

Background: HIV-1 infectivity is decreased by specific mutations that alter the hydrophobicity level in the HIV-1 gp41 loop core.
Results: Antibody recognition, disulfide-bond formation, and lipid mixing of loop domain peptides are strongly affected by these mutations.
Conclusion: The hydrophobic core maintains proper function and structure of the loop region.
Significance: A better understanding of the membrane fusion mechanism of HIV and similar viruses is provided.
Keywords: Biophysics, HIV-1, Membrane Fusion, Membrane Proteins, Peptide Conformation, Peptide-Membrane Interaction, Viral Fusion Protein
Abstract
The gp41 disulfide loop region switches from a soluble state to a membrane-bound state during the human immunodeficiency virus type 1 (HIV-1) envelope-mediated membrane fusion process. The loop possesses a hydrophobic core at the center of the region with an unusual basic residue (Lys-601). Furthermore, two loop core mutations, K601A and L602A, are found to inhibit HIV-1 infectivity while keeping wild type-like levels of the envelope, implying that they exert an inhibitory effect on gp41 during the membrane fusion event. Here, we investigated the mode of action of these mutations on the loop region. We show that the K601A mutation, but not the L602A mutation, abolished the binding of a loop-specific monoclonal antibody to a loop domain peptide. Additionally, the K601A, but not the L602A, impaired disulfide bond formation in the peptides. This was correlated with changes in the circular dichroism spectrum imposed by the K601A mutation. In the membrane, however, the L602A, but not the K601A, reduced the lipid mixing ability of the loop peptides, which was correlated with decreased α-helical content of the L602A mutant. The results suggest that the Lys-601 residue provides a moderate hydrophobicity level within the gp41 loop core that contributes to the proper structure and function of the loop inside and outside the membrane. Because basic residues are found between the loop Cys residues of several lentiviral fusion proteins, the findings may contribute to understanding the fusion mechanism of other viruses as well.
Introduction
The membrane fusion process is a fundamental step for viruses to enter their host cells and to start an infectious cycle (1). Viruses utilize the fusion protein of their envelope (ENV)3 to catalyze this process by converting between several ENV conformational changes (2, 3). In the case of the human immunodeficiency virus type 1 (HIV-1), its gp41 fusion protein alternates between at least three conformations during fusion (2–6) as follows. (i) The first is the native, non-fusogenic conformation in which gp41 is sheltered by the surface subunit, gp120. (ii) Upon gp120 binding to CD4 and co-receptor, structural changes occur both in gp120 and gp41 (7), which release gp41 in an extended state allowing the penetration of the fusion peptide into the cell membrane (8, 9). This is an intermediate, pre-hairpin conformation in which the N-heptad repeat (NHR) and the C-heptad repeat (CHR) regions of gp41 are not associated. (iii) Subsequently, gp41 folds into the hairpin conformation that comprises the six-helix bundle. The six-helix bundle is formed by an NHR trimer, which is bound to three CHR regions in an anti-parallel fashion (10, 11). This structure represents a conserved element in the fusion proteins of many viruses and is believed to be essential for membrane pore formation (2, 3).
It is now accepted that other regions outside the six-helix bundle participate in the membrane fusion process through not yet fully understood mechanisms. One example is the gp41 loop region that connects the NHR and the CHR regions in the gp41 hairpin conformation (12, 13). The loop possesses a conserved structure in retroviruses that comprises a hydrophobic core at the center of the region with a disulfide motif (see Refs. 13 and 14 and Fig. 1, A–C). The loop is the third hydrophobic region in gp41 after the fusion peptide and the transmembrane domain (15, 16), and it has the capability to bind and to insert into the membrane (17, 18). Functionally, recent studies propose that the loop region and its cysteines participate in the membrane fusion event (17, 19–22).
FIGURE 1.
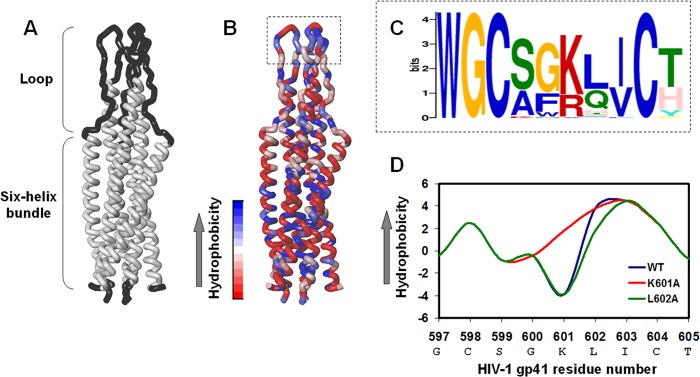
Bioinformatics characterization and mutagenesis of the hydrophobic loop core. A, presentation of the loop structure devoid of the cysteine residues together with the six-helix bundle in the soluble hairpin conformation of the SIV fusion protein. This is the only available structure of the loop. The three-dimensional structure was taken from Caffrey et al. (13), PDB ID 1QCE. B, the location of the hydrophobic core within the loop region. The highest hydrophobic residues are blue, and the lowest hydrophobic residues are red. C, sequence homology in the loop hydrophobic core between HIV and SIV clades shows conservation of the basic residue (Lys or Arg) between the two Cys residues. D, alteration of the hydrophobic level of the gp41 loop core by mutagenesis analysis. Residue numbers correspond to the gp160 HIV-1 HXB2 variant.
An unusual feature in the hydrophobic core of the gp41 loop region is the existence of a basic residue (Lys-601) at the center of the core (CXXKXXC) (Fig. 1C). The existence of a basic residue in that location disrupts the hydrophobic continuity of the core, which might be correlated with the proper function and structure of the loop. Moreover, alanine-scanning mutants of the loop show that two mutations in the loop core, K601A and L602A, decreased HIV-1 infectivity while keeping wild type (WT)-like levels of gp120 and gp41 (23, 24). Therefore, a reasonable possibility is that these mutations affect the gp41-mediated membrane fusion process.
