

Abstract
Francisella tularensis is the causative agent of the debilitating febrile illness tularemia. The severe morbidity associated with F. tularensis infections is attributed to its ability to evade the host immune response. Innate immune activation is undetectable until more than 48 hours after infection. The ensuing inflammatory response is considered pathological, eliciting a septic-like state characterized by hypercytokinemia and cell death. To investigate potential pathological consequences of the innate immune response, mice deficient in a key innate immune signaling molecule, MyD88, were studied. MyD88 knockout (KO) mice were infected with the prototypical virulent F. tularensis strain, Schu S4. MyD88 KO mice succumbed to infection more rapidly than wild-type mice. The enhanced pathogenicity of Schu S4 in MyD88 KO mice was associated with greater bacterial burdens in lungs and distal organs, and the absence of IFN-γ in the lungs, spleens, and sera. Cellular infiltrates were not observed on histological evaluation of the lungs, livers, or spleens of MyD88 KO mice, the first KO mouse described with this phenotype to our knowledge. Despite the absence of cellular infiltration, there was more cell death in the lungs of MyD88 KO mice. Thus, the host proinflammatory response is beneficial, and MyD88 signaling is required to limit bacterial burden and prolong survival during pulmonary infection by virulent F. tularensis.
Inhalation of Francisella tularensis results in pneumonic tularemia, which is the most severe form of disease and associated with mortality rates up to 30% to 60% among untreated individuals.1,2 Additionally, its low infectious dose of <10 colony-forming units (CFU) and high morbidity have led to its incorporation into previous governmental bioweapons programs and prompted the Centers for Disease Control and Prevention to list F. tularensis as a Tier 1 select agent.1–3 There are two clinically relevant subspecies of F. tularensis, F. tularensis subsp. tularensis (type A) and F. tularensis subsp. holarctica (type B), with the former being the most infectious and causing the most severe form of disease.4 Due to the restrictive biocontainment needed to work with these strains, attenuated strains such as F. tularensis subsp. holarctica Live Vaccine Strain (LVS) are often used to model the more virulent strains of F. tularensis.
F. tularensis is considered to be a stealth pathogen due to its ability to evade immune detection.5 Following pulmonary exposure, F. tularensis replicates within macrophages and dendritic cells while simultaneously suppressing their activation and thereby limiting the production of proinflammatory cytokines.6–9 Robust bacterial replication and dissemination to distal organs occur during the early stages of infection while there is not a detectable host response.10 The lack of an innate immune response early during infection is important for the extreme virulence of F. tularensis. Administration of nontypeable Haemophilus influenzae or acai berry polysaccharide before infection with F. tularensis activates the innate immune response and prolongs survival of mice.11,12 Additionally, mutants of F. tularensis that stimulate the host immune response more robustly are less virulent in a pulmonary model of tularemia.13 Taken together, F. tularensis is able to evade the host immune response for at least 48 hours after infection. However, early induction of the host response decreases the morbidity of mice infected with F. tularensis, suggesting that the bacterium is susceptible to an activated host response.
Cytokines and recruited immune cells are readily detected in the lungs by 72 hours after infection, but it is uncertain whether this host response is beneficial or detrimental.6,10,14 The immune response has been characterized as a septic-like state with hypercytokinemia in the organs and blood, bacteremia, and depletion of immune cells.15–17 To control LVS infection, a clear role has been established for a variety of cell populations, cytokines, and signaling pathways of the innate immune response, including tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), Toll-like receptor 2 (TLR2), and myeloid differentiation primary response 88 (MyD88).18,19 However, direct assessments of neutrophils, natural killer cells, IFN-γ, and TNF-α during primary tularemia with virulent F. tularensis strains suggest they do not have a comparable role.14,20,21 Production of host matrix metalloprotease 9, which is important for the recruitment of neutrophils, also exacerbates the pathogenesis of F. tularensis.22 However, the septic-like state also occurs in mice infected with attenuated Francisella strains at nonlethal doses of bacteria,14 suggesting that the correlation with death is not causal. There is also a plateau in the bacterial burden subsequent to immune activation, consistent with the host response restricting bacterial growth.14 Using a convalescent model in which antibiotic administration extends the life of the mice infected with F. tularensis, beneficial contributions of IFN-γ, IL-12, T cells, and B cells were established that could not otherwise be observed in a native infection.23 Investigations of the convalescent model suggest that there is a role for the immune response in combating virulent strains of F. tularensis, which contrasts with previous studies investigating the cytokines and cell populations during F. tularensis infection in naive mice.21 Therefore, additional studies investigating the innate host response against virulent F. tularensis are required to clarify the role for this response and to define its contribution to F. tularensis pathogenesis.
The innate immune response in conjunction with the epithelial barrier provides the first line of defense against pathogens. The Toll/IL-1R–receptor (TIR) domain–containing proteins play a crucial role in innate immune responses.24 The TIR family comprises TLRs and IL-1 family receptors, as well as adaptor proteins required for signal transduction from the receptors to induce proinflammatory cytokines.25 MyD88 is a TIR domain–containing adaptor protein for signaling from IL-1 family receptors and all TLRs except for TLR3, which makes MyD88 an important signaling molecule of the initial host response.26 For many bacterial pathogens, the host requires MyD88 signaling to successfully control and clear the infection. MyD88-dependent signaling is often required to prolong survival,27–30 for cellular recruitment and inflammation,27,29–31 and for restriction of bacterial burden.28,31 In circumstances where MyD88 signaling is required, it is almost universally needed for cytokine and chemokine responses.27–29,31–33 By contrast, there is at least one scenario where MyD88 signaling is actually detrimental to the host response.34 Thus, although MyD88 may be important generally, the phenotypic requirement for MyD88 is specific for each pathogen.
