnormalities in memory B-cell subsets, in line with our hypothesis that most of these patients suffer from plasma-cell survival or homing defects.
CVID and IPH are two partly overlapping conditions. IPH is similar to CVID with respect to infectious complications, but is not the same with respect to non-infectious clinical complications, immunoglobulin levels, distribution of B-cell patterns and memory B-cell counts. Functional immunological studies should focus on plasma-cell differentiation and homing. Clinical follow-up studies of larger numbers of IPH patients should be performed to assess the prognosis, facilitate the development of optimal treatment strategies, and determine the place of IPH in current PID classification systems.
Acknowledgments
The authors would like to thank Mrs. S. De Bruin-Versteeg for assistance with preparing the figures; Mrs. P van Jaarsveld-Bakker, Mrs. M.W. van der Ent and Mrs. E. van Mastrigt for assistance with collecting blood samples and/or clinical data; Mrs. J. van Rhee-Brinkhorst and Mrs. L. Winter of the department of obstetrics of the ErasmusMC for the collection of the cord blood samples.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 (Suppl 1):3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86 [DOI] [PubMed] [Google Scholar]
- 3.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7): 1650–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ESID CVID classification of the European Society for Immunodeficiency Diseases. Available from: www.ESID.org
- 5.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Classification of Diseases, 10th version. Available from: WHO.int/classifications/icd/en
- 7.Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. 2012;130(5):1197–8 e9 [DOI] [PubMed] [Google Scholar]
- 8.Berkowska MA, Driessen GJ, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood. 2011;118(8):2150–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Driessen GJ, van Zelm MC, van Hagen PM, Hartwig NG, Trip M, Warris A, et al. B-cell replication history and somatic hypermutation status identify distinct pathophysiologic backgrounds in common variable immunodeficiency. Blood. 2011;118(26): 6814–23 [DOI] [PubMed] [Google Scholar]
- 10.Borgers H, Moens L, Picard C, Jeurissen A, Raes M, Sauer K, et al. Laboratory diagnosis of specific antibody deficiency to pneumococcal capsular polysaccharide antigens by multiplexed bead assay. Clin Immunol. 2010;134(2):198–205 [DOI] [PubMed] [Google Scholar]
- 11.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85 [DOI] [PubMed] [Google Scholar]
- 12.Rezaei N, Aghamohammadi A, Siadat SD, Moin M, Pourpak Z, Nejati M, et al. Serum bactericidal antibody responses to meningococcal polysaccharide vaccination as a basis for clinical classification of common variable immunodeficiency. Clin Vaccine Immunol. 2008;15(4):607–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rezaei N, Aghamohammadi A, Read RC. Response to polysaccharide vaccination amongst pediatric patients with common variable immunodeficiency correlates with clinical disease. Iran J Allergy Asthma Immunol. 2008;7(4):231–4 [PubMed] [Google Scholar]
- 14.Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. 2012;130 (5): 1197–8 [DOI] [PubMed] [Google Scholar]
- 15.Schatorje EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, van der Burg M, et al. Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. 2011;74(5):502–10 [DOI] [PubMed] [Google Scholar]
- 16.van Gent R, van Tilburg CM, Nibbelke EE, Otto SA, Gaiser JF, Janssens-Korpela PL, et al. Refined characterization and reference values of the pediatric T- and B-cell compartments. Clin Immunol. 2009;133(1):95–107 [DOI] [PubMed] [Google Scholar]
- 17.Piatosa B, Wolska-Kusnierz B, Pac M, Siewiera K, Galkowska E, Bernatowska E. B cell subsets in healthy children: reference values for evaluation of B cell maturation process in peripheral blood. Cytometry B Clin Cytom. 2010;78(6):372–81 [DOI] [PubMed] [Google Scholar]
- 18.Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162(2):271–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huck K, Feyen O, Ghosh S, Beltz K, Bellert S, Niehues T. Memory B-cells in healthy and antibody-deficient children. Clin Immunol. 2009;131(1):50–9 [DOI] [PubMed] [Google Scholar]