Here, we utilized biochemical and biophysical approaches to study the affect of the K601A and L602A mutations on the function and structure of the loop region inside and outside the membrane. For this purpose, peptides corresponding to the loop region were prepared with the mutations K601A and L602A that have an opposite effect on the hydrophobic level of the core (Fig. 1D). We found that outside the membrane, the K601A mutation abolished the ability of the loop to form a disulfide bond, whereas the L602A was not effective. In the membrane, the L602A mutation, but not that of the K601A, decreased the lipid mixing ability of the loop. Moreover, these defects were in correlation with alterations in the structure and proper conformation of the loop as revealed by antibody binding and circular dichroism spectroscopy. The results are discussed in the context of the HIV-1 gp41-mediated membrane fusion event.
EXPERIMENTAL PROCEDURES
Materials
Fmoc amino acids and Fmoc rink amide 4-methylbenzhydrylamine resin were purchased from Novabiochem AG (Laufelfinger, Switzerland). Tris(2-carboxyethyl) phosphine hydrochloride, ethane dithiol, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), phosphatidylcholine (PC) from egg yolk, cholesterol (Chol) and l-α-lysophosphatidylcholine, and sodium dithionite (DTH) were purchased from Sigma. N-(Lissamine rhodamine B sulfonyl)dioleoylphosphatidylethanolamine (Rho-PE) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)dioleoylphosphatidylethanolamine (NBD-PE) were purchased from Molecular Probes (Eugene, OR).
Peptide Synthesis and Purification
Peptides were synthesized with an automatic peptide synthesizer (433A from Applied Biosystems) on rink amide MBHA resin by using the Fmoc strategy as previously described (25). Peptides with cysteine residues were cleaved with a trifluoroacetic acid (TFA): double distilled water (DDW):radical-scavenger:thioanisol:ethane dithiol (92.1:3.9:1.25:1.25:2.5 (v/v)) mixture. Before reverse phase high performance liquid chromatography (RP-HPLC) purification to >95%, the samples were dissolved in 5 mm Tris(2-carboxyethyl) phosphine hydrochloride as a reducing agent, and the peptides were purified under acidic conditions of 0.1% (v/v) TFA to maintain the cysteine residues in a reduced form. The molecular weight of the peptides was confirmed by platform LCA electrospray mass spectrometry.
Kinetics of Cysteine Oxidation
The peptides were maintained as powders and dissolved just before the experiments started (final concentration of 13.3 μm) in PBS (pH 7.4). Oxidation was performed under stirring conditions, and the peptides were exposed to air for 9 h. Samples were taken to RP-HPLC to monitor the oxidation process as previously described (20, 21).
Determination of Thiol Groups by DTNB
Peptides were dissolved in PBS or in lipid suspension of 100 μm large unilamellar vesicles (LUVs) in PBS to give final peptide concentrations of 13.3 μm. The oxidation protocol was performed as described above. Thiol groups were assessed at different time points by taking samples of the peptides followed by the addition DTNB (from a 5 mm fresh stock solution in PBS (pH 7.2) containing 0.1 mm EDTA) at a final concentration of 100 μm. The reaction product (2-nitro-5-thiobenzoate) was quantified in a spectrophotometer by measuring the absorbance of visible light at 412 nm. The absorbance of DTNB without peptides was used as the blank control.
Preparation of LUVs
Thin films were generated after dissolution of the lipids in a 2:1 (v/v) mixture of chloroform/methanol and then dried under a stream of nitrogen gas while they were rotated. Two populations of films were generated: (i) unlabeled, containing PC:Chol (9:1), and (ii) labeled, containing PC:Chol (9:1) and 0.6% m concentrations of NBD-PE and Rho-PE each. The films were lyophilized overnight, sealed with argon gas to prevent oxidation, and stored at −20 °C. Before an experiment, the films were suspended in PBS and vortexed for 1.5 min. The lipid suspension underwent five cycles of freezing-thawing followed by extrusion through polycarbonate membranes with 1- and 0.1-μm diameter pores 21 times to create the LUVs.
Lipid Mixing Assay
Lipid mixing of LUVs was measured using a fluorescence probe dilution assay (26, 27). LUVs were prepared in PBS as described above from unlabeled and labeled films combined to yield a 9:1 molar ratio of a 100-μm final lipid concentration. The emission of NBD was monitored at 530 nm with the excitation set at 467 nm. The basal fluorescence level was measured initially for a 400-μl vesicle mixture. Then the peptides were dissolved in 2 μl of DMSO and added to the mixture. The fluorescence was monitored for several minutes after the peptide was added to ensure a steady state as indicated by a plateau. The increase in fluorescence induced by the peptides was referred as total lipid mixing. The inner leaflet mixing assay was modified from the method developed by Meers et al. (28). The method is based on the fact that DTH reacts more rapidly with NBDs in the outer leaflet than those in the inner leaflet. After the lipid mixing of the peptides, DTH was added to the mixture in a final concentration of 32 mm. This concentration decreased maximum NBD fluorescence in the system, and higher DTH concentrations retained the same effect. The decrease in fluorescence was monitored until a plateau was reached. As a control, DTH was added to the LUVs that was treated only with DMSO. The difference between the steady state fluorescence of the peptide and the DMSO after DTH was added was referred as inner leaflet mixing.