The nearly ubiquitous need for MyD88 signaling during the innate host response to microbial infections makes it a good model to explore the contribution of host activation to F. tularensis pathogenesis. Microbiological, immunological, and natural history outcomes were assessed by comparing wild-type (WT) and MyD88 KO mice infected with the prototypical type A strain of F. tularensis, Schu S4, to identify the possible beneficial and detrimental roles of the innate host response. MyD88 KO mice had reduced survival, greater bacterial burdens, and more cell death in the lungs. Collectively, these data demonstrate a beneficial role for the innate immune response during primary pneumonic tularemia with virulent F. tularensis.
Materials and Methods
Bacterial Culture
Francisella tularensis subsp. tularensis Schu S4 was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, National Institute of Allergy and Infectious Diseases: (FSC237), NR-643. Schu S4 was cultured on chocolate agar plates (prepared as previously described13) at 37°C with 5% CO2. Overnight cultures were incubated at 37°C and shaken at 250 rpm in Mueller Hinton Broth (BD Diagnostics, Sparks, MD) supplemented with IsoVitaleX (BD Diagnostics), 0.025% ferric pyrophosphate (Sigma, St. Louis, MO), and 0.1% glucose (Fisher Scientific, Fair Lawn, NJ).
Animal Infections
C57BL/6 mice and mice with a MyD88-deficient allele were purchased from Jackson Laboratory (Bar Harbor, ME) or provided by Dr. Timothy Billiar, and infected as described previously.13 Briefly, approximately 100 CFU in 50 μL of PBS (Life Technologies, Grand Island, NY) were administered intratracheally by oropharyngeal instillation. Blood was removed from mice by cardiac puncture using a heparinized needle and syringe. The lungs were removed and then crushed in RPMI (Lonza, Walkersville, MD) medium supplemented with 10% fetal bovine serum (Life Technologies). The livers and spleens were removed and then homogenized in trypticase soy broth (BD Diagnostics) supplemented with 0.1% cysteine (Fisher Scientific). To quantify CFU, serial dilutions were performed of the organs and blood in the supplemented trypticase soy broth, and the bacteria were plated on chocolate agar plates. Morbidity of mice was followed using a clinical score incorporating both the appearance and the activity of the mice. The activity of the mice was determined as follows: 1, mild reduction in activity; 2, moderate reduction in activity; and 3, severe reduction in activity. The appearance of the mice was scored as: 1, mild piloerection; 2, moderate piloerection; and 3, severe piloerection. The sum of both scores was used, with 0 being a healthy mouse and 4 requiring the mouse to be euthanized. Mice were monitored daily, but once the mice showed signs of illness, they were assessed every 2 to 8 hours, based on the severity of illness.
Cytokine Analysis
To measure cytokine production, lungs were manually homogenized through a cell strainer in RPMI medium supplemented with 10% fetal bovine serum, centrifuged at 21,000 × g for 3 minutes, filtered, and then treated with 100 μg/mL gentamicin (Life Technologies). Plasma was generated from collected blood by centrifugation at 21,000 × g for 3 minutes and then treated with 100 μg/mL gentamicin and 200 μg/mL ciprofloxacin (Enzo Life Sciences, Farmingdale, NY). After spleens were homogenized for enumeration of CFU, they were centrifuged at 21,000 × g for 3 minutes to remove cellular debris. The resulting supernatants were filtered and treated with 100 μg/mL gentamicin. Cytokines from the lung, plasma, and spleens were measured by enzyme-linked immunosorbent assay following the manufacturer’s instructions: IFN-γ (Duoset; R&D Systems, Minneapolis, MN), IL-12, and TNF-α (matched antibody pairs) (eBiosciences, San Diego, CA), and IL-6 (BD Biosciences, San Jose, CA).
Histological Analysis
After sacrificing the animal, lungs were inflated with 37.5% formaldehyde for 10 minutes, and spleens and livers were placed directly into the fixative. The organs were embedded into paraffin blocks and sectioned by the University of Pittsburgh, School of Medicine Histology Core in the Department of Pathology Development Laboratory. The sections were deparaffinized and stained with H&E (Sigma), or immunohistochemistry was performed on the tissues. TUNEL staining was performed following the manufacturer’s protocol (Roche Applied Science, Indianapolis, IN).
Results
MyD88 Signaling Prolongs Survival and Delays Morbidity
There is robust activation of the innate host immune response during infection with F. tularensis after 72 hours, but the impact of the response on pathogenesis remains uncertain.15 To investigate the importance of this innate immune response, WT C57BL/6 and MyD88 knockout (KO) mice were administered 100 CFU of virulent F. tularensis Schu S4 intratracheally to model pulmonary tularemia. The median time to death (MTD) for WT mice was 5.7 days after infection, with 100% of the mice succumbing to infection by day 6 after infection (Figure 1A). By contrast, the MTD for MyD88 KO mice was 4.7 days after infection, with 100% of these mice succumbing to infection by 5.2 days after infection, before the MTD of the WT mice (Figure 1A). Thus, there was a 1-day decrease in the MTD of the MyD88 KO mice, demonstrating a beneficial role for MyD88 during pulmonary tularemia with virulent F. tularensis.