Binding of gp41 Loop-specific Antibodies Analyzed by ELISA
A 96-well plate was coated with the loop peptides in dose-dependent amounts (maximum of 1 μg/well) in 0.05 m sodium carbonate solution (pH 9.6) at 4 °C overnight. Then the plate was blocked with 5% skim milk for 1 h followed by 1 h of incubation at 37 °C with gp41 loop-specific monoclonal antibodies. The following reagents were obtained through the NIH AIDS Research and Reference Program, Division of AIDS, NIAID, NIH: monoclonal antibodies to HIV-1 gp41 (246-D and 240-D) from Dr. Susan Zolla-Pazner (29, 30) and monoclonal antibody to HIV-1 gp41 (T32) from Dr. Patricia Earl, NIAID (31). For the 246-D and T32 antibodies, the concentrations were 0.5 μg/ml (100 μl/well) and 0.4 μg/ml (100 μl/well), respectively. Next, peroxidase-conjugated secondary antibodies were added for 1 h of incubation. The 3,3′,5,5′-tetramethylbenzidine substrate and H2SO4 (1 m) were added sequentially. The amount of bound monoclonal antibodies was detected by monitoring the absorbance in 450 nm. Several antibody binding constants were determined by nonlinear least squares (NLLSQ) representing a steady state affinity model. The NLLSQ fitting was done using the equation,
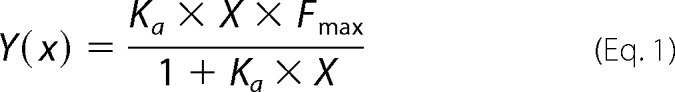 |
where X is the peptide concentration, Y(x) is the antibody binding measured by absorbance (arbitrary units), Fmax is the maximal binding, and Ka is the antibody binding constant.
Bioinformatics Analysis
A database was created from all the universal protein resource (UniProt) entries for a transmembrane protein of the envelope of HIV and SIV. These protein sequences were then run in a motif-based sequence analysis tool (MEME) that analyzes sequences for similarities among them and produces a description (motif) for each pattern it discovers. The motifs found were automatically arranged by their E-value, which is an estimate of the expected number of motifs with the given log likelihood ratio that one would find in a similarly sized set of random sequences. The log likelihood ratio is the logarithm of the ratio of the probability of the occurrences of the motif given the motif model versus their probability given the background model. The background model here is a 0-order Markov model using the background letter frequencies. The y axis is in bits, which is equal to the relative entropy of the motif relative to a uniform background frequency model. The relative entropy of the motif, computed in bits and relative to the background letter frequencies, is equal to the log-likelihood ratio (llr) divided by the number of contributing sites of the motif times 1/ln(2). To better view the sequence, the MEME results were run through WebLogo, which is designed to generate sequence logos as a method for graphical representation of amino acid sequence alignment.
Secondary Structure Determination Utilizing Circular Dichroism (CD) Spectroscopy
CD measurements were performed by using an Applied Photophysics spectropolarimeter. The spectra were scanned using a thermostatic quartz cuvette with a path length of 1 mm. Wavelength scans were performed at 25 °C; the average recording time was 15 s in 1-nm steps in the wavelength range of 190–260 nm. Peptides were scanned at a concentration of 10 μm in solution (5 mm HEPES (pH 7.4)) and in a membrane mimetic environment of 1% lysophosphatidylcholine in HEPES solution.
Membrane Binding
Peptide interactions with membranes were analyzed and quantified using fluorescence anisotropy of their intrinsic Trp residue in the presence of PC:Chol (9:1) LUVs membranes. Excitation and emission wavelengths were set to 280/350 nm, respectively, and 1 μm peptide (in 400 μl of PBS) was titrated with 13.8 mm membrane solution successively. Because Trp is known to change its emission in a hydrophobic environment, a change in its emission represented the amount of peptide bound to membranes. The system reached binding equilibrium (Fmax) at a certain lipid/peptide ratio, allowing us to calculate the affinity constant from the relations between the equilibrium level of Trp emission and the lipid concentration (C) using a steady state affinity model. The affinity constants were determined by NLLSQ fitting (Equation 1), where X is the lipid concentration, Y(x) is the fluorescence emission, Fmax is the maximal difference in the emission of Trp-containing peptide before and after the addition of lipids (it represents the maximum peptide bound to lipid), and Ka is the affinity constant.
RESULTS
Bioinformatics Analysis of Loop Core Sequences in the Fusion Proteins of HIV and SIV
We observed an unusual Lys incorporated in the hydrophobic core of the HIV-1 gp41 loop region (Lys-601) that disrupts the hydrophobic continuity of the core. We examined the conservation of this feature utilizing a bioinformatics approach. A database was created from all reported fusion proteins from HIV and SIV strains. The hydrophobic loop core is the most conserved sequence in the protein with an E-value of 4.0e−609 based on 75 sites contributing to the construction of the motif (Fig. 1C). The conservation of the two Cys residues was already established. Importantly, we report here that a charged residue (Lys or Arg) in between the Cys residue is a further conserved feature in the loop core.
Alterations in the Hydrophobicity Level of the Disulfide Loop Core
Peptides corresponding to the loop regions were prepared with the mutations K601A (termed L27 K601A) and L602A (termed L27 L602A). Both of the mutations decreased HIV-1 cell fusion when introduced to the inact ENV while keeping similar WT levels of the ENV (23). These mutations change the local hydrophobicity in the disulfide core, whereas K601A or L602A increase or decrease hydrophobicity. The effect of the mutations on the overall hydrophobicity of the loop was analyzed by their retention times in the RP-HPLC and by the calculated grand average of hydropathicity (GRAVY) index. Both methods showed that the overall hydrophobicity of the loop was affected by these mutations in a corresponding manner (Table 1 and Fig. 1D).
TABLE 1.
Designation, sequence, and hydrophobicity values of the peptides used in this study as well as the infectivity rate of each of the mutations
| Designation | gp41 location | Sequence | HIV-1 infectivitya | RP-HPLCb | GRAVY |
|---|---|---|---|---|---|
| % | min | ||||
| L27 WTc | Loop | KDQQLLGIWGCSGKLICTTAVPWNASW | 100 | 20.5 | 0.107 |
| L27 K601A | Loop | KDQQLLGIWGCSGALICTTAVPWNASWd | 54 | 22.3 | 0.319 |
| L27 L602A | Loop | KDQQLLGIWGCSGKAICTTAVPWNASW | 2 | 19.3 | 0.033 |
| L42 WT | Loop | LAVERYLKDQQLLGIWGCSGKLICTTAVPWNASWSNKSLEQI | 100 | N.D | −0.012 |
| N27e | NHR | RQLLSGIVQQQNNLLRAIEAQQHLLQK | 100 | N.D | −0.470 |
a Effect of the Env mutations on viral infectivity. Results are presented in percentages from the infectivity of the WT virus and were taken from (23).
b The peptides were eluted with a flow rate of 0.6 ml/min using a linear gradient from CH3CN/H2O 20:80 (v/v) to CH3CN/H2O 80:20 (v/v) in 40 min on an analytical C4 column (5 μm particle size, 0.46 × 25 cm; GraceVydac, Columbia, MD).
c The sequence corresponds to the loop region (amino acids 588–614 of gp160, HIV-1 HXB2).
d The mutation incorporated in the loop sequence is indicated by a boldface and underlined letter.
e The sequence is taken from Wexler-Cohen et al. (37) and corresponds to the NHR region (amino acids 543–568 of gp160, HIV-1 HXB2 with a Lys residue at the C terminus).
The Hydrophobic Core and Its Basic Residue Participate in the Proper Conformation of the Loop as Revealed by Antibody Binding
Structural changes within proteins can make intrinsic epitopes less accessible to antibody binding. We examined the binding capacity of three monoclonal antibodies to the loop peptides and their mutants by ELISA. An N-peptide (termed N27) derived from the gp41 NHR region was used as a control (the sequence is indicated in Table 1). The first antibody (T32) targets the loop sequence corresponding to the disulfide hydrophobic core, whereas the other antibodies (240-D and 246-D) target a loop sequence outside of the core (Table 2). We utilized the WT loop (L27 WT) and a larger segment that also contains stabilizing flanking elements (L42 WT). We then examined if the flanking elements affect the binding of these antibodies by determining their binding constants, derived from the NLLSQ fitting (Equation 1). Overall, the antibodies exhibited comparable binding affinity both to L27 WT and to L42 WT (Fig. 2 and Table 2). Respectively, the affinity constants were: 4.4 × 106 ± 1.5 × 106 m−1 and 7.7 × 106 ± 3.3 × 106 m−1 (for T32), 4.3 × 107 ± 0.8 × 107 m−1 and 10.3 × 107 ± 2.8 × 107 m−1 (for 246-D), and 1.9 × 107 ± 0.4 × 107 m−1 and 4.9 × 107 ± 0.5 × 107 m−1 (for 240-D). Additionally, none of the antibodies was reactive to the control N27 peptides (Fig. 2 and Table 2).
TABLE 2.
Differential binding capacities of loop-specific monoclonal antibodies to the gp41 loop domain and its analogues
| MAb | Epitope | Locationa | Antibody bindingb |
||||
|---|---|---|---|---|---|---|---|
| L27 WT | L27 K601A | L27 L602A | L42 WT | N27 | |||
| T32 | WGCSGKLICTTAVPWNA | 596–612 | + | − | − | + | − |
| 240-D | IWG | 595–597 | + | + | + | + | − |
| 246-D | LLGI | 592–595 | + | − | + | + | − |
a Residue numbers correspond to the HIV-1 HXB2 gp160 variant.
b + represents antibody binding, and − represents no binding.
FIGURE 2.
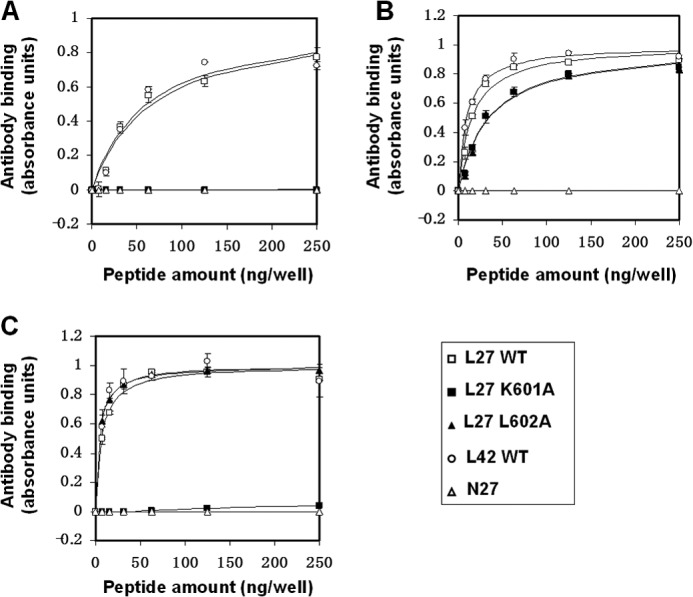
The hydrophobic core and its basic residue participate in the proper conformation of the loop as revealed by antibody binding. Binding capacity of loop-specific monoclonal antibodies to L27 WT loop peptides (□), L27 K601A mutant peptides (■), L27 L602A mutant peptides (▴), L42 WT loop peptides (○), and N27 control peptides (▵) is shown. A, analysis of T32 antibody binding. B, analysis of 240-D antibody binding. C, analysis of 246-D antibody binding. The amount of bound monoclonal antibodies was determined utilizing an ELISA protocol by monitoring the absorbance in 450 nm. Results are the mean ± S.D., n = 2 from a representative experiment out of two experiments. The fitting curves from the NLLSQ model that were used for calculating antibody binding constants are presented.
The mutations K601A and L602A that perturb the epitope sequence of T32 abolished antibody binding probably by altering the local structure of the core, thus preventing direct antibody binding (Fig. 2A). Interestingly, we observed differential binding of the 240-D and 246-D antibodies that target epitopes outside of the core (Fig. 2, B and C, and Table 2). While the 240-D antibody strongly bound the WT loop and its mutants, the 246-D antibody failed to do so. The 246-D antibody bound the WT loop and the L602A but not the K601A mutant. This suggests that increased hydrophobicity resulting from the K601A mutation has a wider effect on the overall conformation of the loop that could prevent the binding of the 246-D antibody.