Figure 1.
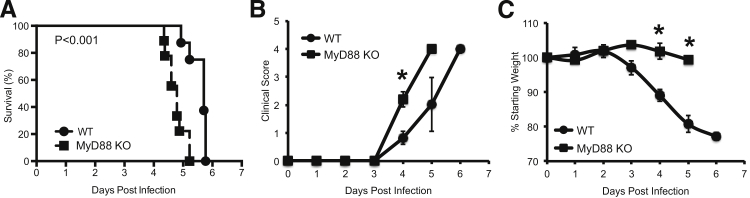
MyD88 prolongs survival during pulmonary tularemia. WT and MyD88 KO mice were infected intratracheally with 100 CFU of Schu S4. A: Survival was assessed as described in Materials and Methods. There were nine mice in the MyD88 KO group and eight mice in the WT group, combined from two independent experiments. Statistical significance was determined using a log-rank test. The morbidity of the mice over the course of the infection was monitored using a clinical score (B) and weight loss (C). These data represent the means ± SEM for two independent experiments. Significance was determined using a two-tailed Student’s t-test to compare clinical score and weight loss. ∗P < 0.05.
The morbidity of mice was used as an additional measure to determine the impact of the innate host response. Mice were monitored over the course of the infection using weight loss and a clinical score, which included both the appearance and activity of the mice. The clinical score for WT or MyD88 KO mice did not change for the first 3 days of the infection. By the fourth day, there was a slight increase in the average clinical score of the WT mice to 0.8 ± 0.2 (Figure 1B). By contrast, there was a significant increase in the morbidity of the MyD88 KO mice by day 4 after infection, resulting in an average clinical score of 2.2 ± 0.3 (Figure 1B). Because of the increased morbidity, 7 of 9 MyD88 KO mice needed to be removed from the study by day 4, but 0 of 8 WT mice needed to be removed before day 5 after infection. In contrast to the clinical score, the weight of MyD88 KO mice did not change (1.7 ± 2.3%), whereas the WT mice lost weight (11 ± 1.7%) by day 4 after infection compared to their starting weights (P < 0.05) (Figure 1C). Overall, these results demonstrate that MyD88 signaling delays the onset of clinical symptoms measured by the clinical score and contributes to weight loss.
The Presence of MyD88 Restricts Bacterial Replication
Activation of the host immune response correlates with stabilization of bacterial burden within the organs of mice.14 To assess the impact of MyD88-dependent processes on bacterial burden, WT and MyD88 KO mice were sacrificed 2 and 4 days after infection for enumeration of CFU. Two days after infection, there was no statistical difference in CFU found in the lungs, livers, spleens, or blood of infected MyD88 KO or WT mice (data not shown). By day 4 after infection, there was over 1 log more CFU in all organs of MyD88 KO mice compared to WT mice (Figure 2). The largest difference was observed in the blood, in which MyD88 KO mice contained 3.5 logs more CFU than WT mice (Figure 2). These data demonstrate that the previously described plateau in bacterial burden is the result of the MyD88-dependent immune responses.
Figure 2.

MyD88-dependent processes restrict bacterial burdens. On day 4 after infection, the lungs, livers, spleens, and blood were harvested from WT and MyD88 KO mice, and bacterial burdens were determined. The data are the means ± SEM for three independent experiments. Significance was determined using a two-tailed Student’s t-test. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
MyD88-Deficient Mice Have an Impaired Cytokine Response
Due to the critical importance of MyD88 for initiating and maintaining a proinflammatory cytokine response, the production of several cytokines was analyzed in the lungs, sera, and spleens of the infected mice. Cytokine concentrations in MyD88 KO and WT mice treated with PBS were not different (data not shown). Additionally, cytokine production on day 2 after infection in Schu S4–infected WT or MyD88 KO mice was similar to PBS-treated WT and MyD88 KO mice (data not shown), which is consistent with previous observations that the inflammatory response is delayed with a Schu S4 infection.10,14,15 Four days after infection, however, there were statistically significant differences in expression of cytokines between WT and MyD88 KO mice (Figure 3). IFN-γ and IL-12 concentrations were at least 1000-fold higher in the lungs, sera, and spleens of WT mice infected with Schu S4 than in uninfected mice or in MyD88 KO mice infected with Schu S4 (Figure 3). IL-6 was readily measured in the lungs, sera, and spleens of both strains of mice after infection, but levels were still 3- to 5-fold lower in the MyD88 KO mice (Figure 3). TNF-α was consistently elevated in the sera of WT mice compared to MyD88 KO mice, but there was not a consistent statistical difference in the spleens or lungs of infected WT or MyD88 KO mice (Figure 3). Overall, cytokine concentrations were higher in the organs and serum of infected WT mice than infected MyD88 KO mice. These data correlate the strength of the proinflammatory response with an increase in the MTD of the mice and a decrease in the bacterial burden during pulmonary tularemia.
Figure 3.

Loss of MyD88 reduces cytokine production in response to F. tularensis. Cytokine response was assessed in the lungs (A), serum (B), and spleens (C) of WT (black bars) and MyD88 KO (grey bars) mice on day 4 after infection. The data are representative of three independent experiments with four to five mice per group for the lungs and sera, and two independent experiments with three mice per group for the spleens. Significance was determined using a Student’s t-test. ∗∗P < 0.01, ∗∗∗P < 0.001. ND, value is below detectable limits.