The Hydrophobic Loop Core and Its Basic Residue Contribute to Disulfide Bond Formation
The WT L27 peptide and its mutants were subjected to oxidation. The amount of free thiol groups was analyzed before and after oxidation by their reactivity to DTNB to analyze the fraction that underwent oxidation. The oxidative process of the loop peptides was also followed by the RP-HPLC. The oxidation process was performed in an aqueous solution or when the L27 peptides were bound to the membrane. The WT L27 peptide was oxidized in an aqueous solution (Fig. 3, A and B). The L602A mutant did not decrease the ability of the loop peptides to form disulfide bonds. However, the incorporation of the mutation K601A decreased the ability of the loop to oxidize (Fig. 3, A and B). Moreover, when the loop peptides were subjected to oxidation in the presence of liposomes, the oxidation process decreased (Fig. 3A). Consequently, the oxidation process of the loop preferably takes place outside the membrane.
FIGURE 3.

The hydrophobic loop core and its basic residue contribute to disulfide bond formation. L27 peptides and their mutants were dissolved in PBS or in a lipid suspension of 100 μm large LUVs to give a concentration of 13.3 μm, and oxidation was monitored for several hours. A, the percentage of oxidation as determined by the difference in DTNB reaction with free thiol groups before and after the oxidation. Black columns represent oxidation in PBS, whereas white columns represent oxidation in the presence of LUVs. Results are the mean ± S.D. (n = 3) of the percentage of the oxidized fraction out of the total amount of the peptide. B, oxidation kinetics were monitored by injecting samples to the RP-HPLC at different time points for the L27 WT loop peptides (■), L27 K601A mutant peptides (▵), and L27 L602A mutant peptides (□). For each time point, the percentage of peptide oxidation (mean ± S.D., n = 3) was determined by calculating the amount of the oxidized peptide divided by the total amount of the peptide injected.
Effect of the Mutations on the Secondary Structure of the Loop Region
We observed differences in antibody binding to the L27 mutant peptides that could result from alteration in the structural organization of the loop imposed by its hydrophobic core. Hence, we examined if such differences existed in the secondary structure of the loop peptides as detected by CD spectroscopy. In HEPES solution, the WT L27 peptide exhibited a random coil structure with a low α-helical content (Fig. 4A). A similar CD spectrum was observed for the L602A mutant peptide. However, a different CD spectrum was detected for the L27 K601A peptide in HEPES solution (Fig. 4A). In a membrane-mimetic environment, the WT L27 peptide adopted an α-helical structure. In this manner, both of the mutant L27 peptides showed differences in the fractional helicity content compared with the WT L27 (Fig. 4B). The L27 (K/A) peptide presented a stronger α-helical structure, whereas the L27 (L/A) peptide exhibited a weaker α-helical structure.
FIGURE 4.
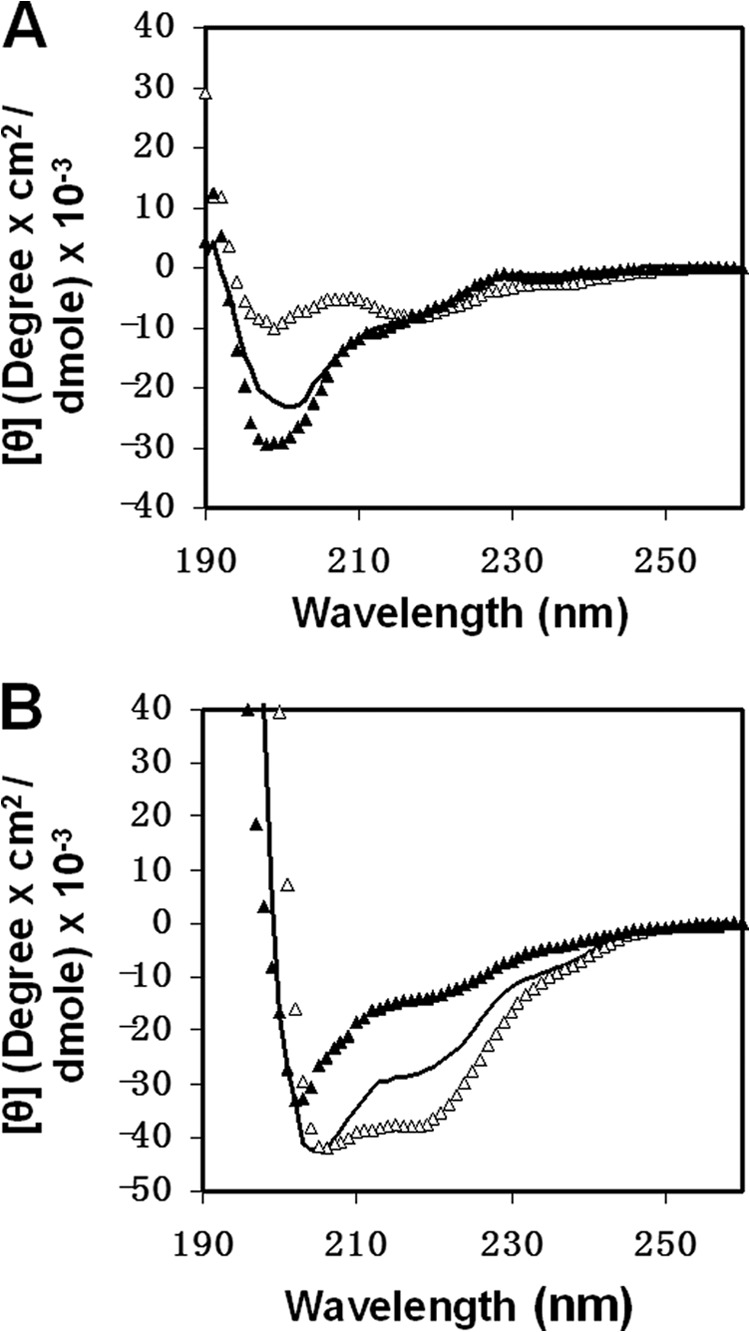
Effect of the mutations on the secondary structure of the loop region utilizing CD spectroscopy. L27 WT peptides (black line), L27 K601A mutant peptides (▵), and L27 L602A mutant peptides (▴) were scanned at a concentration of 10 μm in solution (HEPES 5 mm, pH 7.4) (A) and in a membrane mimetic environment of 1% lysophosphatidylcholine in HEPES (B).