MyD88 Is Required for the Recruitment of Inflammatory Cells into the Lungs after Infection
A histopathological hallmark of infection with Francisella is the formation of inflammatory foci within the lungs and livers of infected mice.15,21,35–38 To assess the formation of these foci and the recruitment of immune cells into the lungs, livers, and spleens of infected mice, H&E staining was performed on tissue sections collected 4 days after infection. There was no difference in the appearance of lungs, spleens, or livers from mice treated with PBS (Figure 4, A–F). Robust recruitment of immune cells to the alveolar spaces was observed in the lungs of WT mice infected with F. tularensis (Figure 4G), which is consistent with previous studies.15 In contrast to the WT mice, MyD88 KO mice did not have cells visible within the alveolar spaces of the lung (Figure 4J). The lack of cells recruited to the alveolar spaces was consistent with the lower cytokine levels measured in these lungs (Figure 3A). Similar to previous observations,38 the spleens of WT mice were pale on gross pathology, and depletion of the red pulp was visible by H&E staining (Figure 4H). Additionally, there was a loss of white pulp organization, in which there was deterioration in the architecture associated with the marginal zone and follicle (Figure 4H). The regions of red pulp in the spleens of infected WT mice showed the presence of white cells (Figure 4H). By contrast, the spleens of the MyD88 KO mice looked normal in color on gross inspection (data not shown). Despite the normal gross appearance, there was a loss of the marginal zones, but the centers of the follicles appeared mostly intact (Figure 4K). Additionally, there did not appear to be white cells present throughout the red pulp as was observed for WT infected mice; rather, there were more intact erythrocytes present in the MyD88 KO mice compared to WT mice (Figure 4K). Inflammation in the livers of WT mice was consistent with previous reports in which the livers of WT infected mice had small inflammatory foci scattered throughout,38 but MyD88 KO mice did not have similar infiltrates (Figure 4, I and L). In MyD88 KO mice, there were large lesions readily visible throughout the livers of infected MyD88 KO mice (Figure 4L). These lesions comprised particles that stained with hematoxylin, consistent with the size of F. tularensis. These data suggest that the recruitment of inflammatory cells to organs during infection with F. tularensis required MyD88, and without MyD88, there was an altered pathology associated with increased mortality in MyD88 KO mice.
Figure 4.

Altered organ pathology in MyD88 KO mice during F. tularensis infection. Lungs (A, D, G, and J), spleens (B, E, H, and K), and livers (C, F, I, and L) were harvested from MyD88 KO and WT mice on day 4 after infection, fixed in formalin, and then embedded in paraffin. H&E staining was performed to assess histopathological changes within the organs. The images shown are representative of two lungs, two spleens, and two livers for PBS-treated mice. Six lungs, five spleens, and five livers were collected during three independent experiments for Schu S4–treated WT and MyD88 KO mice. Scale bars: 100 μm. Insets are digital magnifications of the boxed areas measuring 50 μm square.
MyD88 Signaling Reduces Cell Death in the Lung
Within the mouse model of pulmonary tularemia, there is depletion of inflammatory cells, particularly NK cells and T cells,14,15 but it is unknown whether the cell death is caused by the bacteria directly or whether it is a result of bystander effects from the host response. To delineate the contribution of the host response to cell death14,15 and the histopathology seen in MyD88 KO mice (Figure 4), tissue sections from mice infected for 4 days with Schu S4 were investigated using a TUNEL assay. Similar to previous findings,15 cell death was observed in the lungs of WT mice infected with F. tularensis (Figure 5, A–D). Both TUNEL staining and F. tularensis were in the same regions of inflammation in the lungs of the WT mice (Figure 5, A–D). Unlike WT mice, F. tularensis antigen was found throughout the lungs of MyD88 KO mice, consistent with the greater bacterial burden (Figure 5, E–H). TUNEL-positive cells were also broadly present throughout the lungs of MyD88 KO mice, including areas near the large airways and structural components of alveoli (Figure 5, E–H). As in the WT, the Francisella antigen staining was associated with regions of TUNEL positivity, though not necessarily colocalized with one another (Figure 5, D and H). Some TUNEL staining in the lungs of both WT and KO mice appeared more diffuse than that of isolated eukaryotic nuclei (Figure 5, D and H). This staining pattern has been observed previously and has been associated with cell death.15
Figure 5.

MyD88 restricts cell death in the lung of F. tularensis–infected mice. The lungs (A–H), spleens (I–P), and livers (Q–X) were harvested from MyD88 KO and WT mice on day 4 after infection, fixed in formalin, and then paraffin embedded. Immunohistochemistry was performed to identify the presence of F. tularensis (red) and cell death by TUNEL staining (green). The images shown are representative of six lungs, five spleens, and five livers collected over three independent experiments. Scale bars: 100 μm. Insets in D and H are digital magnifications measuring 125 μm square. Anti-Ft, F. tularensis antigen staining; DIC, differential interference contrast.