Alterations in the Hydrophobic Core Affect the Binding of the Loop Region to Zwitterionic Membranes
The loop region has the ability to bind and to insert into the membrane. Because the loop core mutations affected the overall hydrophobicity of the peptides, they could modulate the ability of the loop to bind the membrane. Thus, we analyzed the binding affinity of each of the peptides to zwitterionic model membranes of LUVs composed of PC:Chol (9:1) (Fig. 5). Zwitterionic membranes resemble the outer leaflets of both the cell and the viral membranes. The membrane binding constants of the WT L27 peptide, the L27 K601A mutant, and the L27 L602A mutant were 3.8 × 103 ± 0.5 × 103, 7.2 × 103 ± 1.8 × 103, and 1.5 × 103 ± 0.9 × 103 m−1, respectively (results are mean ± S.D., n = 3). The more hydrophobic the peptide, the stronger it binds the membrane.
FIGURE 5.
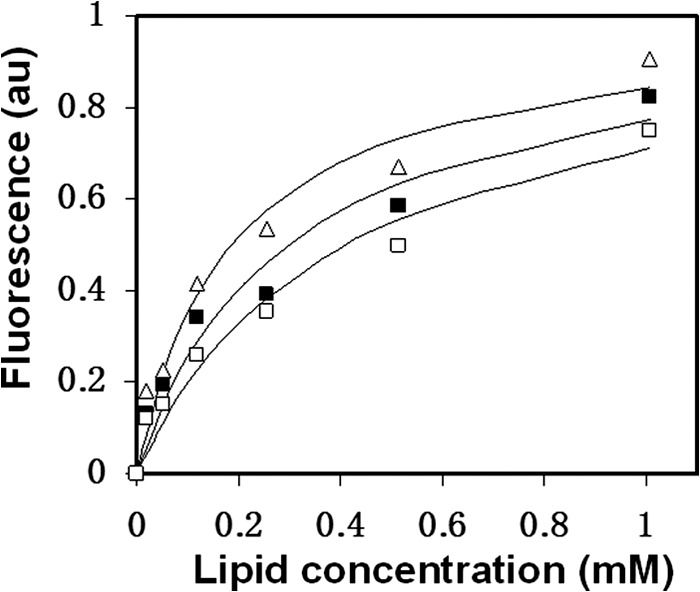
Alterations in the hydrophobic core affect the binding of the loop region to zwitterionic membranes. WT L27 peptides (■), L27 K601A mutant peptides (▵), and L27 L602A mutant peptides (□) were titrated with increasing concentrations of PC:Chol (9:1) LUVs, and changes in fluorescence anisotropy (arbitrary units (au)) of their intrinsic Trp were measured. The fitting curve from the NLLSQ model is presented (Equation 1 under “Experimental Procedures”) that gives the membrane binding affinity constant.
Full Lipid Mixing Mediated by the Loop Region Is Modulated by Its Hydrophobic Core
The ability of the core mutations to interfere with the lipid mixing capacity of the loop peptides was investigated in zwitterionic LUVs composed of PC:Chol (9:1) using a fluorescence probe dilution assay. When the labeled liposomes are fused with unlabeled liposomes to form a new membrane, this results in reduced surface density of the rhodamine energy acceptor. Therefore, the lipid mixing reduces the efficiency of the resonance energy transfer, which is measured by the increase in fluorescence of the NBD serving as an energy donor. We also tracked inner leaflet mixing to analyze whether the increase in fluorescence is due to full lipid mixing or involves mainly outer-leaflet mixing. For this propose, DTH was added to the mixture. The DTH reacts with the NBD in the outer-leaflet resulting in a decrease in the fluorescent signal. Hence, the remaining fluorescent signal after the addition of DTH indicates inner leaflet mixing (Fig. 6, A and B). The results were normalized to the membrane-bound fraction of the peptides that was calculated from the binding curves in Fig. 5.
FIGURE 6.

Full lipid mixing mediated by the loop region is modulated by its hydrophobic core. The loop peptide and its mutants were added to PC:Chol (9:1) LUVs in PBS containing 10% of pre-labeled LUVs (0.6 mol % NBD-PE and Rho-PE) and 90% of unlabeled LUVs. The peptide to lipid molar ratio was 0.05. The increase in NBD fluorescence intensity was measured until a plateau was reached, which represents full lipid mixing capacity and was normalized to the membrane-bound fraction of the peptides. Next, DTH (32 mm) was added to detect the inner leaflet mixing. A, a representative experiment for the loop peptides. B, a representative experiment for the N27 control peptides. C, mean ± S.D. (n = 3) of the percentage of the total lipid mixing of the L27 K601A mutant, L27 L602A mutant, and N27 peptides that was normalized to the WT L27. D, mean ± S.D. (n = 3) of the percentage of inner leaflet mixing out of the total lipid mixing for each of the peptides. au, arbitrary units.
The WT L27 peptide exhibited lipid mixing of zwitterionic LUVs that involves inner leaflet mixing, whereas the control N27 peptide did not induce lipid mixing (Fig. 6). The L27 K601A peptide also induced lipid mixing with inner-leaflet mixing that was 40% higher compared with the WT L27 peptide. However, the L27 L602A peptide had an impaired ability to enhance lipid mixing (Fig. 6, C and D). This suggests that decreased hydrophobicity in the disulfide core alters the ability of the loop region to mediate the direct lipid mixing processes, which are required for membrane fusion.
DISCUSSION
During the early membrane fusion steps, the gp41 loop region is not membrane-embedded (3) and is one of the immunodominant regions of gp41 (32). Only in the late fusion steps, while gp41 acquires the hairpin conformation, is the loop able to insert into the membrane (17, 18, 21). Emerging studies suggest that the loop region and its conserved cysteines participate in the actual lipid mixing process thereby stabilizing the gp41 hairpin conformation (17, 20, 21). We demonstrate a conserved feature in the gp41 loop region that is composed of a basic residue (Lys or Arg) at the center of its hydrophobic disulfide core. We assumed that the existence of such a polar residue in that location allows a certain balance between hydrophobicity and polarity that maintains the proper structure and function of the loop during membrane fusion. We show by mutagenesis analysis that changes in the hydrophobicity of the disulfide core modulate structural and functional properties of loop-derived peptides inside and outside the membrane. This might explain the reduced HIV-1 infectivity that was observed when the corresponding mutations were incorporated in to the viral ENV (23).