In contrast to the lungs of MyD88 KO mice, the livers and spleens of MyD88 KO mice showed levels of cell death similar to WT mice after infection. The spleens of WT mice showed TUNEL positivity in areas where there was F. tularensis antigen, consistent with previous reports (Figure 5, I–L).15 Similar to what occurred in the WT mice, TUNEL-positive cells also localized to areas with F. tularensis antigen in the spleens of MyD88 KO mice (Figure 5, M–P). Scattered TUNEL-positive inflammatory cells were found in the livers of WT mice infected with Schu S4 (Figure 5, Q–T). F. tularensis antigen staining was limited in these mice, and it was localized to areas of inflammation and TUNEL staining (Figure 5, Q–T). By contrast, the amorphous lesions seen on H&E staining of MyD88 KO liver sections (Figure 4L) stained intensely with F. tularensis antigen (Figure 5, U–X). Similar to what occurred in WT mice, some cells were positive for both TUNEL and F. tularensis antigen, but the vast majority of F. tularensis staining in the livers of MyD88 KO mice did not colocalize with TUNEL-positive cells, including the bacteria comprising the amorphous lesions described above (Figure 5, U–X). Overall, discrete Francisella antigen staining did not colocalize with TUNEL in lungs, spleens, and especially livers, indicating that the TUNEL staining was not from bacterial DNA. Together, these data highlight the organ-specific contributions of MyD88 during infection with F. tularensis, and demonstrate that the presence of MyD88 limits the areas where F. tularensis antigen can be found in the lungs and the livers of infected mice. Finally, these data highlight that within the lung, MyD88 has an additional role of limiting cell death associated with F. tularensis pathogenesis.
Discussion
F. tularensis is a formidable pathogen owing to its ability to infect a diverse range of hosts and evade the immune response.39 Immunological studies using the attenuated strains of F. tularensis recapitulate the organ pathology observed with virulent strains, suggesting they are appropriate models.39–41 The induction of pulmonary tularemia in mice using LVS has revealed that TNF-α, IFN-γ, NK cells, and neutrophils are required for early control of LVS, but clearance and resolution of the infection requires the presence of either CD4 or CD8 T cells.39,40 These host defenses, however, have not translated to the virulent type A or B strains of F. tularensis. SCID, B-cell KO, IFNγ KO, NK cell–depleted, and neutrophil-depleted mice infected with virulent strains of F. tularensis do not have altered survival compared to WT controls.20,21 These studies have led to the conclusion that the innate immune response does not contribute to the control of virulent F. tularensis.21 However, recent evidence demonstrates that following immune activation and immune cell infiltration there is a plateau of bacterial burden in the lung, liver, and spleen suggesting that an innate immune response curtails bacterial replication.14 Here, using virulent F. tularensis in a murine model of pulmonary tularemia, we demonstrate for the first time that early host responses depend on MyD88 to limit bacterial replication and prolong survival of infected mice.
Phenotypes associated with a loss of MyD88 are not apparent until the innate immune response can be detected experimentally. Recent investigations of the immune response to F. tularensis have demonstrated that cytokines or recruited cells are not detected until 3 days after infection. Although a host response could function below the limits of detection of standard assays, assessment of F. tularensis infection 2 days after infection in MyD88 KO and WT mice did not reveal differences in CFU, cytokines, or lung pathology (data not shown). Therefore, any response that might be occurring below the limits of detection did not have an impact on CFU. These data are consistent with previous publications demonstrating that the immune response is suppressed until at least 48 hours after infection.10,14,15 The similarities between WT and MyD88 KO mice on day 2 after infection substantiate the conclusion that F. tularensis is a stealth pathogen until days after infection.
Weight loss can be an indicator of morbidity during infection,13 but results reported here suggest it may be an unreliable measure. MyD88 KO mice maintained their weight over the course of infection, despite significant bacterial burdens. By contrast, WT mice lost >20% of their body weight on average (Figure 1C). These results suggest that the weight loss observed during infection with F. tularensis is dependent on the presence of MyD88 and is likely a result of the greater inflammatory response observed during infection of WT mice with F. tularensis. These results are in agreement with a previous report in which WT mice infected with Clostridium difficile lost more weight than MyD88 KO mice.42 In patients with HIV, weight loss is also correlated with activation of the inflammatory response.43 Finally, LPS-induced anorexia is dependent on MyD88 signaling from myeloid cells.44 Thus, weight loss as a clinical indicator of disease during tularemia is associated with the inflammatory response itself rather than the direct effects of F. tularensis.
Immunological mechanisms responsible for cellular recruitment and the role of these cells during virulent F. tularensis infection are not completely understood. Inflammation develops in the lung and liver commensurate with immune activation, and is typically detectable by 72 hours after infection.14,15,35,38 However, methodological differences, such as administering a lower dose of a different type A strain by aerosol, could affect the amount of pulmonary inflammation observed.38 Matrix metalloprotease-9 (MMP-9) KO mice infected with LVS have reduced neutrophil recruitment in the lung compared to WT controls.22 In the same study, MMP-9–deficient mice were less susceptible to infection with either LVS or Schu S4,22 demonstrating that neutrophil recruitment could be detrimental during infection with both attenuated and virulent F. tularensis strains. IFN-γ mediates formation of focal inflammation in the livers of mice infected with LVS.35 However, opposing results have been obtained with virulent F. tularensis. Different studies have concluded that IFN-γ does21 and does not45 contribute to focal hepatic inflammation. In our experiments, inflammation was consistently present in the lungs and livers of WT mice, but not in MyD88 KO mice (Figure 4). These data demonstrate that MyD88 signaling is necessary for the recruitment of inflammatory cells to the lungs and livers of mice infected with virulent F. tularensis. By contrast, focal inflammation was observed in the livers of MyD88 KO mice infected with the attenuated LVS strain, suggesting that different immunological mechanisms contribute to the host response to these closely related bacteria.18 In conclusion, these studies indicate that although neutrophils specifically may be detrimental, the recruitment of inflammatory cells overall is beneficial and may be causal to controlling replication of virulent F. tularensis.
The MyD88 KO mice also provide insight into how the innate immune response restricts the cell range and location of bacterial replication. F. tularensis infections typically culminate with high bacterial loads in the lungs and distal organs. There is a plateau in the bacterial burden coinciding with activation of the host response,14 but the reason for the limit in bacterial replication could be from depleting the replicative niche or activities of the immune system. In MyD88 KO mice, there were significantly higher bacterial burdens reached in the organs, and as high as 1010 CFU in the liver (Figure 2). These high bacterial burdens were achieved in the absence of recruited cells (Figure 4), which have been hypothesized to expand the available replicative niche for F. tularensis.6 Therefore, these data demonstrate that bacterial replication in WT mice is inhibited by activities of the host response rather than depletion of the niche.
The enhanced cell death seen in F. tularensis–infected MyD88 KO mice indicates the host response is beneficial rather than detrimental. Previous work concluded that overactivation and misdirection of the immune response stimulate cell death. Here, however, cell death was the same (spleen and liver) or greater (lung) in MyD88 KO mice compared to WT (Figure 5). Although these results do not rule out the possibility of a pathological role for inflammation later during infection of WT mice, they support the idea that the host response is beneficial. Our findings corroborate those using the convalescent model of F. tularensis in which beneficial contributions of the host response were identified,23 but also show that MyD88 signaling actually contributes to host cell survival within the lung.
A definitive mechanism of MyD88-dependent protection during pulmonary tularemia remains to be defined. MyD88 functions as an adaptor for multiple TIR-containing receptors, including IL-1 family receptors and TLRs.26 Therefore, MyD88-dependent signaling can drive multiple host responses. F. tularensis is resistant to a variety of host antimicrobials, including oxidative stress, bile salts, and host complement, which suggests that this bacterium must contend with a variety of host defenses.46–48 Recently, it was found that NADPH oxidase contributes to resistance against F. tularensis Schu S4 in vivo.20 MyD88 signaling is important for the assembly of the NADPH oxidase complex.49 In this study, however, the phenotypes associated with a loss of MyD88 were greater in magnitude than in previous studies looking at the pathogenesis of F. tularensis in gp91phox−/− KO mice.20 The general importance of MyD8826 and the overall suppression of cytokines (Figure 3) suggests that the requirement for MyD88 is likely going to be multifaceted and could provide an explanation for why attempts to target specific cytokines or immune cell populations have been unsuccessful in altering the course of infection with virulent F. tularensis.
Despite the beneficial role observed for MyD88 during primary tularemia with virulent F. tularensis strains, the innate immune response is still inadequate to combat infection. This is in part due to bacterial effects that restrict MyD88-dependent, proinflammatory responses. Monocytes infected with Schu S4 down-regulate MyD88 gene expression,50 which would limit the proinflammatory response. There is activation of MKP-1 during LVS infection, which restricts the proinflammatory response through TLR2.51 Bacterial inhibition of MyD88 would exacerbate infection, accelerate mortality, and could explain in part why the MTD decreased by only 1 day in the KO mice.
In conclusion, the work presented here demonstrates a positive role for the host innate immune response during a primary pulmonary infection with virulent F. tularensis. MyD88 signaling is needed for production of IFN-γ and IL-12p40, reaching WT levels of IL-6 and TNF, maintaining the survival of host cells, and the recruitment of immune cells to the sites of F. tularensis replication. The immunological functions that depend on MyD88 restrict the replication of F. tularensis and prolong survival of infected mice. These results highlight a previously unappreciated role for the host response during infection with virulent F. tularensis.
Acknowledgments
We thank Timothy Billiar and Debra Williams for reagents and Carolyn Coyne, Jonathan Proto, and Samantha Slight for technical assistance. The following reagent was obtained through BEI Resources, NIAID, NIH: Francisella tularensis subsp. tularensis, Strain SCHU S4 (FSC237), NR-643.
Footnotes
Supported by NIH grants T32AI060525, “Immunology of Infectious Disease” (B.C.R.), T32 AI049820, “Molecular Microbial Persistence and Pathogenesis” (M.J.B.), and institutional funds from the University of Pittsburgh.