Redox changes within the HIV-1 ENV are important for productive membrane fusion (20, 33). Yet studies have failed to directly follow the redox state of the gp41 subunit of the ENV during the membrane fusion process because mutagenesis analysis and reducing agents disrupt upstream events before the fusion step (33, 34). Utilizing loop-derived peptides, we provide evidence that the Lys-601 contributes to disulfide bond formation in the gp41 loop. The K601A mutation increases hydrophobicity in the loop core, thus preventing the ability of the loop peptides to form disulfide bonds. This is due to alterations in the overall conformation of the loop region as observed by the reduced 246-D antibody binding and by the change in the CD spectrum in solution. In the membrane, however, the K601A mutation enhances the ability of the loop to induce lipid mixing of zwitterionic membranes and improves its α-helical structure. Thus, we believe that the increase in hydrophobicity of the loop core, reflected by the K601A mutation, disrupts structural and functional properties of the loop before its insertion into the membrane. Because the K601A mutation reduces but does not abolish viral entry (23), we speculate that the Lys-601 is not the sole contributor for shaping the structure and function of the loop outside the membrane.
On the contrary, we demonstrate that decreased hydrophobicity of the core by the mutation L602A dramatically influences structural and functional properties of the loop region in the membrane. The mutation decreases the α-helical content of the loop in the membrane and diminishes its lipid mixing capacity. Yet no alteration is observed in the ability of the L602A mutant peptides to form disulfide bonds. It is likely that a moderate hydrophobicity level exists in the disulfide loop core. Moderate hydrophobicity allows both oxidation outside the membrane and induction of lipid mixing in the membrane while keeping the proper conformation of the loop in each of the processes. Hence, to generate such moderate hydrophobicity, HIV strategy may be to incorporate a basic residue in the hydrophobic loop core.
Thereupon, the question arises regarding the Lys-601 location during the membrane-bound state of the loop region. The K601A mutation stabilizes the structural and functional properties of the loop in the membrane, suggesting that the insertion of the Lys-601 in the membrane is not favorable. A snorkeling behavior of the Lys-601 might provide the answer. In the late fusion steps the loop region is folded in its looped form, and the helices may lie on or penetrate into the membrane (17). The side chain of lysine can stretch out of the membrane interior to place the charged amino group in the more polar interface region (35). Snorkeling of Arg and Lys lets the peptide penetrate deeper inside the membrane and strongly bind to it (36).
Overall, we suggest that the incorporation of a basic residue in the loop hydrophobic core provides the right balance between hydrophobicity and polarity that contributes to the proper function and structure of the gp41 loop region both outside and inside the membrane. Considering the sequence homology in this feature between different clades of lentiviruses, our findings may be of general utility.
Acknowledgments
We thank Omri Faingold assistance in bioinformatics and Yoel Klug for valuable input.
This work was supported by the Israel Science Foundation (988/09), Science and Technology Planning Project of Guangdong Province, China (2011B050200006), and by the Oversea Study Program of Guangzhou Elite Project, China (a fellowship to J. Q.).
- ENV
- viral envelope protein
- Chol
- cholesterol
- CHR
- C-terminal heptad repeat
- LUV
- large unilamellar vesicle
- NHR
- N-terminal heptad repeat
- PC
- phosphatidylcholine
- SIV
- simian immunodeficiency virus
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- DTH
- sodium dithionite
- Rho-PE
- N-(lissamine rhodamine B sulfonyl)dioleoylphosphatidylethanolamine
- NBD-PE
- N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) dioleoylphosphatidylethanolamine
- NLLSQ
- nonlinear least squares
- RP-HPLC
- reverse phase HPLC.
REFERENCES
- 1. White J. M. (1992) Membrane fusion. Science 258, 917–924 [DOI] [PubMed] [Google Scholar]
- 2. Poranen M. M., Daugelavicius R., Bamford D. H. (2002) Common principles in viral entry. Annu. Rev. Microbiol. 56, 521–538 [DOI] [PubMed] [Google Scholar]
- 3. Eckert D. M., Kim P. S. (2001) Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70, 777–810 [DOI] [PubMed] [Google Scholar]
- 4. Gallo S. A., Finnegan C. M., Viard M., Raviv Y., Dimitrov A., Rawat S. S., Puri A., Durell S., Blumenthal R. (2003) The HIV Env-mediated fusion reaction. Biochim Biophys. Acta 1614, 36–50 [DOI] [PubMed] [Google Scholar]
- 5. Colman P. M., Lawrence M. C. (2003) The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4, 309–319 [DOI] [PubMed] [Google Scholar]
- 6. Jiang S., Lin K., Strick N., Neurath A. R. (1993) HIV-1 inhibition by a peptide. Nature 365, 113. [DOI] [PubMed] [Google Scholar]
- 7. Finzi A., Xiang S. H., Pacheco B., Wang L., Haight J., Kassa A., Danek B., Pancera M., Kwong P. D., Sodroski J. (2010) Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Epand R. M. (2003) Fusion peptides and the mechanism of viral fusion. Biochim Biophys. Acta 1614, 116–121 [DOI] [PubMed] [Google Scholar]
- 9. Freed E. O., Myers D. J., Risser R. (1990) Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. U.S.A. 87, 4650–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan D. C., Fass D., Berger J. M., Kim P. S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263–273 [DOI] [PubMed] [Google Scholar]
- 11. Weissenhorn W., Dessen A., Harrison S. C., Skehel J. J., Wiley D. C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 [DOI] [PubMed] [Google Scholar]