References
- 1.McLendon M.K., Apicella M.A., Allen L.A. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Ann Rev Microbiol. 2006;60:167–185. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennis D.T., Inglesby T.V., Henderson D.A., Bartlett J.G., Ascher M.S., Eitzen E., Fine A.D., Friedlander A.M., Hauer J., Layton M., Lillibridge S.R., McDade J.E., Osterholm M.T., O’Toole T., Parker G., Perl T.M., Russell P.K., Tonat K. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 3.Pohanka M., Skladal P. Bacillus anthracis, Francisella tularensis, and Yersinia pestis. The most important bacterial warfare agents—review. Folia Microbiol (Praha) 2009;54:263–272. doi: 10.1007/s12223-009-0046-1. [DOI] [PubMed] [Google Scholar]
- 4.Petersen J.M., Molins C.R. Subpopulations of Francisella tularensis ssp. tularensis and holarctica: identification and associated epidemiology. Future Microbiol. 2010;5:649–661. doi: 10.2217/fmb.10.17. [DOI] [PubMed] [Google Scholar]
- 5.Sjostedt A. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 2006;8:561–567. doi: 10.1016/j.micinf.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Hall J.D., Woolard M.D., Gunn B.M., Craven R.R., Taft-Benz S., Frelinger J.A., Kawula T.H. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4. LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlson P.E., Jr., Carroll J.A., O’Dee D.M., Nau G.J. Modulation of virulence factors in Francisella tularensis determines human macrophage responses. Microb Pathogs. 2007;42:204–214. doi: 10.1016/j.micpath.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosio C.M., Bielefeldt-Ohmann H., Belisle J.T. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 9.Telepnev M., Golovliov I., Grundstrom T., Tarnvik A., Sjostedt A. Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell Microbiol. 2003;5:41–51. doi: 10.1046/j.1462-5822.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 10.Cowley S.C. Editorial: proinflammatory cytokines in pneumonic tularemia: too much too late? J Leukoc Biol. 2009;86:469–470. doi: 10.1189/jlb.0309119. [DOI] [PubMed] [Google Scholar]
- 11.Evans S.E., Scott B.L., Clement C.G., Larson D.T., Kontoyiannis D., Lewis R.E., Lasala P.R., Pawlik J., Peterson J.W., Chopra A.K., Klimpel G., Bowden G., Hook M., Xu Y., Tuvim M.J., Dickey B.F. Stimulated innate resistance of lung epithelium protects mice broadly against bacteria and fungi. Am J Respiratory Cell Mol Biol. 2010;42:40–50. doi: 10.1165/rcmb.2008-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skyberg J.A., Rollins M.F., Holderness J.S., Marlenee N.L., Schepetkin I.A., Goodyear A., Dow S.W., Jutila M.A., Pascual D.W. Nasal Acai polysaccharides potentiate innate immunity to protect against pulmonary Francisella tularensis and Burkholderia pseudomallei infections. PLoS Pathog. 2012;8:e1002587. doi: 10.1371/journal.ppat.1002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo B.C., Horzempa J., O’Dee D.M., Schmitt D.M., Brown M.J., Carlson P.E., Jr., Xavier R.J., Nau G.J. A Francisella tularensis locus required for spermine responsiveness is necessary for virulence. Infect Immun. 2011;79:3665–3676. doi: 10.1128/IAI.00135-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt D.M., O’Dee D.M., Brown M.J., Horzempa J., Russo B.C., Morel P.A., Nau G.J. Role of NK cells in host defense against pulmonary type A Francisella tularensis infection. Microbes Infect. 2013;15:201–211. doi: 10.1016/j.micinf.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma J., Mares C.A., Li Q., Morris E.G., Teale J.M. Features of sepsis caused by pulmonary infection with Francisella tularensis Type A strain. Microb Pathog. 2011;51:39–47. doi: 10.1016/j.micpath.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mares C.A., Sharma J., Ojeda S.S., Li Q., Campos J.A., Morris E.G., Coalson J.J., Teale J.M. Attenuated response of aged mice to respiratory Francisella novicida is characterized by reduced cell death and absence of subsequent hypercytokinemia. PloS One. 2010;5:e14088. doi: 10.1371/journal.pone.0014088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mares C.A., Ojeda S.S., Morris E.G., Li Q., Teale J.M. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect Immun. 2008;76:3001–3010. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collazo C.M., Sher A., Meierovics A.I., Elkins K.L. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 2006;8:779–790. doi: 10.1016/j.micinf.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Leiby D.A., Fortier A.H., Crawford R.M., Schreiber R.D., Nacy C.A. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect Immun. 1992;60:84–89. doi: 10.1128/iai.60.1.84-89.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.KuoLee R., Harris G., Conlan J.W., Chen W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 2011;13:447–456. doi: 10.1016/j.micinf.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Chen W., KuoLee R., Shen H., Conlan J.W. Susceptibility of immunodeficient mice to aerosol and systemic infection with virulent strains of Francisella tularensis. Microb Pathog. 2004;36:311–318. doi: 10.1016/j.micpath.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Malik M., Bakshi C.S., McCabe K., Catlett S.V., Shah A., Singh R., Jackson P.L., Gaggar A., Metzger D.W., Melendez J.A., Blalock J.E., Sellati T.J. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol. 2007;178:1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- 23.Crane D.D., Scott D.P., Bosio C.M. Generation of a convalescent model of virulent Francisella tularensis infection for assessment of host requirements for survival of tularemia. PloS One. 2012;7:e33349. doi: 10.1371/journal.pone.0033349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar H., Kawai T., Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 25.Chong A., Wehrly T.D., Child R., Hansen B., Hwang S., Virgin H.W., Celli J. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy. 2012;8:1342–1356. doi: 10.4161/auto.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssens S., Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–482. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 27.Sukhumavasi W., Egan C.E., Warren A.L., Taylor G.A., Fox B.A., Bzik D.J., Denkers E.Y. TLR adaptor MyD88 is essential for pathogen control during oral Toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–3473. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biondo C., Midiri A., Messina L., Tomasello F., Garufi G., Catania M.R., Bombaci M., Beninati C., Teti G., Mancuso G. MyD88 and TLR2, but not TLR4, are required for host defense against Cryptococcus neoformans. Eur J Immunol. 2005;35:870–878. doi: 10.1002/eji.200425799. [DOI] [PubMed] [Google Scholar]
- 29.Villamon E., Gozalbo D., Roig P., Murciano C., O’Connor J.E., Fradelizi D., Gil M.L. Myeloid differentiation factor 88 (MyD88) is required for murine resistance to Candida albicans and is critically involved in Candida-induced production of cytokines. Eur Cytokine Netw. 2004;15:263–271. [PubMed] [Google Scholar]
- 30.Skerrett S.J., Liggitt H.D., Hajjar A.M., Wilson C.B. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–3381. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 31.Power M.R., Peng Y., Maydanski E., Marshall J.S., Lin T.J. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J Biol Chem. 2004;279:49315–49322. doi: 10.1074/jbc.M402111200. [DOI] [PubMed] [Google Scholar]
- 32.Sporri R., Joller N., Albers U., Hilbi H., Oxenius A. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J Immunol. 2006;176:6162–6171. doi: 10.4049/jimmunol.176.10.6162. [DOI] [PubMed] [Google Scholar]
- 33.Koedel U., Rupprecht T., Angele B., Heesemann J., Wagner H., Pfister H.W., Kirschning C.J. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. 2004;127:1437–1445. doi: 10.1093/brain/awh171. [DOI] [PubMed] [Google Scholar]
- 34.Weighardt H., Kaiser-Moore S., Vabulas R.M., Kirschning C.J., Wagner H., Holzmann B. Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J Immunol. 2002;169:2823–2827. doi: 10.4049/jimmunol.169.6.2823. [DOI] [PubMed] [Google Scholar]
- 35.Bokhari S.M., Kim K.J., Pinson D.M., Slusser J., Yeh H.W., Parmely M.J. NK cells and gamma interferon coordinate the formation and function of hepatic granulomas in mice infected with the Francisella tularensis live vaccine strain. Infect Immun. 2008;76:1379–1389. doi: 10.1128/IAI.00745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conlan J.W., North R.J. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect Immun. 1992;60:5164–5171. doi: 10.1128/iai.60.12.5164-5171.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindgren H., Stenmark S., Chen W., Tarnvik A., Sjostedt A. Distinct roles of reactive nitrogen and oxygen species to control infection with the facultative intracellular bacterium Francisella tularensis. Infect Immun. 2004;72:7172–7182. doi: 10.1128/IAI.72.12.7172-7182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conlan J.W., Chen W., Shen H., Webb A., KuoLee R. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb Pathog. 2003;34:239–248. doi: 10.1016/s0882-4010(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 39.Cowley S.C., Elkins K.L. Immunity to Francisella. Front Microbiol. 2011;2:26. doi: 10.3389/fmicb.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bosio C.M. The subversion of the immune system by Francisella tularensis. Front Microbiol. 2011;2:9. doi: 10.3389/fmicb.2011.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conlan J.W., Chen W., Bosio C.M., Cowley S.C., Elkins K.L. Infection of mice with Francisella as an immunological model. Curr Protoc Immunol. 2011 doi: 10.1002/0471142735.im1914s93. Chapter 19:Unit 19.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarchum I., Liu M., Shi C., Equinda M., Pamer E.G. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rimaniol A.C., Zylberberg H., Zavala F., Viard J.P. Inflammatory cytokines and inhibitors in HIV infection: correlation between interleukin-1 receptor antagonist and weight loss. AIDS. 1996;10:1349–1356. doi: 10.1097/00002030-199610000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Ruud J., Wilhelms D.B., Nilsson A., Eskilsson A., Tang Y.J., Strohle P., Caesar R., Schwaninger M., Wunderlich T., Backhed F., Engblom D., Blomqvist A. Inflammation- and tumor-induced anorexia and weight loss require MyD88 in hematopoietic/myeloid cells but not in brain endothelial or neural cells. FASEB J. 2013;27:1973–1980. doi: 10.1096/fj.12-225433. [DOI] [PubMed] [Google Scholar]
- 45.Wickstrum J.R., Bokhari S.M., Fischer J.L., Pinson D.M., Yeh H.W., Horvat R.T., Parmely M.J. Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect Immun. 2009;77:4827–4836. doi: 10.1128/IAI.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bina X.R., Lavine C.L., Miller M.A., Bina J.E. The AcrAB RND efflux system from the live vaccine strain of Francisella tularensis is a multiple drug efflux system that is required for virulence in mice. FEMS Microbiol Lett. 2008;279:226–233. doi: 10.1111/j.1574-6968.2007.01033.x. [DOI] [PubMed] [Google Scholar]
- 47.Gil H., Platz G.J., Forestal C.A., Monfett M., Bakshi C.S., Sellati T.J., Furie M.B., Benach J.L., Thanassi D.G. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc Natl Acad Sci U S A. 2006;103:12897–12902. doi: 10.1073/pnas.0602582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raynaud C., Meibom K.L., Lety M.A., Dubail I., Candela T., Frapy E., Charbit A. Role of the wbt locus of Francisella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect Immun. 2007;75:536–541. doi: 10.1128/IAI.01429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laroux F.S., Romero X., Wetzler L., Engel P., Terhorst C. Cutting edge: myD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol. 2005;175:5596–5600. doi: 10.4049/jimmunol.175.9.5596. [DOI] [PubMed] [Google Scholar]
- 50.Butchar J.P., Cremer T.J., Clay C.D., Gavrilin M.A., Wewers M.D., Marsh C.B., Schlesinger L.S., Tridandapani S. Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PloS One. 2008;3:e2924. doi: 10.1371/journal.pone.0002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medina E.A., Morris I.R., Berton M.T. Phosphatidylinositol 3-kinase activation attenuates the TLR2-mediated macrophage proinflammatory cytokine response to Francisella tularensis live vaccine strain. J Immunol. 2010;185:7562–7572. doi: 10.4049/jimmunol.0903790. [DOI] [PubMed] [Google Scholar]