- 12. Caffrey M. (2001) Model for the structure of the HIV gp41 ectodomain. Insight into the intermolecular interactions of the gp41 loop. Biochim Biophys. Acta 1536, 116–122 [DOI] [PubMed] [Google Scholar]
- 13. Caffrey M., Cai M., Kaufman J., Stahl S. J., Wingfield P. T., Covell D. G., Gronenborn A. M., Clore G. M. (1998) Three-dimensional solution structure of the 44-kDa ectodomain of SIV gp41. EMBO J. 17, 4572–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schulz T. F., Jameson B. A., Lopalco L., Siccardi A. G., Weiss R. A., Moore J. P. (1992) Conserved structural features in the interaction between retroviral surface and transmembrane glycoproteins? AIDS Res. Hum. Retroviruses 8, 1571–1580 [DOI] [PubMed] [Google Scholar]
- 15. Gallaher W. R., Ball J. M., Garry R. F., Griffin M. C., Montelaro R. C. (1989) A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res. Hum. Retroviruses 5, 431–440 [DOI] [PubMed] [Google Scholar]
- 16. Moreno M. R., Giudici M., Villalaín J. (2006) The membranotropic regions of the endo and ecto domains of HIV gp41 envelope glycoprotein. Biochim Biophys. Acta 1758, 111–123 [DOI] [PubMed] [Google Scholar]
- 17. Pascual R., Moreno M. R., Villalaín J. (2005) A peptide pertaining to the loop segment of human immunodeficiency virus gp41 binds and interacts with model biomembranes. Implications for the fusion mechanism. J. Virol. 79, 5142–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ashkenazi A., Faingold O., Kaushansky N., Ben-Nun A., Shai Y. (2013) A highly conserved sequence associated with the HIV gp41 loop region is an immunomodulator of antigen-specific T cells in mice. Blood 121, 2244–2252 [DOI] [PubMed] [Google Scholar]
- 19. Bär S., Alizon M. (2004) Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J. Virol. 78, 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashkenazi A., Viard M., Wexler-Cohen Y., Blumenthal R., Shai Y. (2011) Viral envelope protein folding and membrane hemifusion are enhanced by the conserved loop region of HIV-1 gp41. FASEB J. 25, 2156–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashkenazi A., Merklinger E., Shai Y. (2012) Intramolecular interactions within the human immunodeficiency virus-1 gp41 loop region and their involvement in lipid merging. Biochemistry 51, 6981–6989 [DOI] [PubMed] [Google Scholar]
- 22. Peisajovich S. G., Blank L., Epand R. F., Epand R. M., Shai Y. (2003) On the interaction between gp41 and membranes. The immunodominant loop stabilizes gp41 helical hairpin conformation. J. Mol. Biol. 326, 1489–1501 [DOI] [PubMed] [Google Scholar]
- 23. Jacobs A., Sen J., Rong L., Caffrey M. (2005) Alanine scanning mutants of the HIV gp41 loop. J. Biol. Chem. 280, 27284–27288 [DOI] [PubMed] [Google Scholar]
- 24. Maerz A. L., Drummer H. E., Wilson K. A., Poumbourios P. (2001) Functional analysis of the disulfide-bonded loop/chain reversal region of human immunodeficiency virus type 1 gp41 reveals a critical role in gp120-gp41 association. J. Virol. 75, 6635–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merrifield R. B., Vizioli L. D., Boman H. G. (1982) Synthesis of the antibacterial peptide cecropin A (1–33). Biochemistry 21, 5020–5031 [DOI] [PubMed] [Google Scholar]
- 26. Struck D. K., Hoekstra D., Pagano R. E. (1981) Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20, 4093–4099 [DOI] [PubMed] [Google Scholar]
- 27. Lev N., Shai Y. (2007) Fatty acids can substitute the HIV fusion peptide in lipid merging and fusion. An analogy between viral and palmitoylated eukaryotic fusion proteins. J. Mol. Biol. 374, 220–230 [DOI] [PubMed] [Google Scholar]
- 28. Meers P., Ali S., Erukulla R., Janoff A. S. (2000) Novel inner monolayer fusion assays reveal differential monolayer mixing associated with cation-dependent membrane fusion. Biochim Biophys. Acta 1467, 227–243 [DOI] [PubMed] [Google Scholar]
- 29. Xu J. Y., Gorny M. K., Palker T., Karwowska S., Zolla-Pazner S. (1991) Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J. Virol. 65, 4832–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson W. E., Jr., Gorny M. K., Xu J. Y., Mitchell W. M., Zolla-Pazner S. (1991) Two immunodominant domains of gp41 bind antibodies which enhance human immunodeficiency virus type 1 infection in vitro. J. Virol. 65, 4169–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Earl P. L., Broder C. C., Doms R. W., Moss B. (1997) Epitope map of human immunodeficiency virus type 1 gp41 derived from 47 monoclonal antibodies produced by immunization with oligomeric envelope protein. J. Virol. 71, 2674–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zolla-Pazner S. (2004) Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 4, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Markovic I., Stantchev T. S., Fields K. H., Tiffany L. J., Tomiç M., Weiss C. D., Broder C. C., Strebel K., Clouse K. A. (2004) Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry. Blood 103, 1586–1594 [DOI] [PubMed] [Google Scholar]
- 34. Syu W. J., Lee W. R., Du B., Yu Q. C., Essex M., Lee T. H. (1991) Role of conserved gp41 cysteine residues in the processing of human immunodeficiency virus envelope precursor and viral infectivity. J. Virol. 65, 6349–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strandberg E., Killian J. A. (2003) Snorkeling of lysine side chains in transmembrane helices. How easy can it get? FEBS Lett. 544, 69–73 [DOI] [PubMed] [Google Scholar]
- 36. Granseth E., von Heijne G., Elofsson A. (2005) A study of the membrane-water interface region of membrane proteins. J. Mol. Biol. 346, 377–385 [DOI] [PubMed] [Google Scholar]
- 37. Wexler-Cohen Y., Ashkenazi A., Viard M., Blumenthal R., Shai Y. (2010) Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J. 24, 4196–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]